
Primary Bone Lymphoma: A Review of the Literature with Emphasis on Histopathology and Histogenesis

 ,
,  ,
, 
 , and
, and 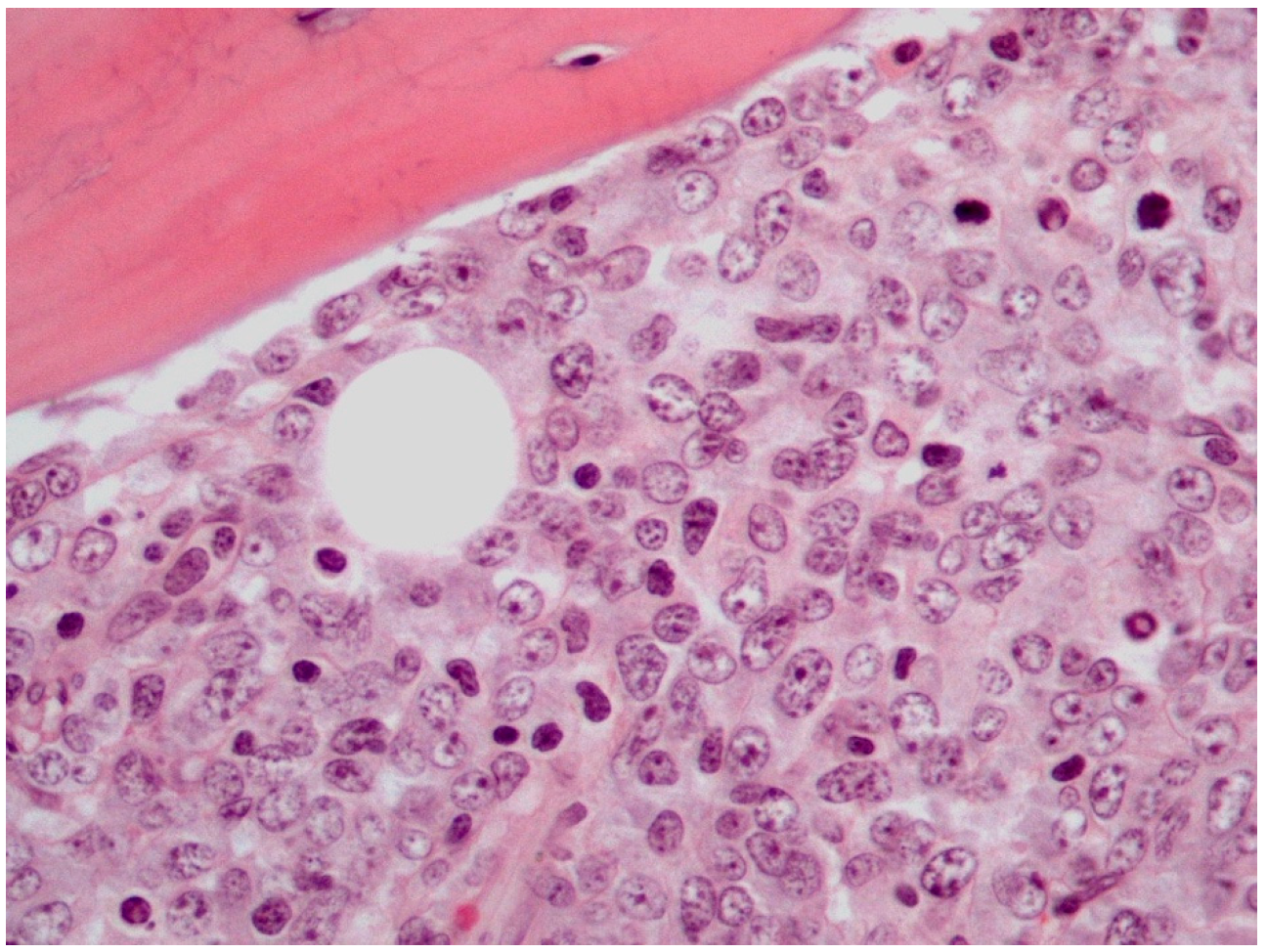{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology
3. Clinical Findings
4. Radiological Findings
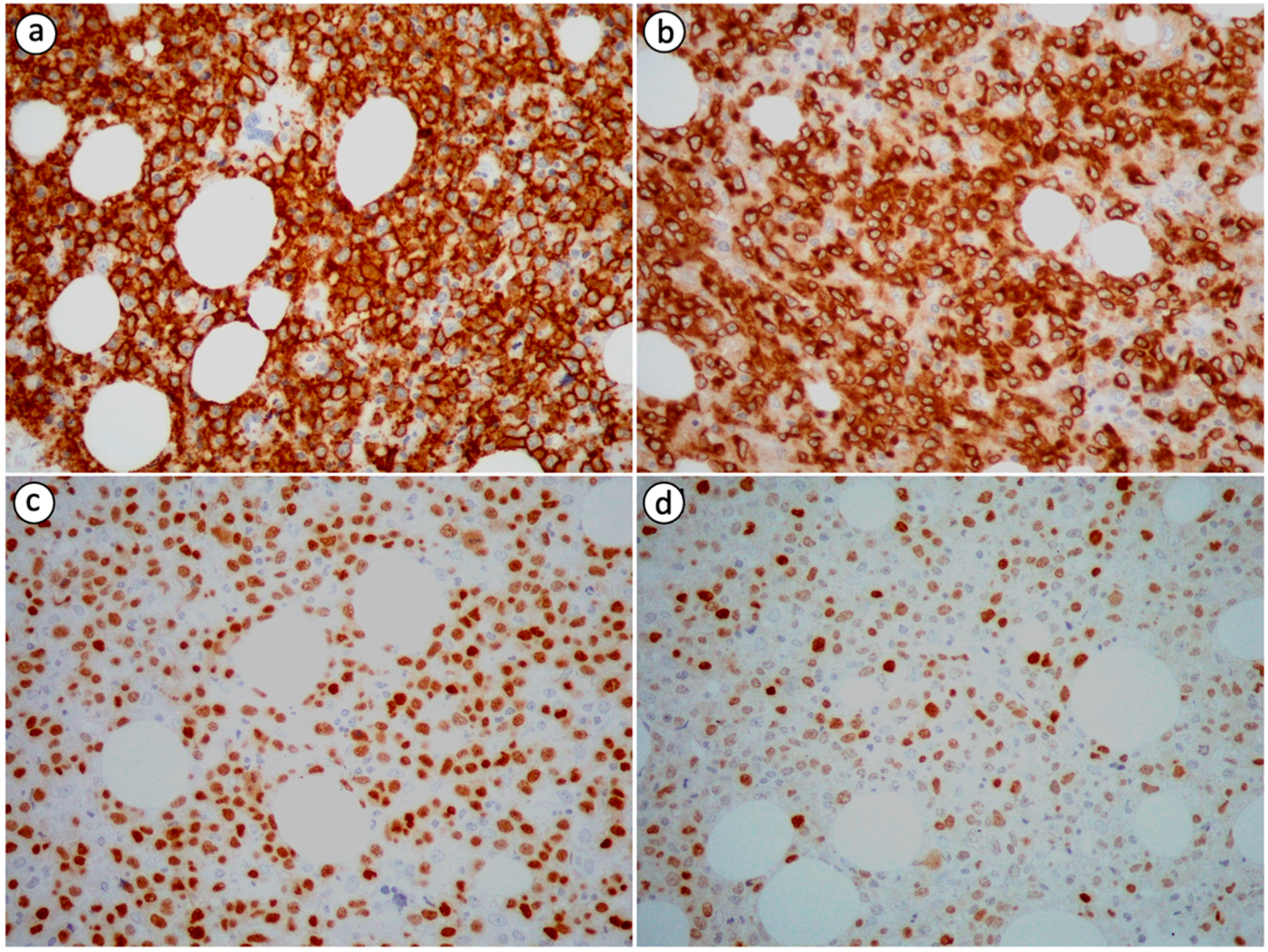
5. Histological Findings
6. Etiology
7. Histogenesis
8. Treatment and Prognosis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centre International de Recherche Sur le Cancer. Soft Tissue and Bone Tumours. In World Health Organization Classification of Tumours, 5th ed.; OMS: Geneva, Switzerland, 2020; ISBN 978-92-832-4502-5. [Google Scholar]
- Cunha, G.; Alçada, M.; Mestre, A.; Duarte, M.B.; Roque, F. Primary Bone Lymphoma: A Rare Cause of Chronic Back Pain. Cureus 2022, 14, e21147. [Google Scholar] [CrossRef] [PubMed]
- Yeom, J.A.; Song, Y.S.; Lee, I.S.; Choi, K.U.; Kim, J.I. A Rare Manifestation of Solitary Primary Bone Lymphoma of the Finger: A Case Report. Investig. Magn. Reason. Imaging 2018, 22, 240. [Google Scholar] [CrossRef]
- Yu, S.; Xu, J. Imaging Features of Primary T Cell Lymphoma in Bone: A Case Report and Review of Literature. Front. Oncol. 2021, 11, 690819. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Bui, M.M.; Leston, D.G.; Shao, H.; Sokol, L.; Sotomayor, E.M.; Zhang, L. Clinical Characteristics and Prognostic Factors of Bone Lymphomas: Focus on the Clinical Significance of Multifocal Bone Involvement by Primary Bone Large B-Cell Lymphomas. BMC Cancer 2014, 14, 900. [Google Scholar] [CrossRef]
- Fletcher, C.D.M. World Health Organization, International Agency for Research on Cancer. WHO Classification of Tumours of Soft Tissue and Bone. In World Health Organization Classification of Tumours, 4th ed.; IARC Press: Lyon, France, 2013; ISBN 978-92-832-2434-1. [Google Scholar]
- Müller, A.; Dreyling, M.; Roeder, F.; Baur-Melnyk, A.; Knösel, T.; Klein, A.; Birkenmaier, C.; Jansson, V.; Dürr, H.R. Primary Bone Lymphoma: Clinical Presentation and Therapeutic Considerations. J. Bone Oncol. 2020, 25, 100326. [Google Scholar] [CrossRef]
- Papageorgiou, S.; Katsikas, T.; Voukelatou, P.; Vrettos, I.; Papanikolaou, A.; Bouchla, A.; Pappa, V.; Kalliakmanis, A. Multiple Osteolytic Lesions Due to Double-Expressor Primary Non-Hodgkin Lymphoma of the Bone. Autops. Case Rep. 2020, 10, e2020141. [Google Scholar] [CrossRef]
- Batia, T.; Yassin, M.A.; Mudawi, D.S.; Hamid, O.A.; Abdalhadi, A.M.A. Primary Bone Lymphoma in Axial Skeleton in a Middle-Aged Female Presented as Recurrent Anemia. Case Rep. Oncol. 2020, 13, 276–280. [Google Scholar] [CrossRef]
- Yang, X.-Y.; He, X.; Zhao, Y. Nomogram-Based Prediction of Overall and Cancer-Specific Survival in Patients with Primary Bone Diffuse Large B-Cell Lymphoma: A Population-Based Study. Evid. Based Complement. Alternat. Med. 2022, 2022, 1566441. [Google Scholar] [CrossRef]
- Ayesh Haj Yousef, M.H.; Audat, Z.; Al-Shorafat, D.M.; Al-Khatib, S.; Daoud, A.K. Primary Diffuse Large B Cell Lymphoma of Bone: A Single-Center Experience. J. Blood Med. 2022, 13, 143–149. [Google Scholar] [CrossRef]
