
An Effective Deep Learning-Based Architecture for Prediction of N7-Methylguanosine Sites in Health Systems



Abstract
:1. Introduction
2. Methods and Materials
2.1. Benchmark Dataset
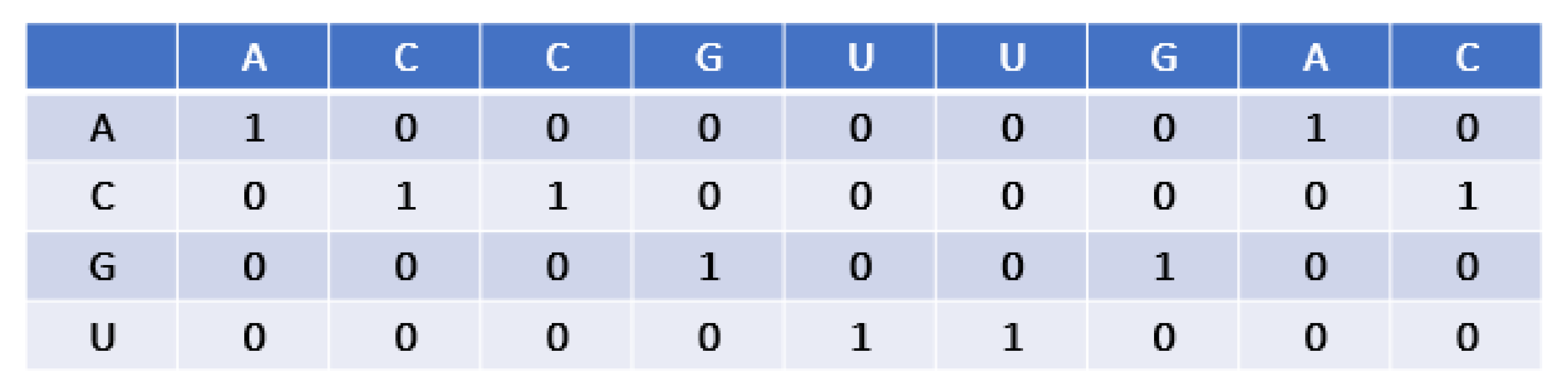
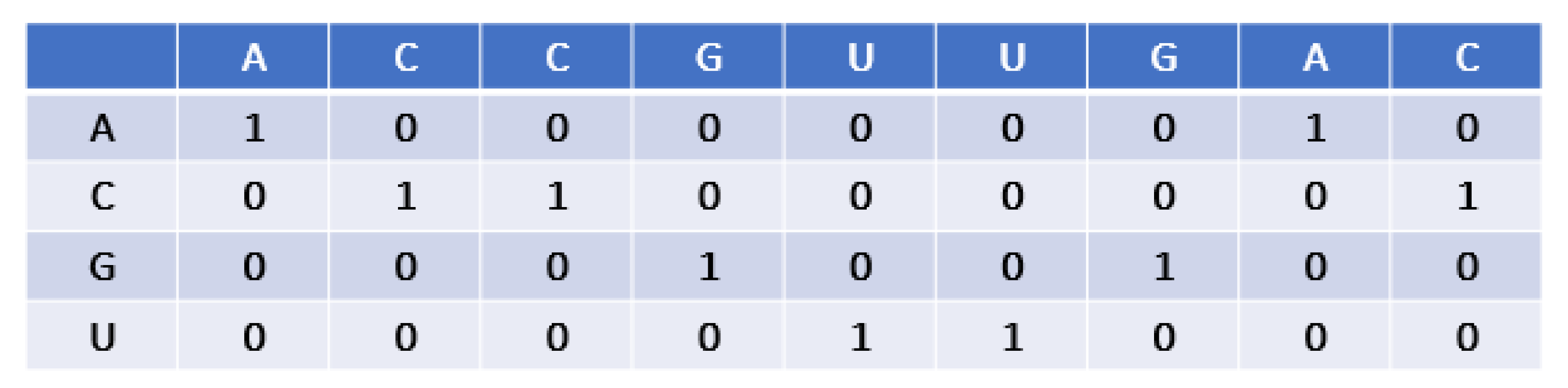
2.2. Encoding Scheme
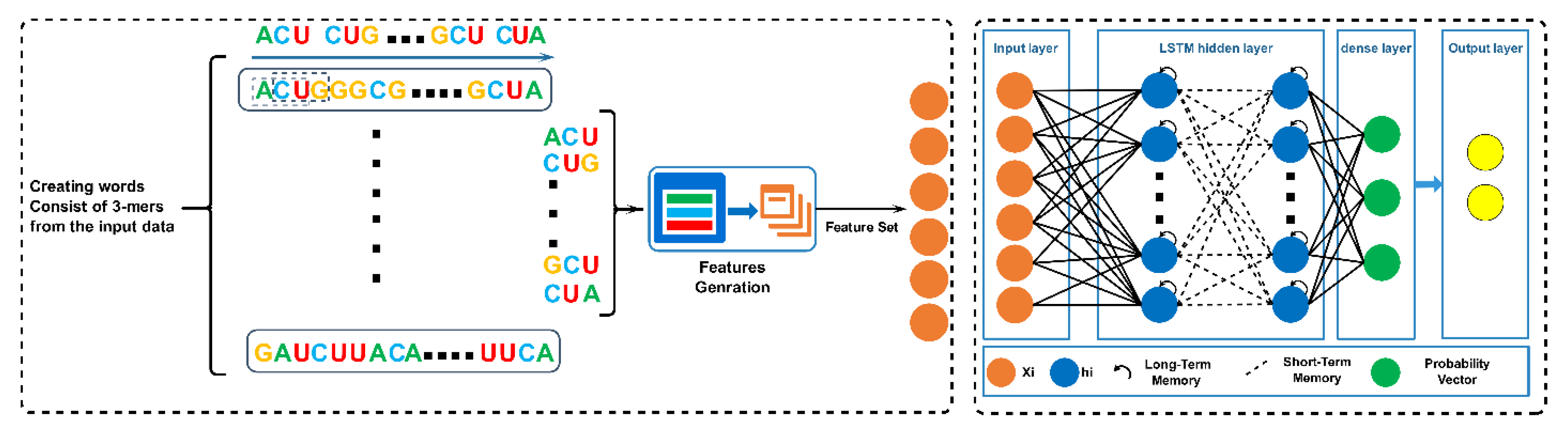
2.3. Distributed Attributes Formulation
2.4. Convolutional Neural Networks (CNN)
2.5. Long Short-Term Memory Layer (LSTM)
2.6. Evaluation Parameters
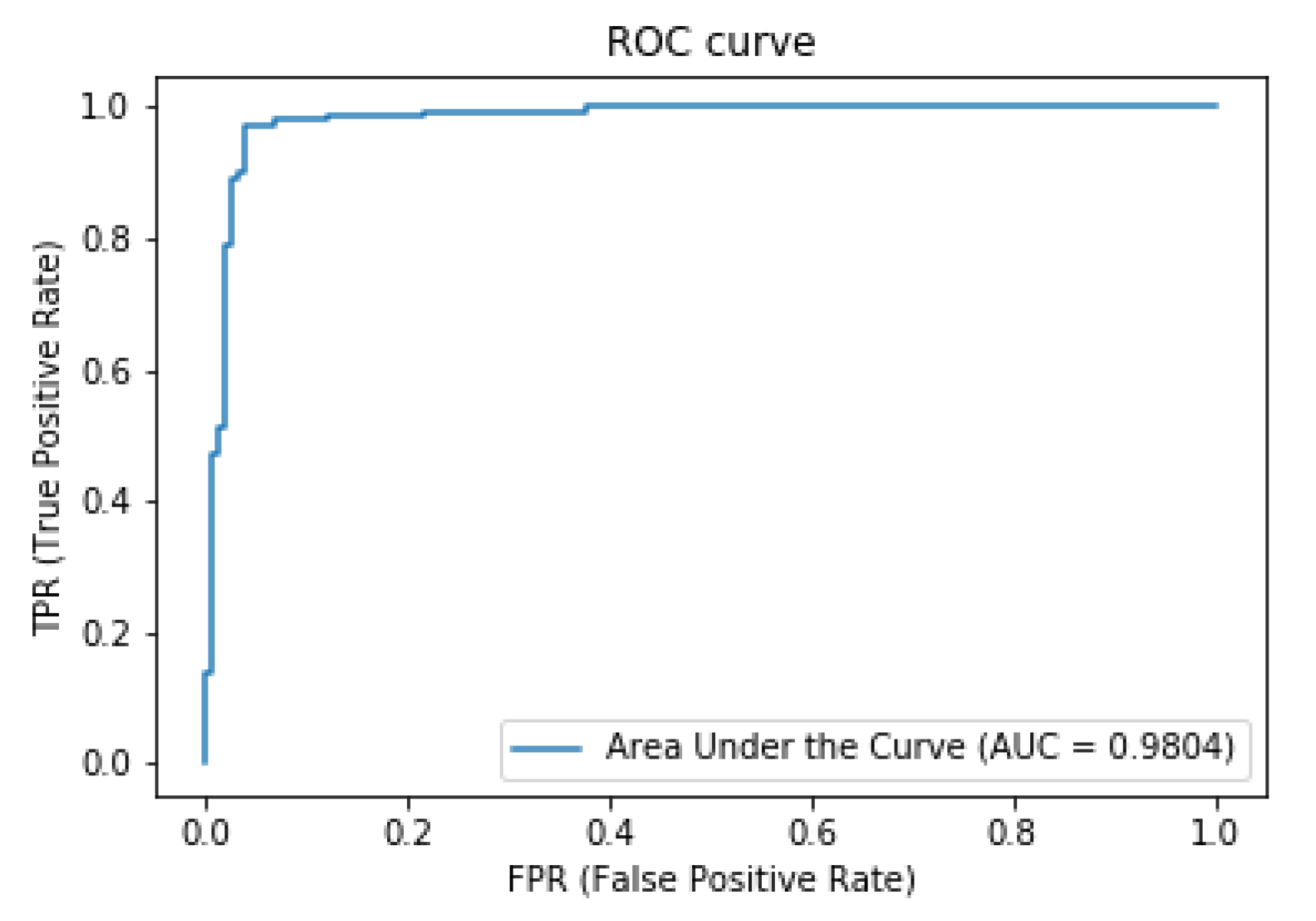
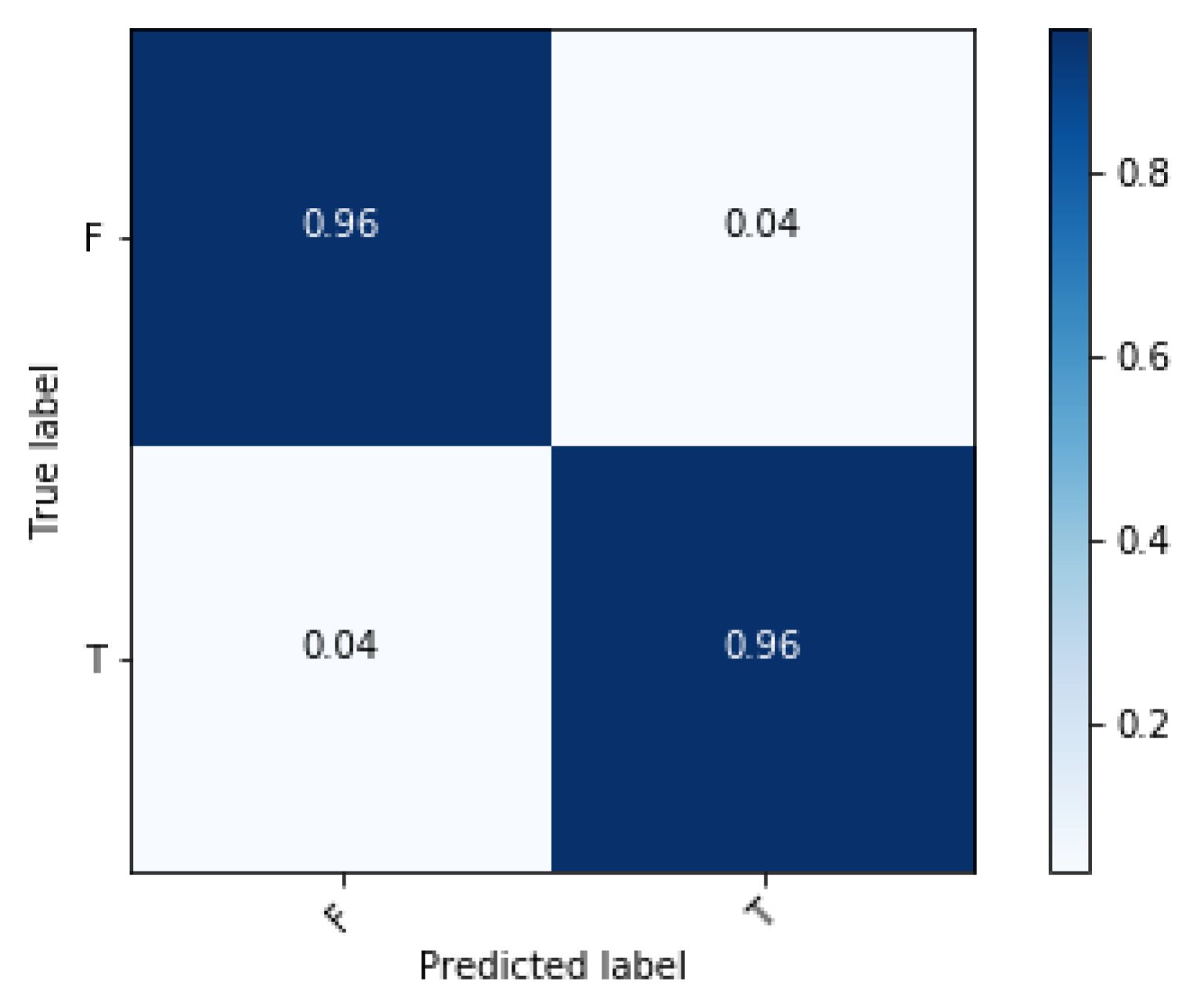
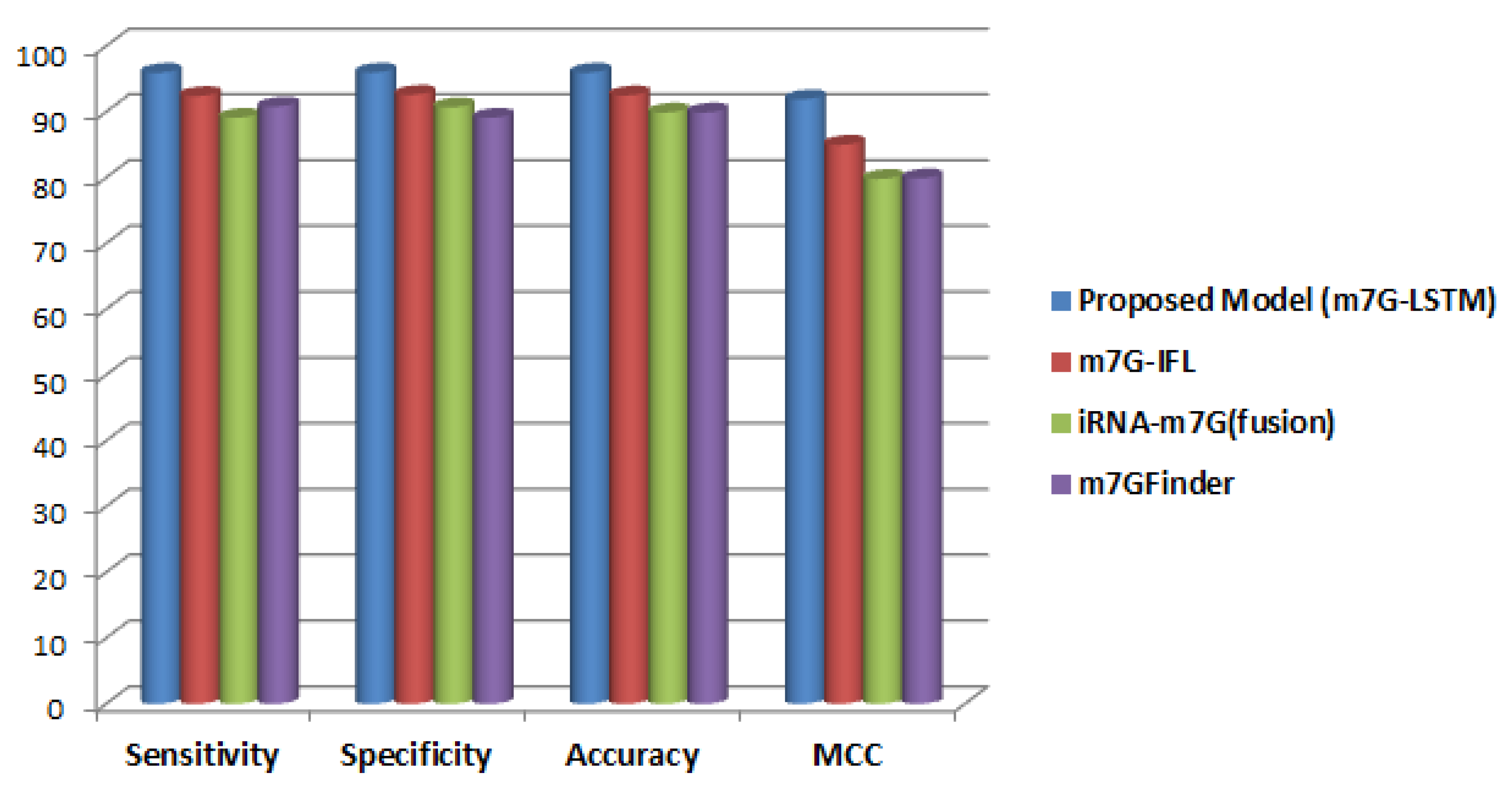
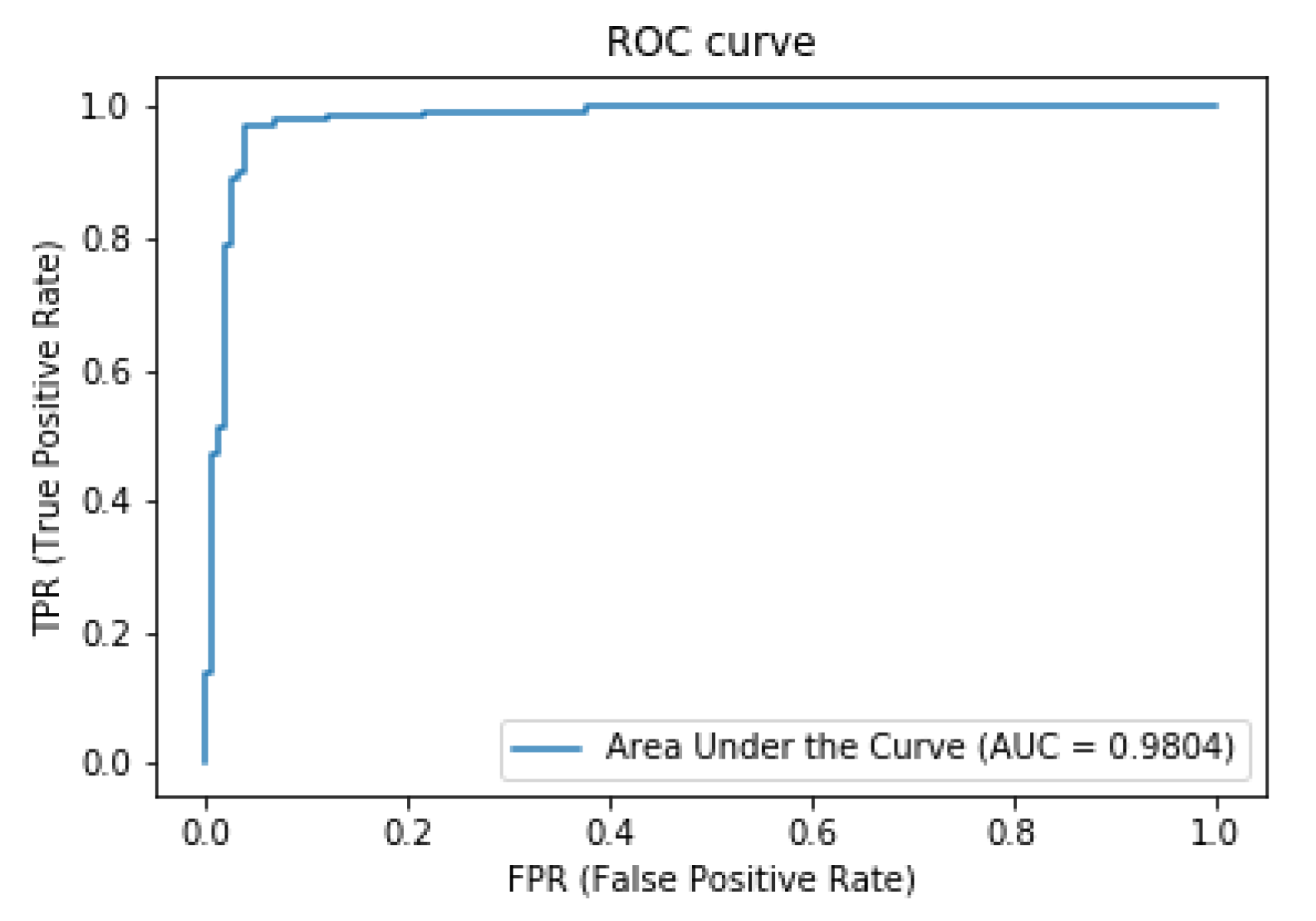
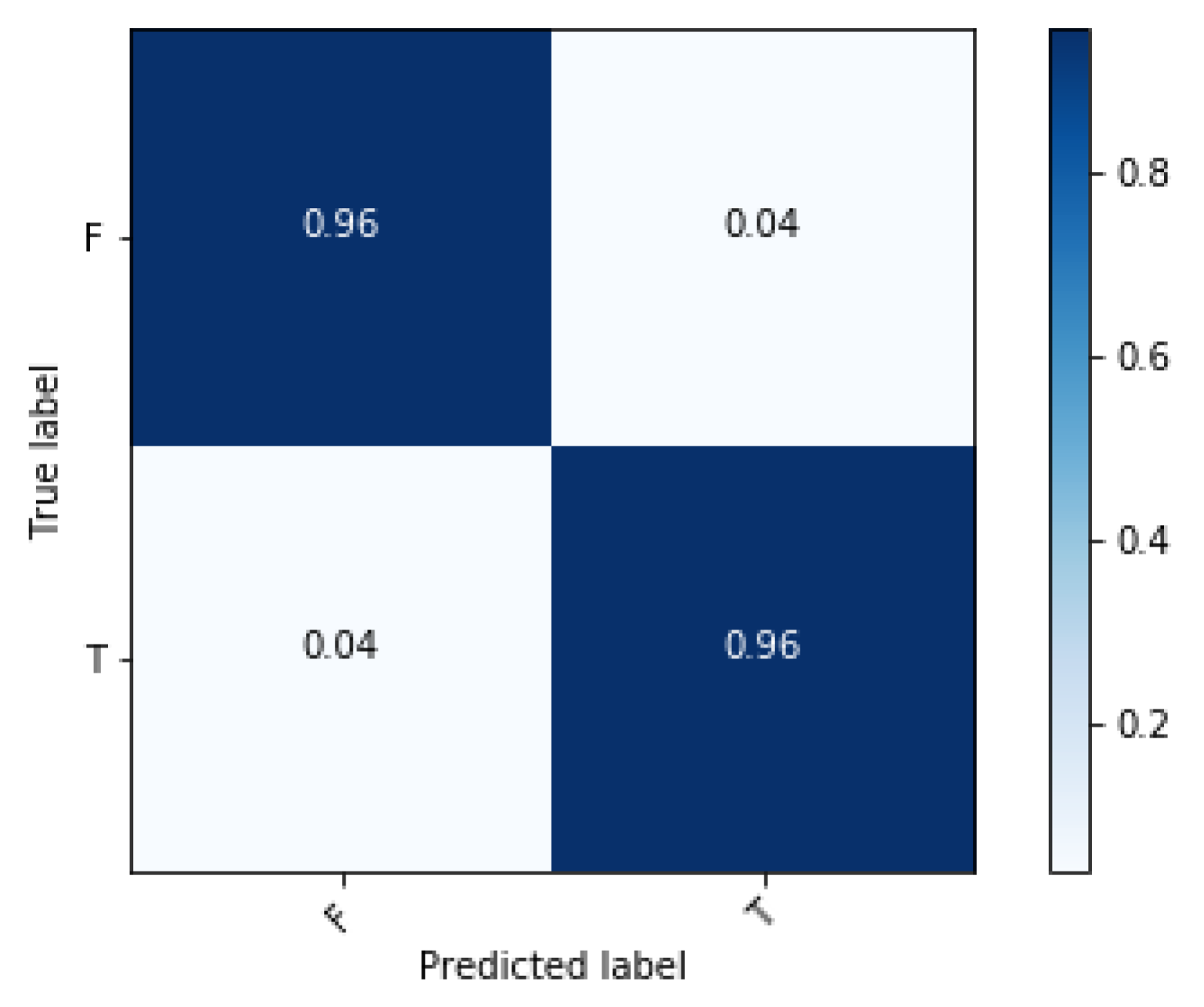
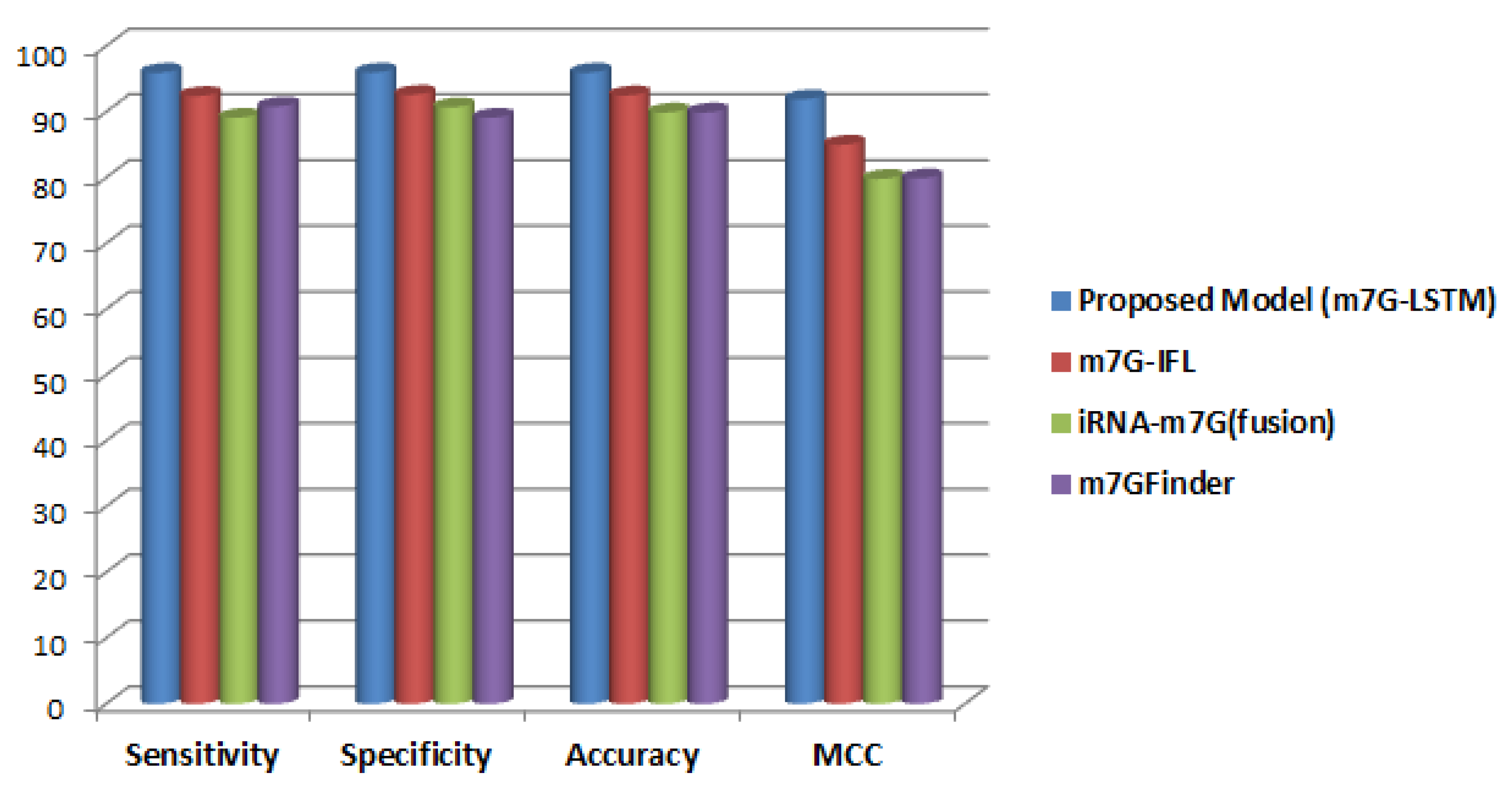
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roost, C.; Lynch, S.R.; Batista, P.J.; Qu, K.; Chang, H.Y.; Kool, E.T. Structure and thermodynamics of N6-methyladenosine in RNA: A spring-loaded base modification. J. Am. Chem. Soc. 2015, 137, 2107–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G. N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H. Regulation of mRNA cap methylation. Biochem. J. 2010, 425, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Zago, E.; Molin, A.D.; Dimitri, G.M.; Xumerle, L.; Pirazzini, C.; Bacalini, M.G.; Maturo, M.G.; Azevedo, T.; Spasov, S.; Gómez-Garre, P. Early downregulation of hsa-miR-144-3p in serum from drug-naïve Parkinson’s disease patients. Sci. Rep. 2022, 12, 1330. [Google Scholar] [CrossRef]
- Marchand, V.; Ayadi, L.; Ernst, F.G.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.; Motorin, Y. AlkAniline-Seq: Profiling of m7G and m3C RNA Modifications at Single Nucleotide Resolution. Angew. Chem. Int. Ed. 2018, 57, 16785–16790. [Google Scholar] [CrossRef]
- Zhang, L.-S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.-L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X. Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol. Cell 2019, 74, 1304–1316.e1308. [Google Scholar] [CrossRef]
