Characterizing the Urban Mine—Challenges of Simplified Chemical Analysis of Anthropogenic Mineral Residues

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Origin and Physical Sample Preparation
2.1.1. Battery Ash Sample (BATT)
2.1.2. Mining Waste Sample (MIN)
2.2. Chemical Analysis
2.2.1. General Approach
2.2.2. Element Selection
2.2.3. Wet-Chemical Analysis
2.2.4. Wavelength Dispersive X-ray Fluorescence Spectrometry (WD-XRF)
2.2.5. Energy Dispersive X-ray Fluorescence Spectrometry (ED-XRF)
2.3. Data Analysis
2.3.1. Test of Sample Homogeneity
- Null hypothesis H0: all subsample mean values are equal
- Level of significance α = 0.01
- BATT: n = 5, υ1 = 6 and υ2 = 28 (degrees of freedom for the F distribution)
- MIN: n = 3, υ1 = 7 and υ2 = 16
2.3.2. Method Validation: Accuracy and Precision
2.3.3. Relative and Significant Differences Between Chemical Analysis Methods
- Null hypothesis H0: mean values of both samples are equal
- Level of significance α = 0.01
- heterogeneity of variances
3. Results and Discussion
3.1. Sample Homogeneity
3.2. Elemental Composition Determined with the Validated Method
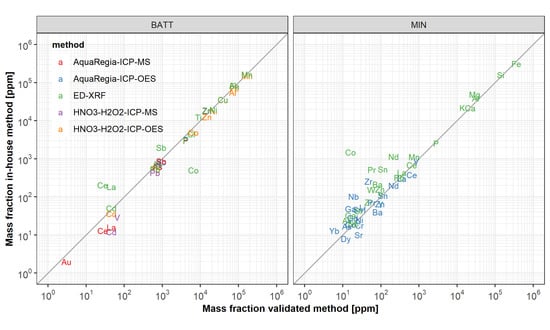
3.3. Applicability of Wet-Chemical In-House Methods
3.3.1. Acid Digestion
3.3.2. Element Detection with ICP-OES
3.3.3. Element Detection with ICP-MS
3.4. Applicability of ED-XRF Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eurostat. Waste Statistics; Eurostat: Brussels, Belgium, 2019; Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php/Waste_statistics#Total_waste_generation (accessed on 7 March 2019).
- Commission of the European Communities. The Raw Materials Initiative—Meeting Our Critical Needs for Growth and Jobs in Europe COM; Commission of the European Communities: Brussels, Belgium, 2008; Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52008DC0699&from=EN (accessed on 28 February 2019).
- European Commission. Report on Critical Raw Materials for the EU Critical Raw Materials Profiles; European Commission: Brussels, Belgium, 2017. [Google Scholar]
- Normenausschuss Wasserwesen (NAW) im DIN. Characterization of Waste—Digestion for Subsequent Determination of Aqua Regia Soluble Portion of Elements; Deutsche Fassung EN 13657:2002; Beuth Verlag GmbH: Berlin, Germany, 2003. [Google Scholar]
- Normenausschuss Wasserwesen (NAW) im DIN. Soil Quality—Determination of Trace Elements in Extracts of Soil by Inductively Coupled Plasma—Atomic Emission Spectrometry (ICP-AES); German Version ISO 22036:2008; Beuth Verlag GmbH: Berlin, Germany, 2009. [Google Scholar]
- Normenausschuss Wasserwesen (NAW) im DIN. Deutsches Institut Für Normung E. V. Soil Quality—Determination of Cadmium, Chromium, Cobalt, Copper, Lead, Manganese, Nickel and Zinc in Aqua Regia Extracts of Soil—Flame and Electrothermal Atomic Absorption Spectrometric Methods; German Version ISO 11047:1998; Beuth Verlag GmbH: Berlin, Germany, 2003. [Google Scholar]
- Normenausschuss Wasserwesen (NAW) im DIN. Characterization of Waste—Microwave Assisted Digestion with Hydrofluoric (Hf), Nitric (Hno3) and Hydrochloric (Hcl) Acid Mixture for Subsequent Determination of Elements, German version EN 13656:2002; Beuth Verlag GmbH: Berlin, Germany, 2003. [Google Scholar]
- Bock, R. Handbuch Der Analytisch-Chemischen Aufschlussmethoden; WILEY-VCH Verlag GmbH: Weinheim, Baden-Wutenberg, Germany, 2001; ISBN 352729791X. [Google Scholar]