- Behzadi, A.H.; Raza, S.I.; Carrino, J.A.; Kosmas, C.; Gholamrezanezhad, A.; Basques, K.; Matcuk, G.R.; Patel, J.; Jadvar, H. Applications of PET/CT and PET/MR Imaging in Primary Bone Malignancies. PET Clin. 2018, 13, 623–634. [Google Scholar] [CrossRef]
- Jawad, M.U.; Schneiderbauer, M.M.; Min, E.S.; Cheung, M.C.; Koniaris, L.G.; Scully, S.P. Primary Lymphoma of Bone in Adult Patients. Cancer 2010, 116, 871–879. [Google Scholar] [CrossRef]
- Jain, A.; Alam, K.; Maheshwari, V.; Khan, R.; Nobin, H.; Narula, V. Primary Bone Lymphomas-Clinical Cases and Review of Literature. J. Bone Oncol. 2013, 2, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-P.; Tseng, H.-H.; Chang, S.-T.; Fu, T.-Y.; Lu, C.-L.; Chuang, S.-S. Primary Non-Hodgkin’s Lymphoma of Bone: A Rare Disorder with High Frequency of T-Cell Phenotype in Southern Taiwan. Leuk. Lymphoma 2006, 47, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Aozasa, K.; Ohsawa, M.; Yoshikawa, H.; Uchida, A.; Ono, K.; Matsumoto, K. Malignant Lymphomas of Bone in Japan. Cancer 1989, 64, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Poggio, A.D.; Facchetti, L.; Ranza, A.; Facchetti, F.; Pazzaglia, U.; Bondioni, M.P. Primary Lymphoma of the Distal Radius of a Child: Imaging Features. Radiol. Case Rep. 2018, 13, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, M.; Cortina, B.; Gómez, V.; Alvaro-Gracia, J.M.; Reina, T.; Castañeda, S. Aggressive Transformation of a Quiescent Primary Bone Lymphoma Simulating Paget’s Disease. Clin. Exp. Rheumatol. 2008, 26, 133–135. [Google Scholar] [PubMed]
- Kenan, S.; Kahn, L.; Edelman, M.; Redner, A.; Kenan, S. Epiphyseal Primary Diffuse Large B-Cell Lymphoma of Bone. Case Rep. Pathol. 2018, 2018, 4160925. [Google Scholar] [CrossRef]
- Otsuka, Y.; Nakano, Y.; Omura, D.; Hasegawa, K.; Otsuka, F. Primary Bone Lymphoma Presenting as Fever of Unknown Origin. J. Gen. Fam. Med. 2022, 23, 280–281. [Google Scholar] [CrossRef]
- Davies, S.; Sergot, L.; Qamhia, N.; Pawade, J.; Chakraverty, J. Case Report: Primary Bone Lymphoma Presenting as a Painful Supraclavicular Lump. Radiol. Case Rep. 2021, 16, 871–873. [Google Scholar] [CrossRef]
- Bindal, P.; Desai, A.; Delasos, L.; Mulay, S.; Vredenburgh, J. Primary Bone Lymphoma: A Case Series and Review of Literature. Case Rep. Hematol. 2020, 2020, 4254803. [Google Scholar] [CrossRef]
- Inklab, M.; Steingart, R.H.; Freeman, J.K. Primary Lymphoma of Bone Presenting as Spindle Cell Neoplasm of the Vertebral Body: A Case Report and Review of the Literature. Case Rep. Hematol. 2015, 2015, 518307. [Google Scholar] [CrossRef] [PubMed]
- Fyllos, A.; Zibis, A.; Markou, A.; Karantanas, A. Clinical and Imaging Features of Primary Bone Lymphoma: A Pictorial Essay. Hell. J. Radiol. 2021, 6. [Google Scholar] [CrossRef]
- Jadidi, J.; Behzadi, F.; Sighary, M.; Alshal, M.; Kolla, S.; Lehto, S.A. Primary Bone Lymphoma of Patella: A Case Report and Review of Literature. Radiol. Case Rep. 2019, 14, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Gao, Y.; Han, J.; Han, X.; Shi, Y.; Liu, C.; Li, J. Diffuse Large B-Cell Lymphoma of the Mandible Diagnosed by Metagenomic Sequencing: A Case Report. Front. Med. 2021, 8, 752523. [Google Scholar] [CrossRef] [PubMed]