- Malbec, L.; Zhang, T.; Chen, Y.-S.; Zhang, Y.; Sun, B.-F.; Shi, B.-Y.; Zhao, Y.-L.; Yang, Y.; Yang, Y.-G. Dynamic methylome of internal mRNA N 7-methylguanosine and its regulatory role in translation. Cell Res. 2019, 29, 927–941. [Google Scholar] [CrossRef]
- Zhao, Y.; Kong, L.; Pei, Z.; Li, F.; Li, C.; Sun, X.; Shi, B.; Ge, J. m7G methyltransferase METTL1 promotes post-ischemic angiogenesis via promoting VEGFA mRNA translation. Front. Cell Dev. Biol. 2021, 9, 1376. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Z.; Mao, X.; Li, Q. m7GPredictor: An improved machine learning-based model for predicting internal m7G modifications using sequence properties. Anal. Biochem. 2020, 609, 113905. [Google Scholar] [CrossRef]
- Bi, Y.; Xiang, D.; Ge, Z.; Li, F.; Jia, C.; Song, J. An interpretable prediction model for identifying N7-methylguanosine sites based on XGBoost and SHAP. Mol. Ther.-Nucleic Acids 2020, 22, 362–372. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Basith, S.; Pitti, T.; Lee, G.; Manavalan, B. THRONE: A New Approach for Accurate Prediction of Human RNA N7-Methylguanosine Sites. J. Mol. Biol. 2022, 434, 167549. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Yin, Z. m7G-DPP: Identifying N7-methylguanosine sites based on dinucleotide physicochemical properties of RNA. Biophys. Chem. 2021, 279, 106697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, X.; Liu, M.; Liu, G.; Ren, Y. BERT-m7G: A Transformer Architecture Based on BERT and Stacking Ensemble to Identify RNA N7-Methylguanosine Sites from Sequence Information. Comput. Math. Methods Med. 2021, 2021, 7764764. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-H.; Ma, C.; Wang, J.-S.; Yang, H.; Ding, H.; Han, S.-G.; Li, Y.-W. Prediction of N7-methylguanosine sites in human RNA based on optimal sequence features. Genomics 2020, 112, 4342–4347. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Song, X.; Lv, H.; Lin, H. iRNA-m7G: Identifying N7-methylguanosine sites by fusing multiple features. Mol. Ther.-Nucleic Acids 2019, 18, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Feng, P.; Cui, L.; Su, R.; Chen, W.; Wei, L. Iterative feature representation algorithm to improve the predictive performance of N7-methylguanosine sites. Brief. Bioinform. 2020, 22, bbaa278. [Google Scholar] [CrossRef]
- Ning, Q.; Sheng, M. m7G-DLSTM: Intergrating directional Double-LSTM and fully connected network for RNA N7-methlguanosine sites prediction in human. Chemom. Intell. Lab. Syst. 2021, 217, 104398. [Google Scholar] [CrossRef]
- Wei, L.; Su, R.; Luan, S.; Liao, Z.; Manavalan, B.; Zou, Q.; Shi, X. Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics 2019, 35, 4930–4937. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Wang, M.; Webb, G.I.; Zhang, Y.; Whisstock, J.C.; Song, J. GlycoMine: A machine learning-based approach for predicting N-, C-and O-linked glycosylation in the human proteome. Bioinformatics 2015, 31, 1411–1419. [Google Scholar] [CrossRef]
- Uriarte-Arcia, A.V.; López-Yáñez, I.; Yáñez-Márquez, C. One-hot vector hybrid associative classifier for medical data classification. PLoS ONE 2014, 9, e95715. [Google Scholar] [CrossRef] [Green Version]
- Khanal, J.; Nazari, I.; Tayara, H.; Chong, K.T. 4mCCNN: Identification of N4-methylcytosine sites in prokaryotes using convolutional neural network. IEEE Access 2019, 7, 145455–145461. [Google Scholar] [CrossRef]
- Lv, Z.; Ding, H.; Wang, L.; Zou, Q. A Convolutional Neural Network Using Dinucleotide One-hot Encoder for identifying DNA N6-Methyladenine Sites in the Rice Genome. Neurocomputing 2021, 422, 214–221. [Google Scholar] [CrossRef]
- Zeng, F.; Fang, G.; Yao, L. A deep neural network for identifying DNA N4-methylcytosine sites. Front. Genet. 2020, 11, 209. [Google Scholar] [CrossRef] [Green Version]