- Meshram, P.; Abhilash; Pandey, B.D.; Mankhand, T.R.; Deveci, H. Acid baking of spent lithium ion batteries for selective recovery of major metals: A two-step process. J. Ind. Eng. Chem. 2016, 43, 117–126. [Google Scholar] [CrossRef]
- Meshram, P.; Pandey, B.D.; Mankhand, T.R. Hydrometallurgical processing of spent lithium ion batteries (LIBs) in the presence of a reducing agent with emphasis on kinetics of leaching. Chem. Eng. J. 2015, 281, 418–427. [Google Scholar] [CrossRef]
- Meshram, P.; Pandey, B.D.; Mankhand, T.R. Recovery of valuable metals from cathodic active material of spent lithium ion batteries: Leaching and kinetic aspects. Waste Manag. 2015, 45, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Swain, B.; Jeong, J.; Lee, J.-C.; Lee, G.-H.; Sohn, J.-S. Hydrometallurgical process for recovery of cobalt from waste cathodic active material generated during manufacturing of lithium ion batteries. J. Power Sources 2007, 167, 536–544. [Google Scholar] [CrossRef]
- Kang, J.; Senanayake, G.; Sohn, J.; Shin, S.M. Recovery of cobalt sulfate from spent lithium ion batteries by reductive leaching and solvent extraction with Cyanex 272. Hydrometallurgy 2010, 100, 168–171. [Google Scholar] [CrossRef]
- Chen, L.; Tang, X.; Zhang, Y.; Li, L.; Zeng, Z.; Zhang, Y. Process for the recovery of cobalt oxalate from spent lithium-ion batteries. Hydrometallurgy 2011, 108, 80–86. [Google Scholar] [CrossRef]
- Lee, C.K.; Rhee, K.-I. Reductive leaching of cathodic active materials from lithium ion battery wastes. Hydrometallurgy 2003, 68, 5–10. [Google Scholar] [CrossRef]
- Golmohammadzadeh, R.; Rashchi, F.; Vahidi, E. Recovery of lithium and cobalt from spent lithium-ion batteries using organic acids: Process optimization and kinetic aspects. Waste Manag. 2017, 64, 244–254. [Google Scholar] [CrossRef]
- Li, L.; Ge, J.; Chen, R.; Wu, F.; Chen, S.; Zhang, X. Environmental friendly leaching reagent for cobalt and lithium recovery from spent lithium-ion batteries. Waste Manag. 2010, 30, 2615–2621. [Google Scholar] [CrossRef]
- Moyle, P.R.; Causey, J.D. Chemical Composition of Samples Collected from Waste Rock Dumps and Other Mining-Related Features at Selected Phosphate Mines in Southeastern Idaho, Western Wyoming, and Northern Utah; US Geological Survey: Reston, VA, USA, 2001. Available online: https://pubs.usgs.gov/of/2001/0411/pdf/OF01-411.pdf (accessed on 24 August 2018).
- Pant, D.; Dolker, T. Green and facile method for the recovery of spent Lithium Nickel Manganese Cobalt Oxide (NMC) based Lithium ion batteries. Waste Manag. 2017, 60, 689–695. [Google Scholar] [CrossRef]
- Zheng, X.; Gao, W.; Zhang, X.; He, M.; Lin, X.; Cao, H.; Zhang, Y.; Sun, Z. Spent lithium-ion battery recycling—Reductive ammonia leaching of metals from cathode scrap by sodium sulphite. Waste Manag. 2017, 60, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, Y.; Yan, X.; Tian, M.; Xiao, W.; Tang, H. A sustainable route from fly ash to silicon nanorods for high performance lithium ion batteries. Chem. Eng. J. 2017, 330, 1052–1059. [Google Scholar] [CrossRef]
- ALS Minerals. Portable XRF in the Mining Industry. 2014. Available online: https://www.alsglobal.com/-/…/als/…/portable-xrf-analysis-technical-note-2014.pdf (accessed on 21 June 2018).