- Flatow-Trujillo, L.; Win, K.; Jencks, A.; Andritsos, L.; Arana Yi, C. Spontaneous Resolution of Untreated Diffuse Large B-Cell Lymphoma of Maxillary Bone after Incisional Biopsy. Clin. Case Rep. 2019, 7, 2082–2086. [Google Scholar] [CrossRef]
- Parker, J.R.; López-Terrada, D.; Gresik, M.V.; Vogel, H.; Baumgartner, J.E.; Finegold, M.J. Neutrophil-Rich Anaplastic Large Cell Lymphoma of the Skull Presenting after Head Trauma. Pediatr. Dev. Pathol. 2001, 4, 397–401. [Google Scholar] [CrossRef]
- Tagawa, M.; Momita, S.; Irie, J. Primary B cell lymphoma of the skull following head trauma: A case report. Rinsho Ketsueki 1987, 28, 589–593. [Google Scholar]
- Kinjo, T.; Satoh, T. A case of malignant lymphoma in the skull after head injury associated with multiple bone tumors. No Shinkei Geka 1985, 13, 1191–1196. [Google Scholar]
- Liu, C.-X.; Xu, T.-Q.; Xu, L.; Wang, P.-P.; Cao, C.; Gao, G.-X.; Zheng, Y.-H. Primary Lymphoma of Bone: A Population-Based Study of 2558 Patients. Ther. Adv. Hematol. 2020, 11, 2040620720958538. [Google Scholar] [CrossRef]
- Krishnan, A.; Shirkhoda, A.; Tehranzadeh, J.; Armin, A.R.; Irwin, R.; Les, K. Primary Bone Lymphoma: Radiographic-MR Imaging Correlation. Radiographics 2003, 23, 1371–1383; discussion 1384–1387. [Google Scholar] [CrossRef]
- Mulligan, M.E.; McRae, G.A.; Murphey, M.D. Imaging Features of Primary Lymphoma of Bone. AJR Am. J. Roentgenol. 1999, 173, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.G.; Gokan, T.; O’Keefe, R.J.; Totterman, S.M.; Fultz, P.J.; Judkins, A.R.; Meyers, S.P.; Rubens, D.J.; Sickel, J.Z.; Rosier, R.N. Primary Lymphoma of Bone. Correlation of Magnetic Resonance Imaging Features with Cytokine Production by Tumor Cells. Cancer 1995, 75, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. The Role of 18F-FDG PET/CT in Staging and Restaging Primary Bone Lymphoma. Nucl. Med. Commun. 2017, 38, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Wu, H.-B.; Wang, M.; Han, Y.-J.; Li, H.-S.; Zhou, W.-L.; Wang, Q.-S. Utility of F-18 FDG PET/CT on the Evaluation of Primary Bone Lymphoma. Eur. J. Radiol. 2015, 84, 2275–2279. [Google Scholar] [CrossRef]
- Stemberga, V.; Dobi-Babić, R.; Bosnar, A.; Cuculić, D.; Fuckar, D.; Stifter, S.; Kusec, R.; Marusić-Vrsalović, M.; Jonjić, N. Primary Non-Hodgkin Lymphoma of the Humerus Following Traumatic Injury: Case Report. Hematol. Oncol. 2003, 21, 109–114. [Google Scholar] [CrossRef]
- Pettit, C.K.; Zukerberg, L.R.; Gray, M.H.; Ferry, J.A.; Rosenberg, A.E.; Harmon, D.C.; Harris, N.L. Primary Lymphoma of Bone. A B-Cell Neoplasm with a High Frequency of Multilobated Cells. Am. J. Surg. Pathol. 1990, 14, 329–334. [Google Scholar] [CrossRef]
- Jaffe, E.S. International Agency for Research on Cancer. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. In World Health Organization Classification of Tumours; IARC Press: Lyon, France, 2001; ISBN 978-92-832-2411-2. [Google Scholar]
- Bacci, G.; Jaffe, N.; Emiliani, E.; Van Horn, J.; Manfrini, M.; Picci, P.; Bertoni, F.; Gherlinzoni, F.; Campanacci, M. Therapy for Primary Non-Hodgkin’s Lymphoma of Bone and a Comparison of Results with Ewing’s Sarcoma. Ten Years’ Experience at the Istituto Ortopedico Rizzoli. Cancer 1986, 57, 1468–1472. [Google Scholar] [CrossRef]
