
ZVI (Fe0) Desalination: Stability of Product Water

Abstract
:1. Introduction
- Gas-pressured static diffusion technology [2].
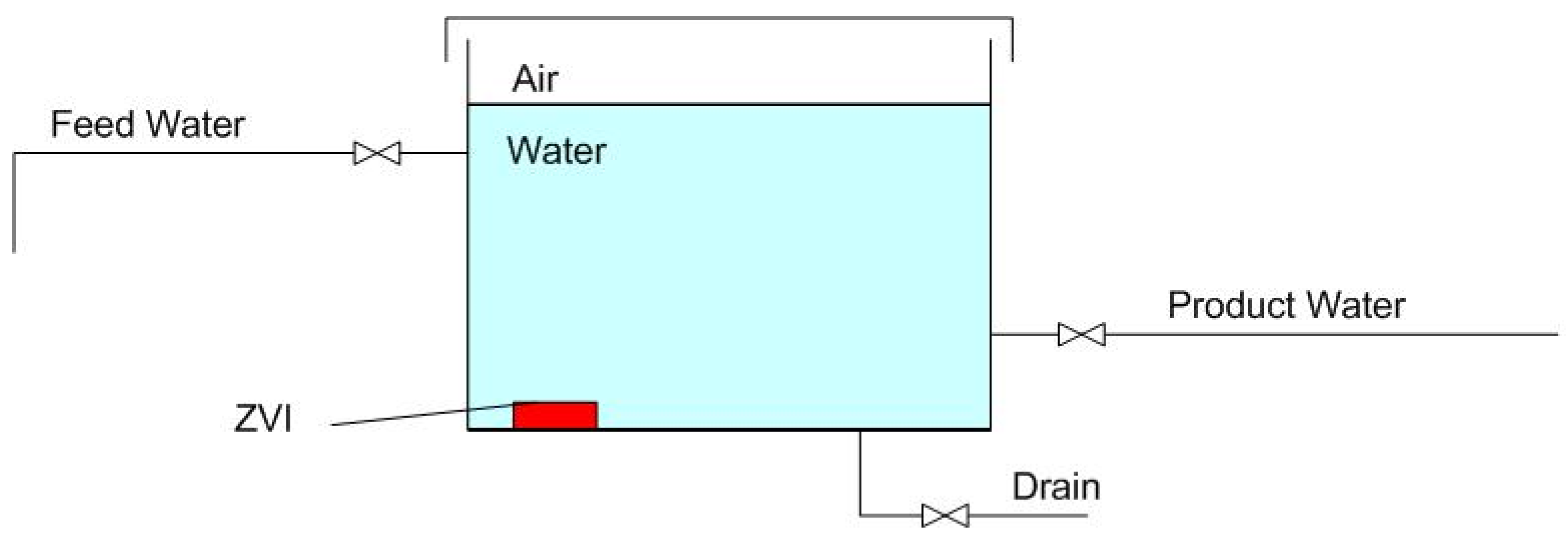
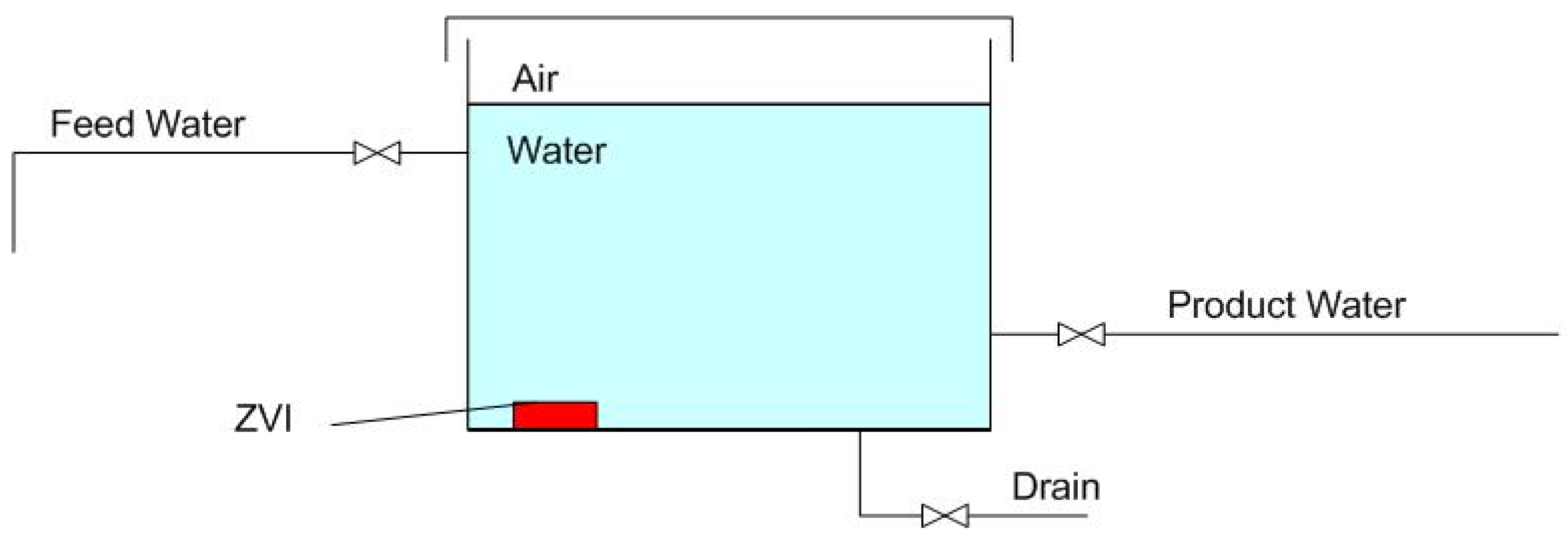