- Tahir, M.; Tayara, H.; Hayat, M.; Chong, K.T. kDeepBind: Prediction of RNA-Proteins binding sites using convolution neural network and k-gram features. Chemom. Intell. Lab. Syst. 2021, 208, 104217. [Google Scholar] [CrossRef]
- Shao, Y.; Chou, K.-C. pLoc_Deep-mVirus: A CNN Model for Predicting Subcellular Localization of Virus Proteins by Deep Learning. Nat. Sci. 2020, 12, 388–399. [Google Scholar] [CrossRef]
- Zeng, W.; Wu, M.; Jiang, R. Prediction of enhancer-promoter interactions via natural language processing. BMC Genom. 2018, 19, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Oubounyt, M.; Louadi, Z.; Tayara, H.; Chong, K.T. Deep learning models based on distributed feature representations for alternative splicing prediction. IEEE Access 2018, 6, 58826–58834. [Google Scholar] [CrossRef]
- Choi, J.; Oh, I.; Seo, S.; Ahn, J. G2Vec: Distributed gene representations for identification of cancer prognostic genes. Sci. Rep. 2018, 8, 13729. [Google Scholar] [CrossRef]
- Nazari, I.; Tahir, M.; Tayara, H.; Chong, K.T. iN6-Methyl (5-step): Identifying RNA N6-methyladenosine sites using deep learning mode via Chou’s 5-step rules and Chou’s general PseKNC. Chemom. Intell. Lab. Syst. 2019, 193, 103811. [Google Scholar] [CrossRef]
- Tan, K.K.; Le, N.Q.K.; Yeh, H.-Y.; Chua, M.C.H. Ensemble of deep recurrent neural networks for identifying enhancers via dinucleotide physicochemical properties. Cells 2019, 8, 767. [Google Scholar] [CrossRef] [Green Version]
- Tayara, H.; Soo, K.G.; Chong, K.T. Vehicle detection and counting in high-resolution aerial images using convolutional regression neural network. IEEE Access 2017, 6, 2220–2230. [Google Scholar] [CrossRef]
- Bai, S.; Kolter, J.Z.; Koltun, V. An empirical evaluation of generic convolutional and recurrent networks for sequence modeling. arXiv 2018, arXiv:1803.01271. [Google Scholar]