- Bacci, G.; Ferraro, A.; Casadei, R.; Barbieri, E.; Avella, M.; Ferrari, S.; Bertoni, F.; Picci, P.; Ruggieri, P.; Biagini, R. Primary Lymphoma of Bone: Long Term Results in Patients Treated with Vincristine--Adriamycin--Cyclophosphamide and Local Radiation. J. Chemother. 1991, 3, 189–193. [Google Scholar] [CrossRef]
- Mendenhall, N.P.; Jones, J.J.; Kramer, B.S.; Hudson, T.M.; Carter, R.L.; Enneking, W.F.; Marcus, R.B.; Million, R.R. The Management of Primary Lymphoma of Bone. Radiother. Oncol. 1987, 9, 137–145. [Google Scholar] [CrossRef]
- Baar, J.; Burkes, R.L.; Bell, R.; Blackstein, M.E.; Fernandes, B.; Langer, F. Primary Non-Hodgkin’s Lymphoma of Bone. A Clinicopathologic Study. Cancer 1994, 73, 1194–1199. [Google Scholar] [CrossRef]
- Fairbanks, R.K.; Bonner, J.A.; Inwards, C.Y.; Strickler, J.G.; Habermann, T.M.; Unni, K.K.; Su, J. Treatment of Stage IE Primary Lymphoma of Bone. Int. J. Radiat. Oncol. Biol. Phys. 1994, 28, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Dubey, P.; Ha, C.S.; Besa, P.C.; Fuller, L.; Cabanillas, F.; Murray, J.; Hess, M.A.; Cox, J.D. Localized Primary Malignant Lymphoma of Bone. Int. J. Radiat. Oncol. Biol. Phys. 1997, 37, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Dosoretz, D.E.; Murphy, G.F.; Raymond, A.K.; Doppke, K.P.; Schiller, A.L.; Wang, C.C.; Suit, H.D. Radiation Therapy for Primary Lymphoma of Bone. Cancer 1983, 51, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Clayton, F.; Butler, J.J.; Ayala, A.G.; Ro, J.Y.; Zornoza, J. Non-Hodgkin’s Lymphoma in Bone. Pathologic and Radiologic Features with Clinical Correlates. Cancer 1987, 60, 2494–2501. [Google Scholar] [CrossRef]
- Suryanarayan, K.; Shuster, J.J.; Donaldson, S.S.; Hutchison, R.E.; Murphy, S.B.; Link, M.P. Treatment of Localized Primary Non-Hodgkin’s Lymphoma of Bone in Children: A Pediatric Oncology Group Study. J. Clin. Oncol. 1999, 17, 456–459. [Google Scholar] [CrossRef]
- Dosoretz, D.E.; Raymond, A.K.; Murphy, G.F.; Doppke, K.P.; Schiller, A.L.; Wang, C.C.; Suit, H.D. Primary Lymphoma of Bone: The Relationship of Morphologic Diversity to Clinical Behavior. Cancer 1982, 50, 1009–1014. [Google Scholar] [CrossRef]
- Jones, D.; Kraus, M.D.; Dorfman, D.M. Lymphoma Presenting as a Solitary Bone Lesion. Am. J. Clin. Pathol. 1999, 111, 171–178. [Google Scholar] [CrossRef]
- Heyning, F.H.; Hogendoorn, P.C.; Kramer, M.H.; Hermans, J.; Kluin-Nelemans, J.C.; Noordijk, E.M.; Kluin, P.M. Primary Non-Hodgkin’s Lymphoma of Bone: A Clinicopathological Investigation of 60 Cases. Leukemia 1999, 13, 2094–2098. [Google Scholar] [CrossRef]
- Yuste, A.L.; Segura, A.; López-Tendero, P.; Gironés, R.; Montalar, J.; Gómez-Codina, J. Primary Lymphoma of Bone: A Clinico-Pathological Review and Analysis of Prognostic Factors. Leuk. Lymphoma 2004, 45, 853–855. [Google Scholar] [CrossRef]
- Luna-Ortiz, K.; Cervera-Ceballos, E.; Dominguez-Malagon, H.; Labardini-Mendez, J.; De la Garza-Salazar, J.; Herrera-Gomez, A.; Barrera-Franco, J.L. Primary Lymphoma of Bone. Rev. Invest. Clin. 2003, 55, 502–506. [Google Scholar]
- Brousse, C.; Baumelou, E.; Morel, P. Primary Lymphoma of Bone: A Prospective Study of 28 Cases. Jt. Bone Spine 2000, 67, 446–451. [Google Scholar]