1.1. Static Diffusion ZVI Partial Desalination
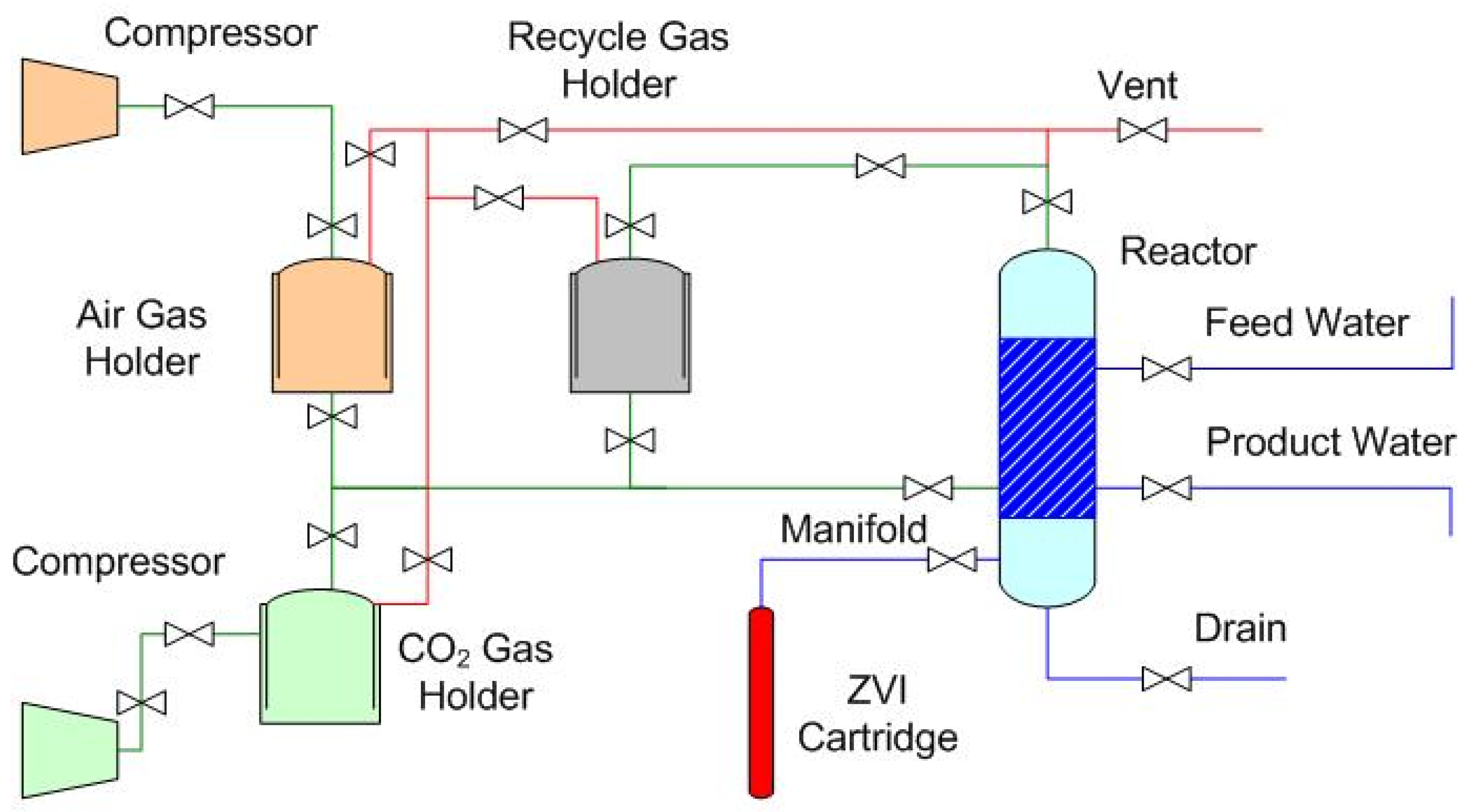
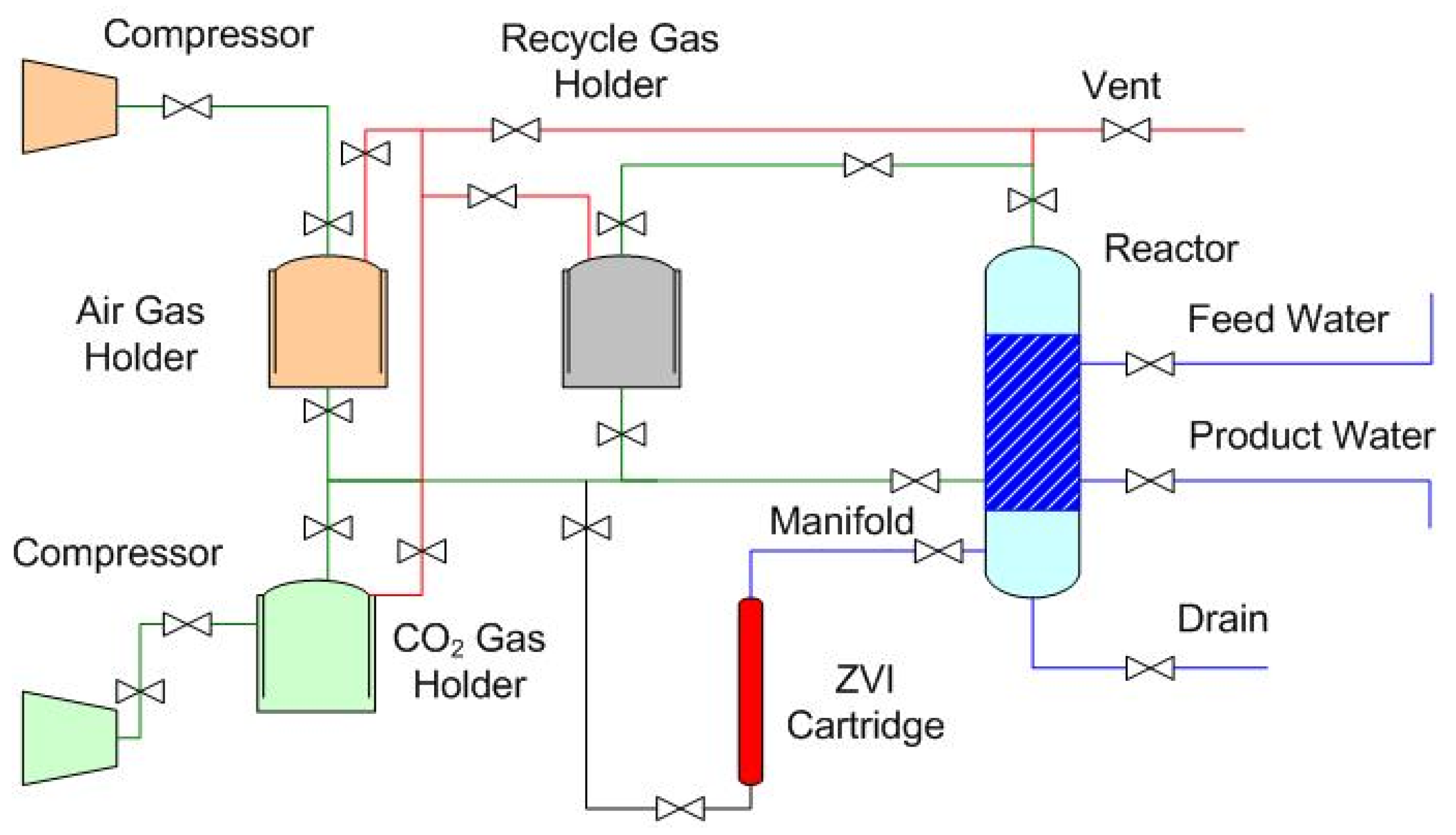
1.2. Gas-Pressured Static Diffusion ZVI Partial Desalination