- Tahir, M.; Tayara, H.; Chong, K.T. iRNA-PseKNC (2methyl): Identify RNA 2′-O-methylation sites by convolution neural network and Chou’s pseudo components. J. Theor. Biol. 2019, 465, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Tayara, H.; Chong, K.T. iPseU-CNN: Identifying RNA pseudouridine sites using convolutional neural networks. Mol. Ther.-Nucleic Acids 2019, 16, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayara, H.; Tahir, M.; Chong, K.T. Identification of prokaryotic promoters and their strength by integrating heterogeneous features. Genomics 2020, 112, 1396–1403. [Google Scholar] [CrossRef]
- Saini, V.K.; Kumar, R.; Mathur, A.; Saxena, A. Short term forecasting based on hourly wind speed data using deep learning algorithms. In Proceedings of the 2020 3rd International Conference on Emerging Technologies in Computer Engineering: Machine Learning and Internet of Things (ICETCE), Jaipur, India, 7–8 February 2020; pp. 1–6. [Google Scholar]
- Sharma, A.K.; Saxena, A.; Soni, B.P.; Gupta, V. Voltage stability assessment using artificial neural network. In Proceedings of the 2018 IEEMA Engineer Infinite Conference (eTechNxT), New Delhi, India, 13–14 March 2018; pp. 1–5. [Google Scholar]
- Tahir, M.; Hayat, M.; Chong, K.T. A convolution neural network-based computational model to identify the occurrence sites of various RNA modifications by fusing varied features. Chemom. Intell. Lab. Syst. 2021, 211, 104233. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Khan, S.; Chong, K.T. Prediction of piwi-interacting RNAs and their functions via Convolutional Neural Network. IEEE Access 2021, 9, 54233–54240. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Ullah, I.; Chong, K.T. A deep learning-based computational approach for discrimination of dna n6-methyladenosine sites by fusing heterogeneous features. Chemom. Intell. Lab. Syst. 2020, 206, 104151. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Chong, K.T. Prediction of n6-methyladenosine sites using convolution neural network model based on distributed feature representations. Neural Netw. 2020, 129, 385–391. [Google Scholar] [CrossRef]
