
Catheter Intervention in a Patient with Intracranial Aneurysms and Glanzmann Thrombasthenia Caused by a Novel Homozygous Likely Pathogenic Variant in the ITGA2B Gene

 , ,
, ,  , and
, and {kind=link}
Abstract
1. Introduction
2. Case Presentation
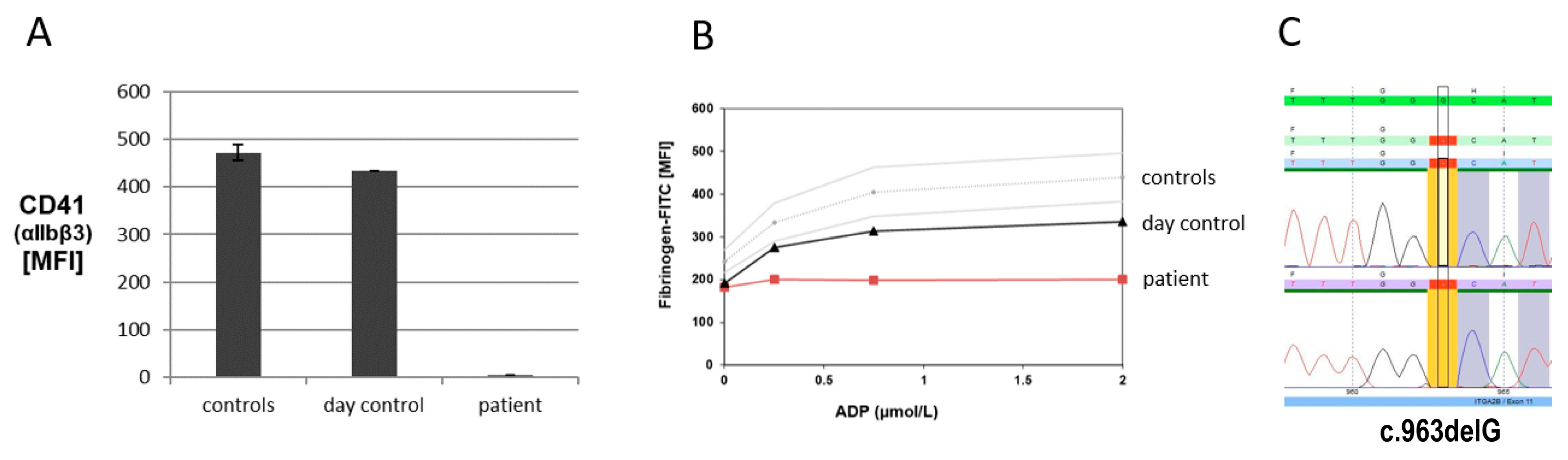
2.1. Platelet Function Analyses and Molecular Genetic Analysis
2.2. Catheter Intervention
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nurden, A.T.; Pillois, X.; Fiore, M.; Alessi, M.C.; Bonduel, M.; Dreyfus, M.; Goudemand, J.; Gruel, Y.; Benabdallah-Guerida, S.; Latger-Cannard, V.; et al. Expanding the Mutation Spectrum Affecting alphaIIbbeta3 Integrin in Glanzmann Thrombasthenia: Screening of the ITGA2B and ITGB3 Genes in a Large International Cohort. Hum. Mutat. 2015, 36, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Sandrock-Lang, K.; Oldenburg, J.; Wiegering, V.; Halimeh, S.; Santoso, S.; Kurnik, K.; Fischer, L.; Tsakiris, D.A.; Sigl-Kraetzig, M.; Brand, B.; et al. Characterisation of patients with Glanzmann thrombasthenia and identification of 17 novel mutations. Thromb. Haemost. 2015, 113, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T. Glanzmann thrombasthenia. Orphanet J. Rare Dis. 2006, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Toogeh, G.; Sharifian, R.; Lak, M.; Safaee, R.; Artoni, A.; Peyvandi, F. Presentation and pattern of symptoms in 382 patients with Glanzmann thrombasthenia in Iran. Am. J. Hematol. 2004, 77, 198–199. [Google Scholar] [CrossRef] [PubMed]
- Botero, J.P.; Lee, K.; Branchford, B.R.; Bray, P.F.; Freson, K.; Lambert, M.P.; Luo, M.; Mohan, S.; Ross, J.E.; Bergmeier, W.; et al. Glanzmann thrombasthenia: Genetic basis and clinical correlates. Haematologica 2020, 105, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, M.Y.J.; Boeckelmann, D.; Naz, A.; Imran, A.; Ahmed, S.; Najmuddin, A.; Zieger, B. Glanzmann Thrombasthenia in Pakistani Patients: Identification of 7 Novel Pathogenic Variants in the Fibrinogen Receptor alphaIIbbeta3. Cells 2023, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.E.; Zhang, B.M.; Lee, K.; Mohan, S.; Branchford, B.R.; Bray, P.; Dugan, S.N.; Freson, K.; Heller, P.G.; Kahr, W.H.A.; et al. Specifications of the variant curation guidelines for ITGA2B/ITGB3: ClinGen Platelet Disorder Variant Curation Panel. Blood Adv. 2021, 5, 414–431. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.; Firah, N.; Pillois, X.; Nurden, P.; Heilig, R.; Nurden, A.T. Natural history of platelet antibody formation against alphaIIbbeta3 in a French cohort of Glanzmann thrombasthenia patients. Haemoph. Off. J. World Fed. Hemoph. 2012, 18, e201–e209. [Google Scholar]
- Siddiq, S.; Clark, A.; Mumford, A. A systematic review of the management and outcomes of pregnancy in Glanzmann thrombasthenia. Haemoph. Off. J. World Fed. Hemoph. 2011, 17, e858–e869. [Google Scholar] [CrossRef] [PubMed]
- Friend, B.D.; Roach, G.D.; Kempert, P.H.; Moore, T.B. Successful Use of Hematopoietic Stem Cell Transplantation for 2 Pediatric Cases of Glanzmann Thrombasthenia and Review of the Literature. J. Pediatr. Hematol./Oncol. 2020, 42, e521–e526. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boeckelmann, D.; von Dobeneck, L.; Henkes, H.; Eichler, H.; Glonnegger, H.; Zieger, B. Catheter Intervention in a Patient with Intracranial Aneurysms and Glanzmann Thrombasthenia Caused by a Novel Homozygous Likely Pathogenic Variant in the ITGA2B Gene. Diseases 2024, 12, 136. https://doi.org/10.3390/diseases12070136
Boeckelmann D, von Dobeneck L, Henkes H, Eichler H, Glonnegger H, Zieger B. Catheter Intervention in a Patient with Intracranial Aneurysms and Glanzmann Thrombasthenia Caused by a Novel Homozygous Likely Pathogenic Variant in the ITGA2B Gene. Diseases. 2024; 12(7):136. https://doi.org/10.3390/diseases12070136
Chicago/Turabian StyleBoeckelmann, Doris, Lara von Dobeneck, Hans Henkes, Hermann Eichler, Hannah Glonnegger, and Barbara Zieger. 2024. "Catheter Intervention in a Patient with Intracranial Aneurysms and Glanzmann Thrombasthenia Caused by a Novel Homozygous Likely Pathogenic Variant in the ITGA2B Gene" Diseases 12, no. 7: 136. https://doi.org/10.3390/diseases12070136
APA StyleBoeckelmann, D., von Dobeneck, L., Henkes, H., Eichler, H., Glonnegger, H., & Zieger, B. (2024). Catheter Intervention in a Patient with Intracranial Aneurysms and Glanzmann Thrombasthenia Caused by a Novel Homozygous Likely Pathogenic Variant in the ITGA2B Gene. Diseases, 12(7), 136. https://doi.org/10.3390/diseases12070136