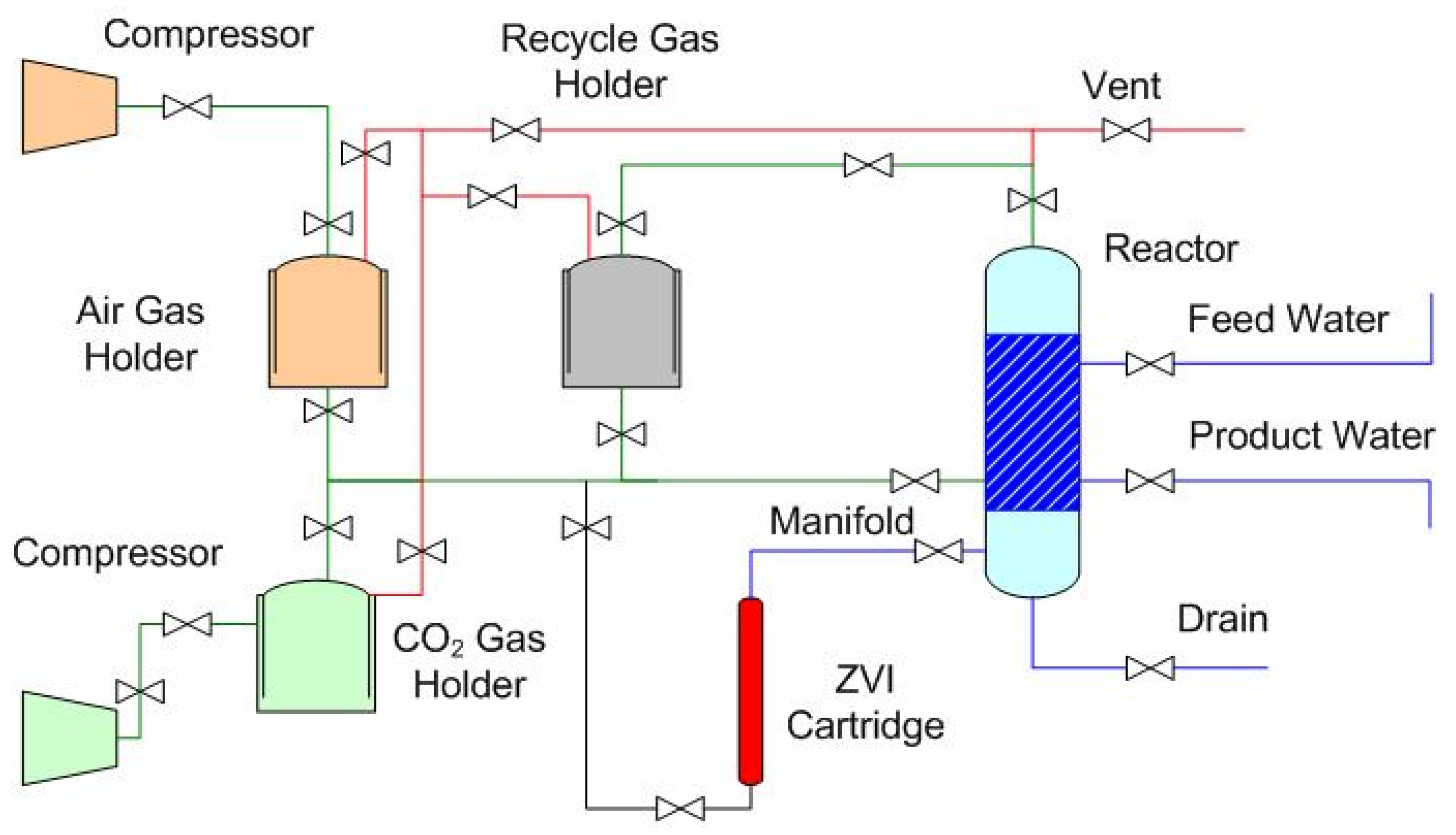
1.2.1. Indicative Dimensions and Costs of a Gas-Pressured Reactor with a Batch Capacity of 10 m3.
- the periodic replacement of ZVI;
- the provision of air and the provision of CO2 [2].