- Wang, L.; Zhong, X.; Wang, S.; Zhang, H.; Liu, Y. A novel end-to-end method to predict RNA secondary structure profile based on bidirectional LSTM and residual neural network. BMC Bioinform. 2021, 22, 169. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Q.; Liu, B. DeepDRBP-2L: A new genome annotation predictor for identifying DNA binding proteins and RNA binding proteins using convolutional neural network and long short-term memory. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiao, S.; Ji, S.; Li, Y. DeepSite: Bidirectional LSTM and CNN models for predicting DNA–protein binding. Int. J. Mach. Learn. Cybern. 2020, 11, 841–851. [Google Scholar] [CrossRef]
- Dutta, A.; Dalmia, A.; Athul, R.; Singh, K.K.; Anand, A. Using the Chou’s 5-steps rule to predict splice junctions with interpretable bidirectional long short-term memory networks. Comput. Biol. Med. 2020, 116, 103558. [Google Scholar] [CrossRef] [PubMed]
- Niu, K.; Luo, X.; Zhang, S.; Teng, Z.; Zhang, T.; Zhao, Y. iEnhancer-EBLSTM: Identifying Enhancers and Strengths by Ensembles of Bidirectional Long Short-Term Memory. Front. Genet. 2021, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- di Gangi, M.; Bosco, G.L.; Rizzo, R. Deep learning architectures for prediction of nucleosome positioning from sequences data. BMC Bioinform. 2018, 19, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A. Grey forecasting models based on internal optimization for Novel Corona virus (COVID-19). Appl. Soft Comput. 2021, 111, 107735. [Google Scholar] [CrossRef]
- van Houdt, G.; Mosquera, C.; Nápoles, G. A review on the long short-term memory model. Artif. Intell. Rev. 2020, 53, 5929–5955. [Google Scholar] [CrossRef]