- Chen, C.-S.; Shih, Y.-J.; Huang, Y.-H. Recovery of lead from smelting fly ash of waste lead-acid battery by leaching and electrowinning. Waste Manag. 2016, 52, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.; Andre, M.-L.; Navarro Amador, R.; Hyvrard, F.; Borrini, J.; Carboni, M.; Meyer, D. Recovery of metals from simulant spent lithium-ion battery as organophosphonate coordination polymers in aqueous media. J. Hazard. Mater. 2016, 317, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yokoyama, T.; Itabashi, O.; Suzuki, T.M.; Inoue, K. Hydrometallurgical process for recovery of metal values from spent lithium-ion secondary batteries. Hydrometallurgy 1998, 47, 259–271. [Google Scholar] [CrossRef]
- Li, L.; Ge, J.; Wu, F.; Chen, R.; Chen, S.; Wu, B. Recovery of cobalt and lithium from spent lithium ion batteries using organic citric acid as leachant. J. Hazard. Mater. 2010, 176, 288–293. [Google Scholar] [CrossRef] [PubMed]
- ALS Minerals. Fire Assay Technical Note 2012. 2012. Available online: https://alsglobal.azureedge.net/-/media/als/resources/services-and-products/geochemistry/technical-notes/fire-assay-technical-note-2012.pdf?rev=a63520e2ed72416eb5d55d3ce6bc8cde (accessed on 26 July 2019).
- ALS Minerals. Technical Note: Super Trace Methods for Soil Samples. 2012. Available online: https://alsglobal.azureedge.net/-/media/als/resources/services-and-products/geochemistry/technical-notes/super-trace-methods-for-soil-samples.pdf?rev=774caa7f48184edf9624d7a1f3a37856 (accessed on 28 May 2018).
- Yang, Y.; Huang, G.; Xu, S.; He, Y.; Liu, X. Thermal treatment process for the recovery of valuable metals from spent lithium-ion batteries. Hydrometallurgy 2016, 165, 390–396. [Google Scholar] [CrossRef]
- Götze, R.; Astrup, T.F. Optimal acid digestion for multi-element analysis of different waste matrices. In Proceedings of the 5th Conference on Engineering for Waste and Biomass Valorisation, Rio de Janeiro, Brazil, 25 August 2014. [Google Scholar]
- Korf, N.; Løvik, A.N.; Figi, R.; Schreiner, C.; Kuntz, C.; Mählitz, P.M.; Rösslein, M.; Wäger, P.; Rotter, V.S. Multi-element chemical analysis of printed circuit boards—Challenges and pitfalls. Waste Manag. 2019, 92, 124–136. [Google Scholar] [CrossRef] [PubMed]
- VARTA Microbattery GmbH. Safety Data Sheet MSDS 2.001.020: Lithium Thionyl Chloride Cylindrical Cell; VARTA Microbattery GmbH: Elwangen, Germany, 2016; Available online: https://products.varta-microbattery.com/applications/mb_data/documents/material_safety_data_sheets/MSDS_20_Primary_Lithium_Cylindrical_Series_ER_en.pdf (accessed on 26 July 2018).
- Berndt, D.; Spahrbier, D. Batteries. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2011; pp. 41–93. [Google Scholar]
- Länderarbeitsgemeinschaft Abfall (LAGA). Guideline for the Procedure for Physical, Chemical and Biological Investigations in Connection with the Recycling/Disposal of Waste to the Guideline for Sampling (LAGA PN98); Notice M32. German Version; Mitteilungen der Länderarbeitsgemeinschaft Abfall (LAGA), 2001; Available online: https://www.laga-online.de/Publikationen-50-Mitteilungen.html (accessed on 25 June 2019).