- Misgeld, E.; Wehmeier, A.; Krömeke, O.; Gattermann, N. Primary Non-Hodgkin’s Lymphoma of Bone: Three Cases and a Short Review of the Literature. Ann. Hematol. 2003, 82, 440–443. [Google Scholar] [CrossRef] [PubMed]
- De Leval, L.; Braaten, K.M.; Ancukiewicz, M.; Kiggundu, E.; Delaney, T.; Mankin, H.J.; Harris, N.L. Diffuse Large B-Cell Lymphoma of Bone: An Analysis of Differentiation-Associated Antigens with Clinical Correlation. Am. J. Surg. Pathol. 2003, 27, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Carrillo, G.; Ascani, S.; Barbieri, E.; Tani, M.; Paulli, M.; Stefoni, V.; Sabattini, E.; Alinari, L.; Binazzi, R.; et al. Primary Bone Lymphoma: Experience with 52 Patients. Haematologica 2003, 88, 280–285. [Google Scholar]
- Deshmukh, C.; Bakshi, A.; Parikh, P.; Nair, R.; Pai, V.; Gupta, S.; Shaikh, A.; Muckaden, M.; Naresh, K.; Saikia, T. Primary Non-Hodgkin’s Lymphoma of the Bone: A Single Institution Experience. Med. Oncol. 2004, 21, 263–267. [Google Scholar] [CrossRef]
- Gianelli, U.; Patriarca, C.; Moro, A.; Ponzoni, M.; Giardini, R.; Massimino, M.; Alfano, R.M.; Armiraglio, E.; Nuciforo, P.; Bosari, S.; et al. Lymphomas of the Bone: A Pathological and Clinical Study of 54 Cases. Int. J. Surg. Pathol. 2002, 10, 257–266. [Google Scholar] [CrossRef]
- Charousset, C.; Anract, P.; Carlioz, B.; Babinet, A.; Tomeno, B. Primary bone lymphoma. Retrospective immunohistochemical study of 22 cases. Rev. Chir. Orthop. Reparatrice. Appar. Mot. 2002, 88, 439–448. [Google Scholar]
- Ostrowski, M.L.; Unni, K.K.; Banks, P.M.; Shives, T.C.; Evans, R.G.; O’Connell, M.J.; Taylor, W.F. Malignant Lymphoma of Bone. Cancer 1986, 58, 2646–2655. [Google Scholar] [CrossRef]
- Edeiken-Monroe, B.; Edeiken, J.; Kim, E.E. Radiologic Concepts of Lymphoma of Bone. Radiol. Clin. N. Am. 1990, 28, 841–864. [Google Scholar] [CrossRef]
- Pileri, S.A.; Montanari, M.; Falini, B.; Poggi, S.; Sabattini, E.; Baglioni, P.; Bacchini, P.; Bertoni, F. Malignant Lymphoma Involving the Mandible. Clinical, Morphologic, and Immunohistochemical Study of 17 Cases. Am. J. Surg. Pathol. 1990, 14, 652–659. [Google Scholar] [CrossRef]
- Chan, J.K.; Ng, C.S.; Hui, P.K.; Leung, W.T.; Sin, V.C.; Lam, T.K.; Chick, K.W.; Lam, W.Y. Anaplastic Large Cell Ki-1 Lymphoma of Bone. Cancer 1991, 68, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.L.; Inwards, C.Y.; Strickler, J.G.; Witzig, T.E.; Wenger, D.E.; Unni, K.K. Osseous Hodgkin Disease. Cancer 1999, 85, 1166–1178. [Google Scholar] [CrossRef]
- Ozdemirli, M.; Fanburg-Smith, J.C.; Hartmann, D.P.; Shad, A.T.; Lage, J.M.; Magrath, I.T.; Azumi, N.; Harris, N.L.; Cossman, J.; Jaffe, E.S. Precursor B-Lymphoblastic Lymphoma Presenting as a Solitary Bone Tumor and Mimicking Ewing’s Sarcoma: A Report of Four Cases and Review of the Literature. Am. J. Surg. Pathol. 1998, 22, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Iravani, S.; Singleton, T.P.; Ross, C.W.; Schnitzer, B. Precursor B Lymphoblastic Lymphoma Presenting as Lytic Bone Lesions. Am. J. Clin. Pathol. 