- Shewalkar, A. Performance evaluation of deep neural networks applied to speech recognition: RNN, LSTM and GRU. J. Artif. Intell. Soft Comput. Res. 2019, 9, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Zazo, R.; Lozano-Diez, A.; Gonzalez-Dominguez, J.; Toledano, D.T.; Gonzalez-Rodriguez, J. Language identification in short utterances using long short-term memory (LSTM) recurrent neural networks. PLoS ONE 2016, 11, e0146917. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.-C. iRNA-AI: Identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget 2017, 8, 4208. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Feng, P.-M.; Lin, H.; Chou, K.-C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Li, K.; Huang, D.-S.; Chou, K.-C. iEnhancer-EL: Identifying enhancers and their strength with ensemble learning approach. Bioinformatics 2018, 34, 3835–3842. [Google Scholar] [CrossRef]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K.-C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2019, 111, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, F.; Huang, D.-S.; Chou, K.-C. iPromoter-2L: A two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 2017, 34, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, X.; Xiong, N.; Shao, J. Learning sparse representation with variational auto-encoder for anomaly detection. IEEE Access 2018, 6, 33353–33361. [Google Scholar] [CrossRef]
- Yi, B.; Shen, X.; Liu, H.; Zhang, Z.; Zhang, W.; Liu, S.; Xiong, N. Deep matrix factorization with implicit feedback embedding for recommendation system. IEEE Trans. Ind. Inform. 2019, 15, 4591–4601. [Google Scholar] [CrossRef]
- Qu, Y.; Xiong, N. RFH: A resilient, fault-tolerant and high-efficient replication algorithm for distributed cloud storage. In Proceedings of the 2012 41st International Conference on Parallel Processing, Pittsburgh, PA, USA, 10–13 September 2012; pp. 520–529. [Google Scholar]