- Normenausschuss Wasserwesen (NAW) im DIN. Characterization of Waste—Calculation of Dry Matter by Determination of Dry Residue or Water Content, German version EN 14346:2006; Beuth Verlag GmbH: Berlin, Germany, 2007. [Google Scholar]
- Luke, E. Technical Report on the Grängesberg Iron Mine Resource Estimate; RPA: Bergslagen, Sweden, 2014; Available online: http://angleseymining.co.uk/projects/grangesberg/29092014-release.pdf (accessed on 17 March 2019).
- Geological Survey of Finland (GTK); Geological Survey of Norway (NGU); Geological Survey of Russia (VSEGEI); Geological Survey of Sweden (SGU); SC Mineral. FODD: Fennoscandian Ore Deposit Database; Geological Survey of Finland: Esbo, Finland, 2011; Available online: http://en.gtk.fi/ExplorationFinland/fodd (accessed on 30 November 2018).
- Eilu, P.; Boyd, R.; Hallberg, A.; Korsakova, M.; Krasotkin, S.; Nurmi, P.A.; Ripa, M.; Stromov, V.; Tontti, M. Mining History of Fennoscandia; Geological Survey of Finland: Esbo, Finland, 2012; Volume 53, pp. 19–32. [Google Scholar]
- Geological Survey of Finland (GTK). Fennoscandian Mineral Deposits Application, Ore Deposits Database and Maps: Essential Information on a Still Under-Explored Region; Geological Survey of Finland: Esbo, Finland, 2017; Available online: http://en.gtk.fi/informationservices/databases/fodd/ (accessed on 27 May 2018).
- European Commission. List of Critical Raw Materials for the EU 2017; European Commission: Brussels, Belgium, 2017; Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52017DC0490&from=EN (accessed on 24 August 2018).
- Normenausschuss Qualitätsmanagement, Statistik und Zertifizierungsgrundlagen (NQSZ) im DIN. General Requirements for the Competence of Testing and Calibration Laboratories; (ISO/IEC DIS 17025:2016); Normenausschuss Qualitätsmanagement, Statistik und Zertifizierungsgrundlagen (NQSZ) im DIN: Berlin, Germany, 2017. [Google Scholar]
- Natural Resources Canada. Certificate of Analysis REE-1. Certified Reference Material for Rare Earth Elements, Zirconium and Niobium; Natural Resources Canada: Ottawa, ON, Canada, 2014. Available online: http://www.nrcan.gc.ca/sites/www.nrcan.gc.ca/files/mineralsmetals/files/pdf/tech-tech/ree-1-2a-en.pdf (accessed on 28 May 2018).
- Anton Paar GmbH. High Pressure Asher; Anton Paar GmbH: Osterfelden, Germany, 2016; Available online: https://www.anton-paar.com/?eID=documentsDownload&document=837&L=0 (accessed on 29 August 2018).
- DIN-Normenausschuss Wasserwesen (NAW). Characterization of Waste—Halogen and Sulfur Content—Oxygen Combustion in Closed Systems and Determination Methods; DIN EN 14582; Beuth Verlag GmbH: Berlin, Germany, 2007. [Google Scholar]
- Thermo Scientific. Slag Analysis with the Thermo Scientific Niton XL3 GOLDD Series XRF Analyzer. In Fast, Lab-Quality Analysis on the Production Floor; Thermo Scientific: Waltham, MA, USA, 2009; Available online: https://assets.thermofisher.com/TFS-Assets/CAD/Application-Notes/Slag-Analysis-Application-Summary.pdf (accessed on 29 March 2019).
- Hall, G.E.M.; Bonham-Carter, G.F.; Buchar, A. Evaluation of portable X-ray fluorescence (pXRF) in exploration and mining: Phase 1, control reference materials. Geochemistry: Exploration, Environment. Analysis 2014, 14, 99–123. [Google Scholar] [CrossRef]
- Aldrian, A.; Ledersteger, A.; Pomberger, R. Monitoring of WEEE plastics in regards to brominated flame retardants using handheld XRF. Waste Manag. 2014, 36, 297–304. [Google Scholar] [CrossRef]
- Dimitrakakis, E.; Janz, A.; Bilitewski, B.; Gidarakos, E. Determination of heavy metals and halogens in plastics from electric and electronic waste. Waste Manag. 2009, 29, 2700–2706. [Google Scholar] [CrossRef] [PubMed]
- Guzzonato, A.; Puype, F.; Harrad, S.J. Improving the accuracy of hand-held X-ray fluorescence spectrometers as a tool for monitoring brominated flame retardants in waste polymers. Chemosphere 2016, 159, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Thermo Scientific. Thermo Scientific. Thermo Scientific Niton XL3t GOLDD+Series Mining Analyzers. In Elemental Limits of Detection in SiO2 and SRM Matrices Using Mining Analysis; Thermo Scientific: Waltham, MA, USA, 2010; Available online: https://www.thermofisher.com/order/catalog/product/10131166 (accessed on 28 May 2018).