1.2.2. Stored Product Water
1.3. Appendices
- Appendix A: Ion Analyses;
- Appendix B: Summary of Reaction Route A;
- Appendix C: Summary of Reaction Route B;
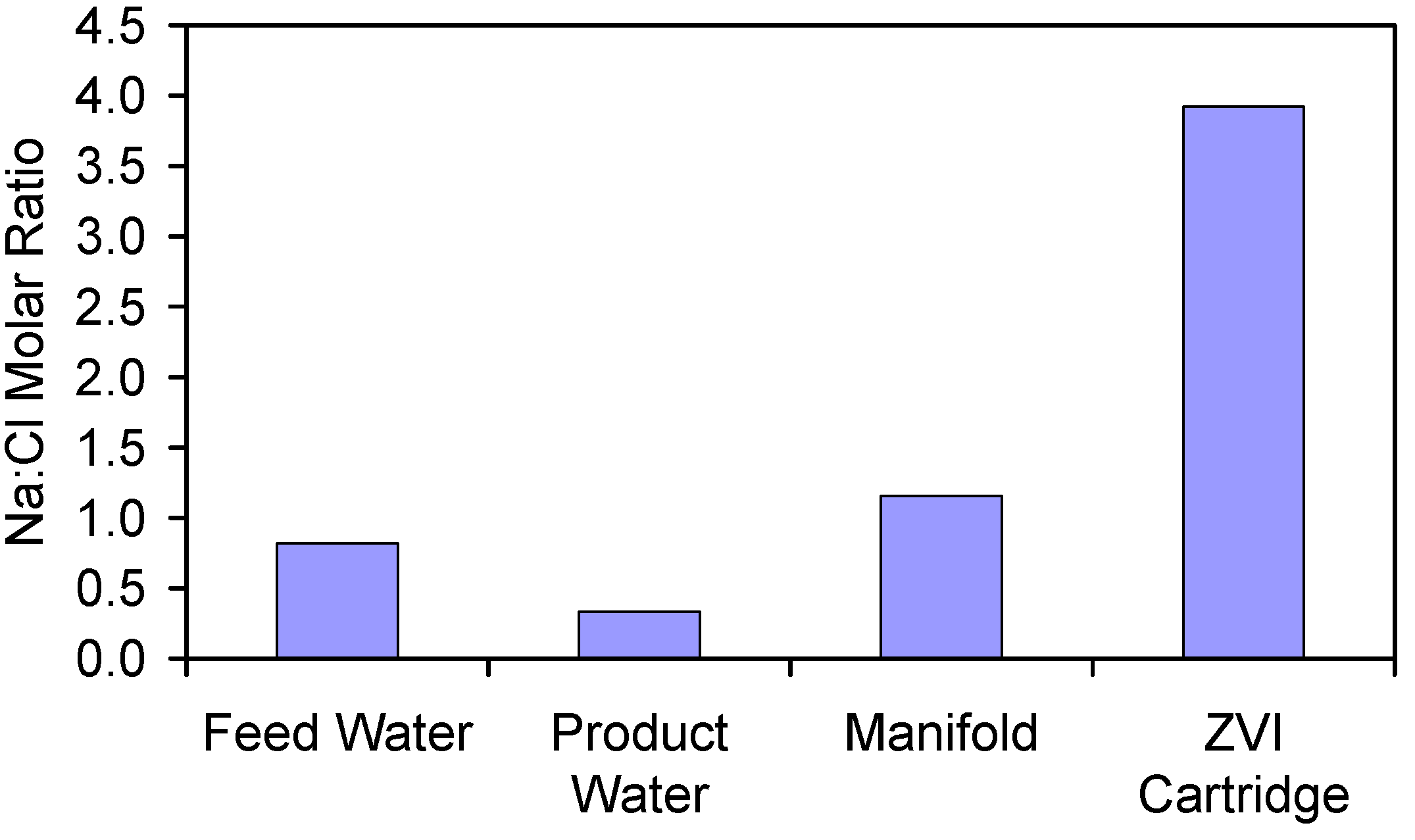