- Lin, B.; Zhu, F.; Zhang, J.; Chen, J.; Chen, X.; Xiong, N.N.; Mauri, J.L. A time-driven data placement strategy for a scientific workflow combining edge computing and cloud computing. IEEE Trans. Ind. Inform. 2019, 15, 4254–4265. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, J.; Liu, R.W.; Xiong, N.; Wu, K.; Kim, T.-H. A dimensionality reduction-based multi-step clustering method for robust vessel trajectory analysis. Sensors 2017, 17, 1792. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Yao, X.; Zhao, X.; Yin, J.; Xiong, N. A stochastic control approach to maximize profit on service provisioning for mobile cloudlet platforms. IEEE Trans. Syst. Man Cybern. Syst. 2016, 48, 522–534. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| List of Parameters | Word2vec Model |
|---|---|
| Training Method | CBOW |
| Corpus | Human Genome |
| Context words | 3-mer |
| Vector size | 100 |
| Window size | 5 |
| Minimum Count | 5 |
| Negative Sampling | 5 |
| Epochs | 20 |
| Models | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| LSTM | 95.94 | 95.97 | 95.95 | 0.919 |
| CNN | 96.62 | 93.28 | 94.94 | 0.899 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahir, M.; Hayat, M.; Khan, R.; Chong, K.T. An Effective Deep Learning-Based Architecture for Prediction of N7-Methylguanosine Sites in Health Systems. Electronics 2022, 11, 1917. https://doi.org/10.3390/electronics11121917
Tahir M, Hayat M, Khan R, Chong KT. An Effective Deep Learning-Based Architecture for Prediction of N7-Methylguanosine Sites in Health Systems. Electronics. 2022; 11(12):1917. https://doi.org/10.3390/electronics11121917
Chicago/Turabian StyleTahir, Muhammad, Maqsood Hayat, Rahim Khan, and Kil To Chong. 2022. "An Effective Deep Learning-Based Architecture for Prediction of N7-Methylguanosine Sites in Health Systems" Electronics 11, no. 12: 1917. https://doi.org/10.3390/electronics11121917
APA StyleTahir, M., Hayat, M., Khan, R., & Chong, K. T. (2022). An Effective Deep Learning-Based Architecture for Prediction of N7-Methylguanosine Sites in Health Systems. Electronics, 11(12), 1917. https://doi.org/10.3390/electronics11121917