- Wilcox, R.R. Basic Statistics: Understanding Conventional Methods and Modern Insights; Oxford University Press: Oxford, UK, 2009; ISBN 978-0-19-531510-3. [Google Scholar]
- ICH. Validation of Analytical Procedures: Text and Methodology Q2(R1); ICH: Geneva, Switzerland, 2005; Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed on 28 August 2018).
- Li, H.; Sharp, G.; Pilkington, C.; Pifat, D.; Petteway, S. GLP-compliant assay validation studies: Considerations for implementation of regulations and audit of studies. Qual. Assur. J. 2006, 10, 92–100. [Google Scholar] [CrossRef]
- Das, S.; Ting, Y.-P. Evaluation of Wet Digestion Methods for Quantification of Metal Content in Electronic Scrap Material. Resources 2017, 6, 64. [Google Scholar] [CrossRef]
- ALS Minerals. Technical Note Geochemical Soil and Sediment Sampling/Analysis Considerations. Available online: https://alsglobal.azureedge.net/-/media/als/resources/services-and-products/geochemistry/technical-notes/geochemical-soil-and-sediment-sampling.pdf?rev=60a26aede15b473998b74c94528041b5 (accessed on 28 May 2018).
- Park, C.-S.; Shin, H.S.; Oh, H.; Cho, H.; Cheong, A.C.-S. Trace element analysis of whole-rock glass beads of geological reference materials by Nd: YAG UV 213 nm LA-ICP-MS. J. Anal. Sci. Technol. 2016, 7, 1693. [Google Scholar] [CrossRef][Green Version]
- Kim, J.; Anawati, J.; Azimi, G. Matrix complexity effect on platinum group metals analysis using inductively coupled plasma optical emission spectrometry. J. Anal. At. Spectrom. 2018, 33, 1310–1321. [Google Scholar] [CrossRef]
- Byers, H.L.; McHenry, L.J.; Grundl, T.J. XRF techniques to quantify heavy metals in vegetables at low detection limits. Food Chem. X 2019, 1, 100001. [Google Scholar] [CrossRef]
- Madian, M.; Eychmüller, A.; Giebeler, L. Current Advances in TiO2-Based Nanostructure Electrodes for High Performance Lithium Ion Batteries. Batteries 2018, 4, 7. [Google Scholar] [CrossRef]
- Gallhofer, D.; Lottermoser, B. The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF). Minerals 2018, 8, 320. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element Group | Battery Ash Sample (BATT) | Mining Waste Sample (MIN) |
|---|---|---|
| Ferrous metals | Fe, Mn, Ni, V | Cr, Fe, Mn, Nb c, Ni, V |
| Non-ferrous metals | Al, Co c, Cu, Pb, Ti, Zn | Al, Co c, Mg c, Sn, Zn |
| Precious metals | Au, Pd c | - |
| Specialty metals | As, Cd, Sb c | As, Ba, Ga c, Li, Sr, W, Zr |
| REE | Ce c, La c | Ce c, Dy c, Gd c, La c, Nd c, Pr c, Sm c, Y c, Yb c |
| Other | Cl, P, F *, C *, O * | Ca, K, P c, Rb, Si, C *, O * |
| Validated Method | In-House Method | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Microwave-assisted digestion (MWD) | |||||||||||
| Samples | BATT | MIN | BATT | MIN | |||||||
| Device name | MLS START | MLS START | CEM Mars 5 | CEM MarsExpress | |||||||
| Acid (1): mixture | 7 mL aqua regia (AR) | 7 mL aqua regia (AR) | 7 mL aqua regia (AR) | 10 mL aqua regia (AR) | |||||||