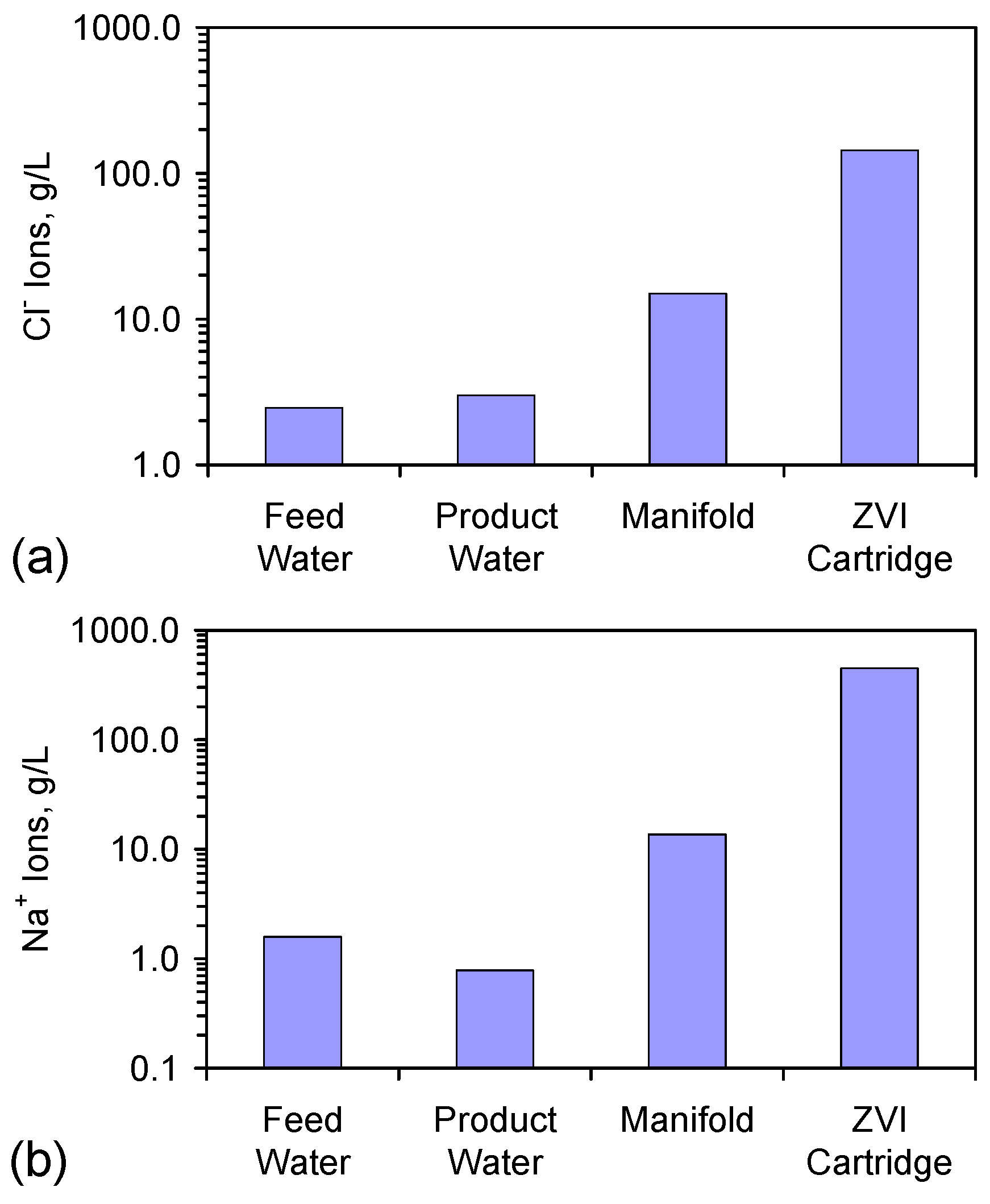
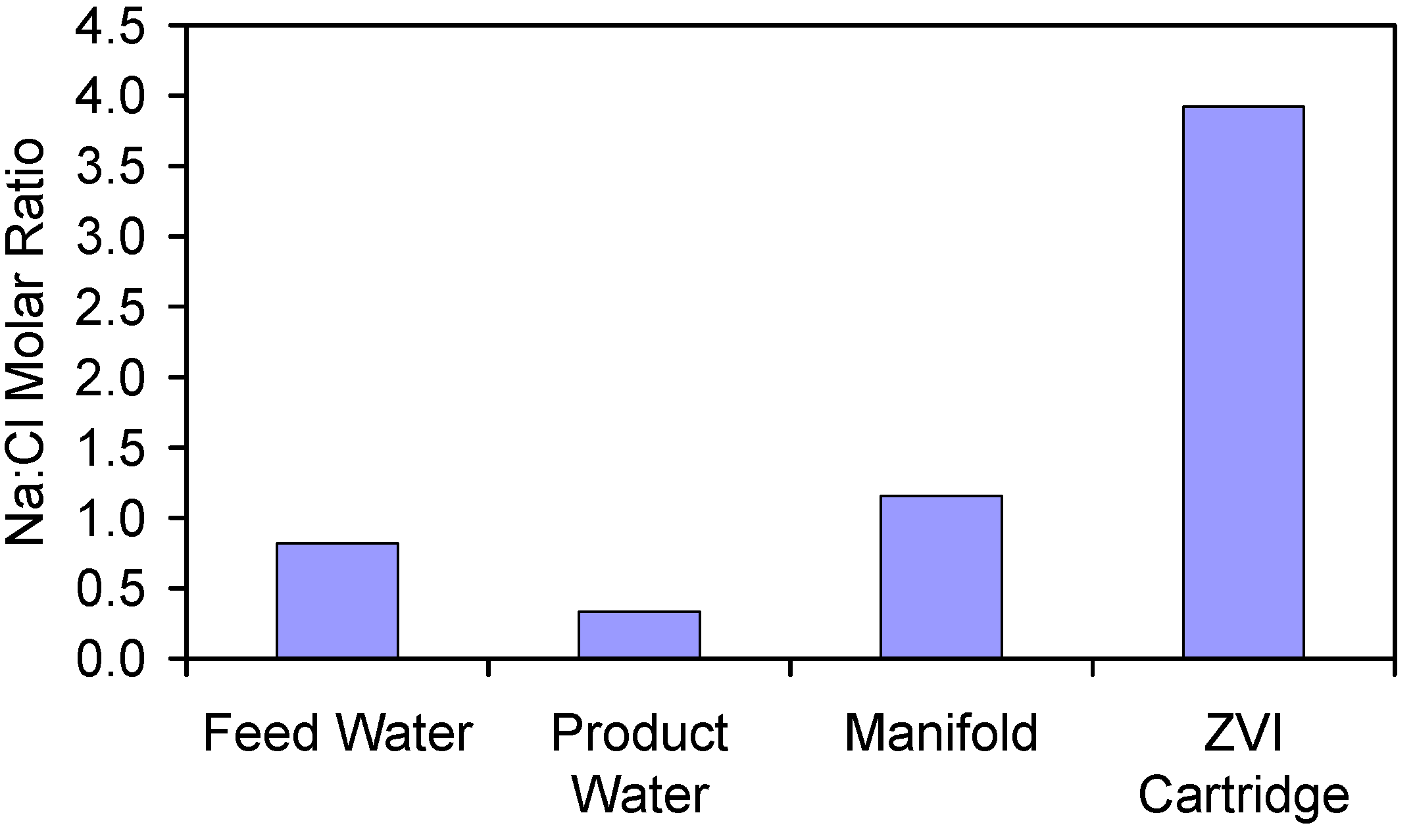
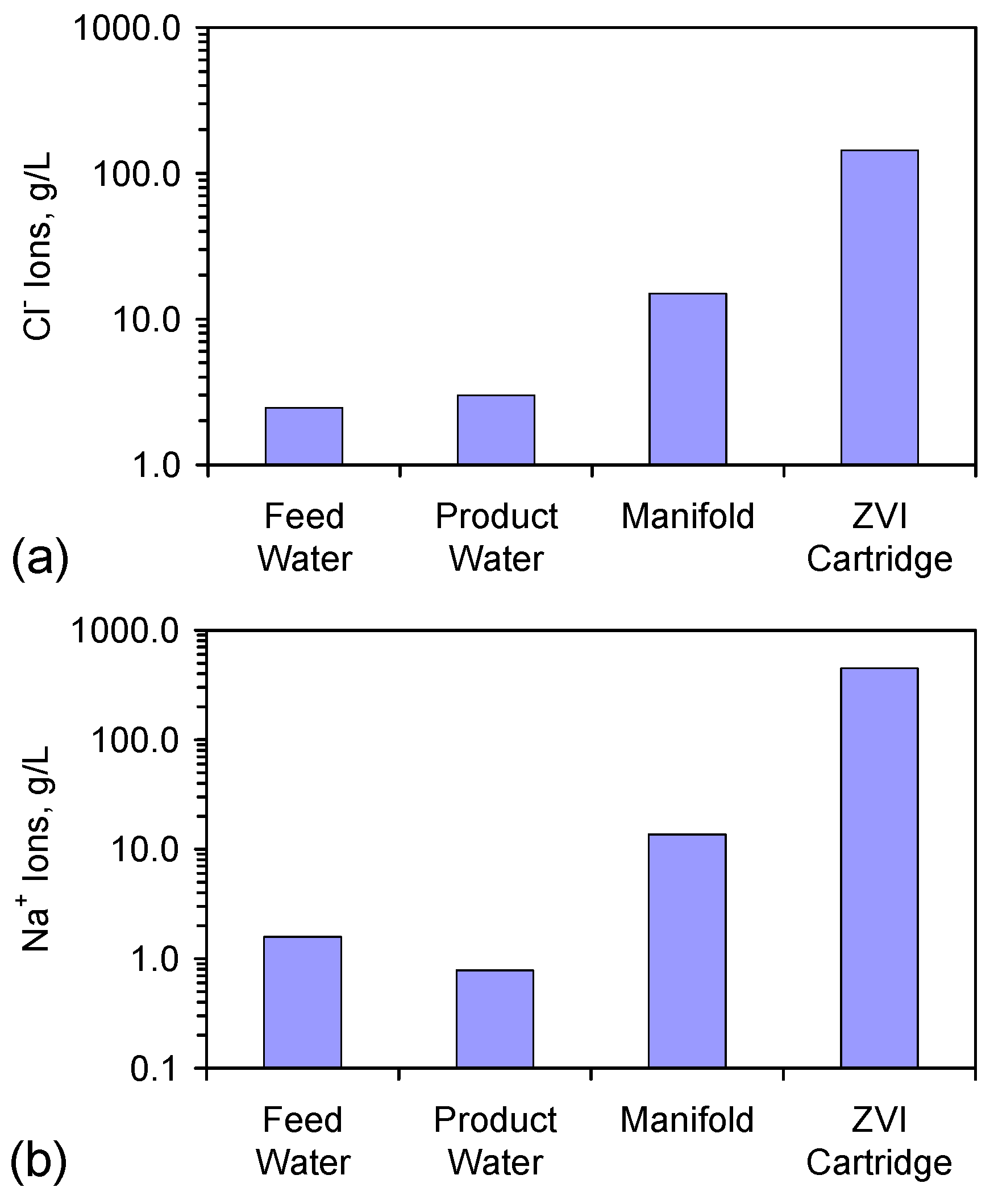
- Appendix D: The Significance of Increased Na+ and Cl− Ion Concentrations in