1999, 112, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Huebner-Chan, D.; Fernandes, B.; Yang, G.; Lim, M.S. An Immunophenotypic and Molecular Study of Primary Large B-Cell Lymphoma of Bone. Mod. Pathol. 2001, 14, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu-Monette, Z.Y.; Yi, S.; Dabaja, B.S.; Manyam, G.C.; Westin, J.; Fowler, N.; Miranda, R.N.; Zhang, M.; Ferry, J.A.; et al. Primary Bone Lymphoma Exhibits a Favorable Prognosis and Distinct Gene Expression Signatures Resembling Diffuse Large B-Cell Lymphoma Derived from Centrocytes in the Germinal Center. Am. J. Surg. Pathol. 2017, 41, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Bhagavathi, S.; Fu, K. Primary Bone Lymphoma. Arch. Pathol. Lab. Med. 2009, 133, 1868–1871. [Google Scholar] [CrossRef]
- Bhagavathi, S.; Micale, M.A.; Les, K.; Wilson, J.D.; Wiggins, M.L.; Fu, K. Primary Bone Diffuse Large B-Cell Lymphoma: Clinicopathologic Study of 21 Cases and Review of Literature. Am. J. Surg. Pathol. 2009, 33, 1463–1469. [Google Scholar] [CrossRef]
- Lima, F.P.; Bousquet, M.; Gomez-Brouchet, A.; de Paiva, G.R.; Amstalden, E.M.I.; Soares, F.A.; Dastugue, N.; Vassallo, J.; Brousset, P. Primary Diffuse Large B-Cell Lymphoma of Bone Displays Preferential Rearrangements of the c-MYC or BCL2 Gene. Am. J. Clin. Pathol. 2008, 129, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, K.M.; Shenkier, T.; Sehn, L.H.; Gascoyne, R.D.; Connors, J.M. A Clinicopathological Retrospective Study of 131 Patients with Primary Bone Lymphoma: A Population-Based Study of Successively Treated Cohorts from the British Columbia Cancer Agency. Ann. Oncol. 2007, 18, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, D.; Watanabe, T.; Beppu, Y.; Kobayashi, Y.; Kim, S.-W.; Tanimoto, K.; Makimoto, A.; Kagami, Y.; Terauchi, T.; Matsuno, Y.; et al. Primary Bone Lymphoma: A New and Detailed Characterization of 28 Patients in a Single-Institution Study. Jpn. J. Clin. Oncol. 2007, 37, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Nayak, H.K.; Nishant, R.; Sinha, N.K.; Daga, M.K. Anaplastic Large T-Cell Lymphoma Presenting as an Isolated Osseous Involvement: A Case Report and Review of the Literature. BMJ Case Rep. 2013, 2013, bcr2013009308. [Google Scholar] [CrossRef]
- Kitsoulis, P.; Vlychou, M.; Papoudou-Bai, A.; Karatzias, G.; Charchanti, A.; Agnantis, N.J.; Bai, M. Primary Lymphomas of Bone. Anticancer Res. 2006, 26, 325–337. [Google Scholar] [PubMed]
- Tian, C.; Wang, Y.; Zhang, Y. ALK-Positive Anaplastic Large Cell Lymphoma with Prominent Bone Involvement. Br. J. Haematol. 2015, 170, 443. [Google Scholar] [CrossRef]
- Mika, J.; Schleicher, I.; Gerlach, U.; Adler, C.-P.; Uhl, M.; Knoeller, S.M. Primary Bone Lymphomas Thought to Be Osteomyelitis Urgently Demand a Rapid Diagnosis in Bone Pathology. Anticancer Res. 2012, 32, 4905–4912. [Google Scholar]
- Romero-Rojas, A.E.; Diaz-Perez, J.A.; Raju, S.; Messa-Botero, O.; Prieto-Bletan, A.; Criollo-Palacios, F. Primary Diffuse Large B-Cell Lymphoma Associated with Chronic Osteomyelitis of the Knee. Knee 2014, 21, 1280–1283. [Google Scholar] [CrossRef]