| Acid (1): elements | As, Au, Ce, La, P, Sb, Ti | As, Ce, Dy, Gd, La, Li, Nd, Pr, Sm, Yb | As, Au, Ce, La, P, Sb | As, Ba, Ce, Co, Cr, Dy, Ga, Gd, La, Li, Nb, Nd, Ni, Pr, Sm, Sn, Sr, V, Y, Yb, Zn, Zr | |||||||
| Acid (2): mixture | 6 mL HNO3 + 1 mL H2O2 | 7 mL aqua regia (AR) + 1.5 mL HF | 6 mL HNO3 + 1 mL H2O2 | - | |||||||
| Acid (2): elements | Al, Cd, Co, Cu, Fe, Li, Mn, Ni, Pb, Pd, V, Zn | Ba, Co, Cr, Ga, Nb, Ni, Sn, Sr, W, Zr | Al, Cd, Co, Cu, Fe, Mn, Ni, Pb, Pd, Zn | - | |||||||
| Acid (3): mixture | 7 mL H2SO4 + 1 mL HNO3 | 7 mL H2SO4 + 1 mL HNO3 | - | - | |||||||
| Acid (3): elements | As | As | - | - | |||||||
| Quality of acids | MERCK HCl 30% (s.p.); MERCK HNO3 65% (s.p.); MERCK H2O2 30% (s.p.); MERCK H2SO4 48% (p.a) | MERCK HCl 30% (s.p.); MERCK HNO3 65% (s.p.); MERCK HF 40% (s.p.); MERCK H2SO4 48% (p.a) | Roth HNO3 69% (s.p.); Roth H2O2 35% (pure) | ||||||||
| Digestion program | Step | Power (W) | Time (min) | Temp. (°C) | Power (W) | Time (min) | Temp. (°C) | Power (W) | Time (min) | Temp. (°C) | Hold. (min) |
| 1 | 700 | 6 | 150 | 700 | 11 | 200 | 1600 | 5 | 150 | 6 | |
| 2 | 800 | 5 | 200 | 800 | 16 | 240 | 1600 | 5 | 200 | 5 | |
| 3 | 800 | 18 | 215 | 0 | 30 | 0 | 1600 | 5 | 215 | 18 | |
| 4 | 0 | 25 | 0 | - | - | - | 1600 | 5 | 0 | 25 | |
| No. of digestions | n = 5 | n = 3 | n = 6 | n = 6 | |||||||
| Sample mass digested | 0.2 g | 0.1 g (0.5 g for Acid (3)) | 0.2 g | 0.2 g | |||||||
| Final volume | 50 mL (PP tube); filled with 2 mol/L HCl s.p (Acid (1)); H2O deionized (Acid (2) and Acid (3)) | 50 mL (PP tube), filled with 2 mol/L HCl s.p. (Acid (1) and Acid (2)); H2O deionized (Acid (3)) | 50 mL (PP tube); filled with 0.5 M HNO3 | ||||||||
| Validation | Element spikes for samples added before digestion. | Element spikes for samples added before digestion. Dilution experiments (1:10, 1:100). Certified reference material CCRMP REE-1. | Element spikes for samples and blinds added before digestion as well as for liquid standards without digestion. | ||||||||
| Analytical determination | |||||||||||
| ICP-OES name | Varian Vista Pro Radial | Varian Vista Pro Radial | Thermo Scientific iCap 6000 Series | ||||||||
| ICP-OES elements | Al, As, Co, Cu, Fe, Li, Mn, Ni, P, Pb, Sb, Ti, Zn | Ce, La | Al, As, Cd, Co, Cu, Fe, Mn, Ni, P, Pb, Sb, Zn | As, Ba, Ce, Co, Cr, Dy, Ga, Gd, La, Li, Nb, Nd, Ni, Pr, Sm, Sn, Sr, V, Y, Yb, Zn, Zr | |||||||
| ICP-MS name | Agilent 8800 QQQ | Agilent 8800 QQQ | Thermo Scientific iCap Q | - | |||||||
| ICP-MS elements | Au (O2), Cd (He), Ce (O2), La (O2), Pd (He), V (O2) | As (He), Ba (O2), Co (O2), Cr (He), Dy (O2), Ga (O2), Gd (O2), Li (He), Nb (O2), Nd (He), Ni (He), Pr (O2), Sm (He), Sn (He), Sr (O2), W (He), Yb (He), Zr (He) | As, Au, Cd, Ce, La, P, Pb, Pd, Sb, Zn | - | |||||||
| ICP-MS gas mode | He (collision), O2 (reaction gas) | He (collision), O2 (reaction gas) | He (collision) | - | |||||||
| Specification | Validated Method |
|---|---|
| Pre-Treatment | |
| Samples | MIN |
| Fusion | 8 g Li₂B₄O₇ and LiBO2 |
| Sample mass | 1 g |
| Muffle furnace | Fluxana Vitriox |
| Temperature | 1200 °C |
| Number of replicates | n = 5 |
| Validation | Certified reference sample CCRMP REE 1 |
| WD-XRF Measurement | |
| WD-XRF device name | Primus IV (Rigaku, Japan) |
| Calibration | Standardless calibration |
| X-ray source | Rhodium source 4 kW |
| Measurement time | 10 min |
| WD-XRF elements | Al, Ca, Fe, K, Mg, Mn, Na, P, Rb, Si, V, Y, Zn |
| LOD | Approximately 0.01% |
| Spiked Sample | Objective | Analysis Procedure | |
|---|---|---|---|
| Liquid standards | RRL | Test of measurement accuracy without the influence of acid digestion or sample matrix. | Multi-element standards (Ag, Al, As, Ba, Cd, Co, Cr, Cu, Fe, Li, Mg, Mn, Mo, Na, Ni, Pb, Sb, Sr, Ti, V, Zn) in different concentrations were dissolved in HNO3 and measured with ICP-OES (Supplementary Materials Table S2). |
| Blind samples | RRB | Test of measurement accuracy with influence caused by digestion acid due to, e.g., volatilization, complex formation, precipitation, etc. but without the influence of the sample matrix. | Element standards for all elements and acids mixtures added before digestion and measured with ICP-OES/MS. |
| Sample matrices (BATT, MIN) | RRS | Test of measurement accuracy with both influences, acid digestion and sample-specific, such as chemical reactions, overlapping of measuring lines (ICP-OES) or element masses (ICP-MS). | Element standards for all elements and acids mixtures added before digestion and measured with ICP-OES/MS. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mählitz, P.M.; Løvik, A.N.; Figi, R.; Schreiner, C.; Kuntz, C.; Korf, N.; Rösslein, M.; Wäger, P.; Rotter, V.S. Characterizing the Urban Mine—Challenges of Simplified Chemical Analysis of Anthropogenic Mineral Residues. Resources 2019, 8, 132. https://doi.org/10.3390/resources8030132
Mählitz PM, Løvik AN, Figi R, Schreiner C, Kuntz C, Korf N, Rösslein M, Wäger P, Rotter VS. Characterizing the Urban Mine—Challenges of Simplified Chemical Analysis of Anthropogenic Mineral Residues. Resources. 2019; 8(3):132. https://doi.org/10.3390/resources8030132
Chicago/Turabian StyleMählitz, Paul Martin, Amund N. Løvik, Renato Figi, Claudia Schreiner, Claudia Kuntz, Nathalie Korf, Matthias Rösslein, Patrick Wäger, and Vera Susanne Rotter. 2019. "Characterizing the Urban Mine—Challenges of Simplified Chemical Analysis of Anthropogenic Mineral Residues" Resources 8, no. 3: 132. https://doi.org/10.3390/resources8030132
APA StyleMählitz, P. M., Løvik, A. N., Figi, R., Schreiner, C., Kuntz, C., Korf, N., Rösslein, M., Wäger, P., & Rotter, V. S. (2019). Characterizing the Urban Mine—Challenges of Simplified Chemical Analysis of Anthropogenic Mineral Residues. Resources, 8(3), 132. https://doi.org/10.3390/resources8030132