- Jellicoe, P.; Hopyan, S. Can Chronic Recurrent Multifocal Osteomyelitis Predispose to Lymphoma of Bone? A Case Report. J. Pediatr. Orthop. B 2008, 17, 329–332. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Hunter, E.; McCord, R.; Ramadass, A.S.; Green, J.; Westra, J.W.; Mundt, K.; Akoulitchev, A. Comparative Molecular Cell-of-Origin Classification of Diffuse Large B-Cell Lymphoma Based on Liquid and Tissue Biopsies. Transl. Med. Commun. 2020, 5, 5. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted Sequencing in DLBCL, Molecular Subtypes, and Outcomes: A Haematological Malignancy Research Network Report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the Molecular Classification of Diffuse Large B-Cell Lymphoma by Immunohistochemistry Using a Tissue Microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Dybkær, K.; Bøgsted, M.; Falgreen, S.; Bødker, J.S.; Kjeldsen, M.K.; Schmitz, A.; Bilgrau, A.E.; Xu-Monette, Z.Y.; Li, L.; Bergkvist, K.S.; et al. Diffuse Large B-Cell Lymphoma Classification System That Associates Normal B-Cell Subset Phenotypes with Prognosis. J. Clin. Oncol. 2015, 33, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- De Groot, F.A.; de Groen, R.A.L.; van den Berg, A.; Jansen, P.M.; Lam, K.H.; Mutsaers, P.G.N.J.; van Noesel, C.J.M.; Chamuleau, M.E.D.; Stevens, W.B.C.; Plaça, J.R.; et al. Biological and Clinical Implications of Gene-Expression Profiling in Diffuse Large B-Cell Lymphoma: A Proposal for a Targeted BLYM-777 Consortium Panel as Part of a Multilayered Analytical Approach. Cancers 2022, 14, 1857. [Google Scholar] [CrossRef]
- De Groen, R.A.L.; van Eijk, R.; Böhringer, S.; van Wezel, T.; Raghoo, R.; Ruano, D.; Jansen, P.M.; Briaire-de Bruijn, I.; de Groot, F.A.; Kleiverda, K.; et al. Frequent Mutated B2M, EZH2, IRF8, and TNFRSF14 in Primary Bone Diffuse Large B-Cell Lymphoma Reflect a GCB Phenotype. Blood Adv. 2021, 5, 3760–3775. [Google Scholar] [CrossRef]
- Zajączkowska, R.; Kocot-Kępska, M.; Leppert, W.; Wordliczek, J. Bone Pain in Cancer Patients: Mechanisms and Current Treatment. Int. J. Mol. Sci. 2019, 20, 6047. [Google Scholar] [CrossRef]
- Mantyh, P.W. Bone Cancer Pain: From Mechanism to Therapy. Curr. Opin. Support. Palliat. Care 2014, 8, 83–90. [Google Scholar] [CrossRef]
- Bruno Ventre, M.; Ferreri, A.J.M.; Gospodarowicz, M.; Govi, S.; Messina, C.; Porter, D.; Radford, J.; Heo, D.S.; Park, Y.; Martinelli, G.; et al. Clinical Features, Management, and Prognosis of an International Series of 161 Patients with Limited-Stage Diffuse Large B-Cell Lymphoma of the Bone (the IELSG-14 Study). Oncologist 2014, 19, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Christie, D.; Dear, K.; Le, T.; Barton, M.; Wirth, A.; Porter, D.; Roos, D.; Pratt, G. Limited Chemotherapy and Shrinking Field Radiotherapy for Osteolymphoma (Primary Bone Lymphoma): Results from the Trans-Tasman Radiation Oncology Group 99.04 and Australasian Leukaemia and Lymphoma Group LY02 Prospective Trial. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Beal, K.; Allen, L.; Yahalom, J. Primary Bone Lymphoma: Treatment Results and Prognostic Factors with Long-Term Follow-up of 82 Patients. Cancer 2006, 106, 2652–2656. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, Y.; Li, Z.; Yan, H.; Xia, L.; Shi, W.; Hu, Y. Role of Radiation Therapy Differs Between Stages in Primary Bone Large B-Cell Lymphoma in Rituximab Era: A Population-Based Analysis. Front. Oncol. 2020, 10, 1157. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Gau, J.-P.; Yu, Y.-B.; Hong, Y.-C.; Yen, C.-C.; Liu, C.-Y.; Chao, T.-C.; Hsiao, L.-T.; Liu, J.-H.; Chiou, T.-J.; et al. Prognostic Factors and Treatment Efficacy in Patients with Primary Diffuse Large B-Cell Lymphoma of the Bone: Single Institute Experience over 11 Years. Intern. Med. 2014, 53, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Messina, C.; Ferreri, A.J.M.; Govi, S.; Bruno-Ventre, M.; Gracia Medina, E.A.; Porter, D.; Radford, J.; Heo, D.S.; Park, H.Y.; Pro, B.; et al. Clinical Features, Management and Prognosis of Multifocal Primary Bone Lymphoma: A Retrospective Study of the International Extranodal Lymphoma Study Group (the IELSG 14 Study). Br. J. Haematol. 2014, 164, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Demircay, E.; Hornicek, F.J.; Mankin, H.J.; Degroot, H. Malignant Lymphoma of Bone: A Review of 119 Patients. Clin. Orthop. Relat. Res. 2013, 471, 2684–2690. [Google Scholar] [CrossRef]
- Horsman, J.M.; Thomas, J.; Hough, R.; Hancock, B.W. Primary Bone Lymphoma: A Retrospective Analysis. Int. J. Oncol. 2006, 28, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Messina, C.; Christie, D.; Zucca, E.; Gospodarowicz, M.; Ferreri, A.J.M. Primary and Secondary Bone Lymphomas. Cancer Treat. Rev. 2015, 41, 235–246. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, L.; Shao, H.; Sokol, L.; Sotomayor, E.; Letson, D.; Bui, M.M. Prognostic Significance of Soft Tissue Extension, International Prognostic Index, and Multifocality in Primary Bone Lymphoma: A Single Institutional Experience. Br. J. Haematol. 2014, 166, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Scoccianti, G.; Rigacci, L.; Puccini, B.; Campanacci, D.A.; Simontacchi, G.; Bosi, A.; Capanna, R. Primary Lymphoma of Bone: Outcome and Role of Surgery. Int. Orthop. 2013, 37, 2437–2442. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanavos, T.; Birbas, E.; Papoudou-Bai, A.; Hatzimichael, E.; Kitsouli, A.; Karpathiou, G.; Kanavaros, P. Primary Bone Lymphoma: A Review of the Literature with Emphasis on Histopathology and Histogenesis. Diseases 2023, 11, 42. https://doi.org/10.3390/diseases11010042
Kanavos T, Birbas E, Papoudou-Bai A, Hatzimichael E, Kitsouli A, Karpathiou G, Kanavaros P. Primary Bone Lymphoma: A Review of the Literature with Emphasis on Histopathology and Histogenesis. Diseases. 2023; 11(1):42. https://doi.org/10.3390/diseases11010042
Chicago/Turabian StyleKanavos, Theofilos, Effrosyni Birbas, Alexandra Papoudou-Bai, Eleftheria Hatzimichael, Aikaterini Kitsouli, Georgia Karpathiou, and Panagiotis Kanavaros. 2023. "Primary Bone Lymphoma: A Review of the Literature with Emphasis on Histopathology and Histogenesis" Diseases 11, no. 1: 42. https://doi.org/10.3390/diseases11010042
APA StyleKanavos, T., Birbas, E., Papoudou-Bai, A., Hatzimichael, E., Kitsouli, A., Karpathiou, G., & Kanavaros, P. (2023). Primary Bone Lymphoma: A Review of the Literature with Emphasis on Histopathology and Histogenesis. Diseases, 11(1), 42. https://doi.org/10.3390/diseases11010042