the Manifold and ZVI Cartridge (Figure D1 and Figure D2);
- Appendix H: Desalination Pathway (Figure H1);
2. Methods and Analysis of Stored Product Water
2.1. Water Samples
2.2. ZVI and Trial Details
2.3. Saline Water Construction
2.4. Salinity Measurement
3. Results
3.1. Principal Operating Differences between Trial Series CSD1, E143, E144
3.2. Initial Salinity Calculation Approach in Trial Series CSD1, E143, E144
3.3. Observed Stored Water Salinities for Trial Series CSD1, E143, E144
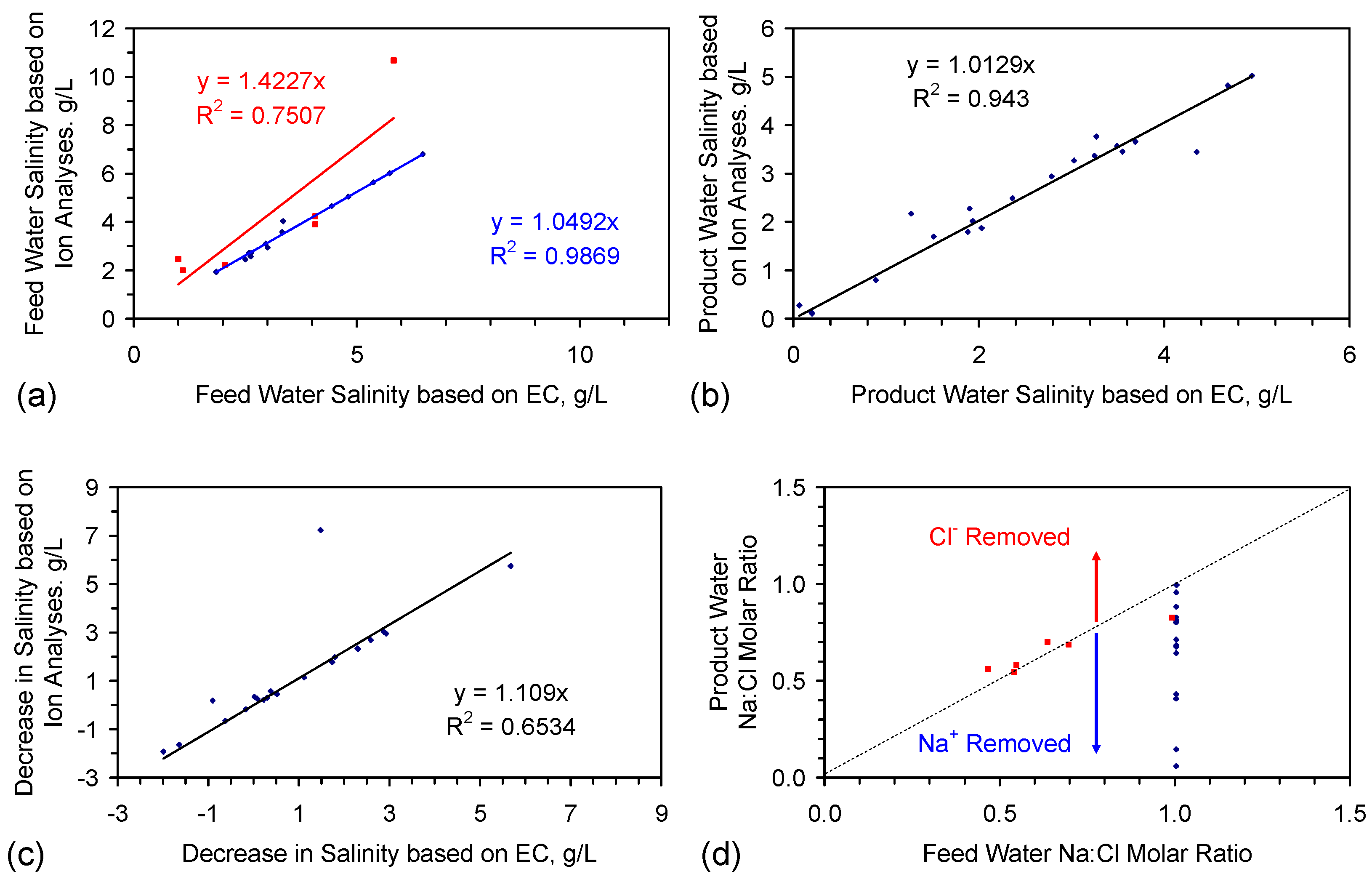
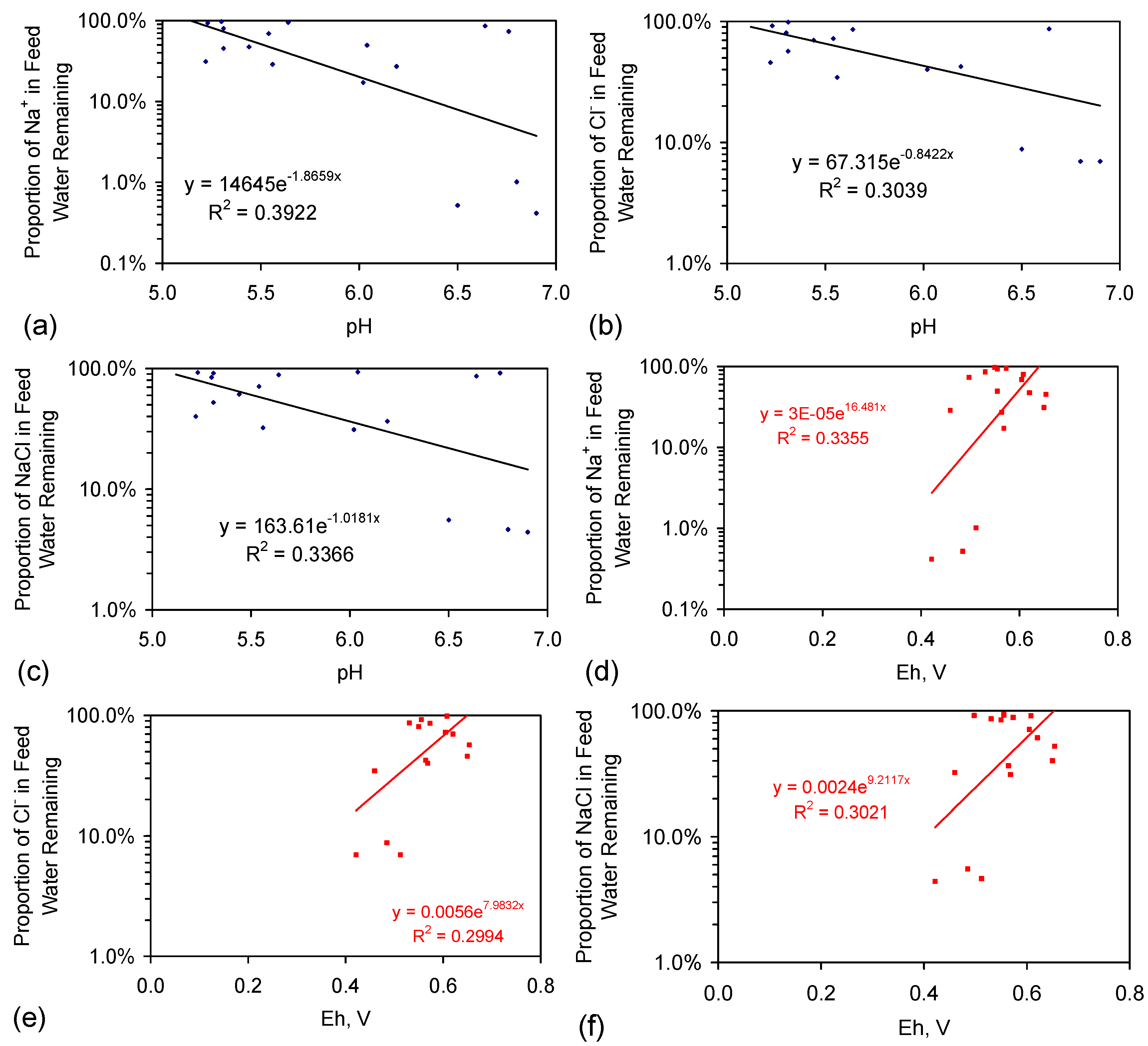
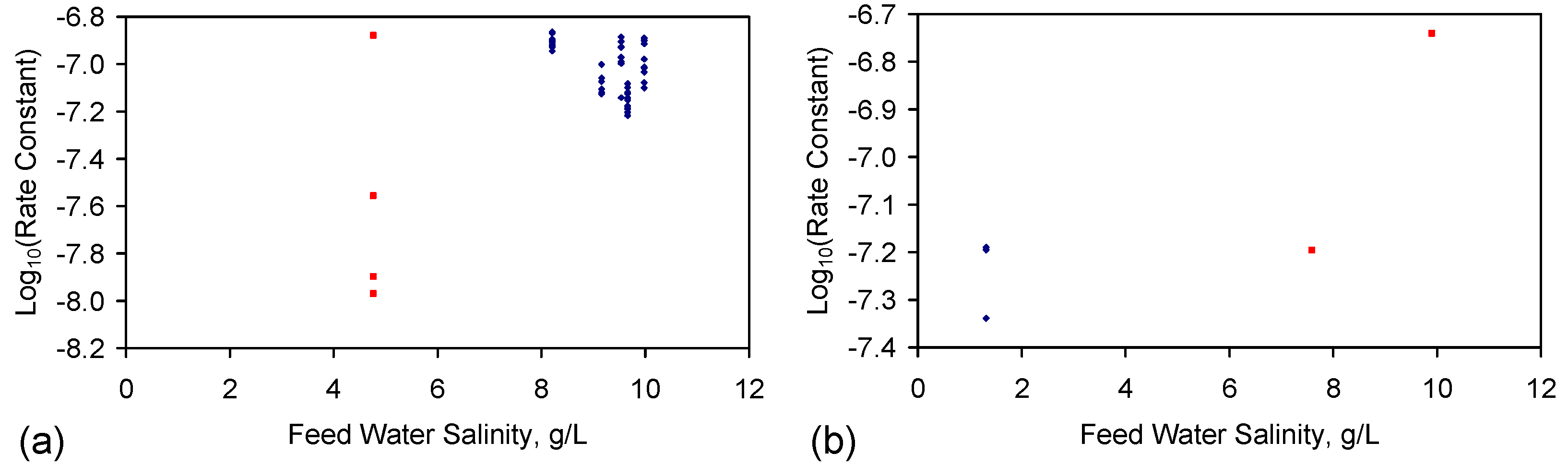
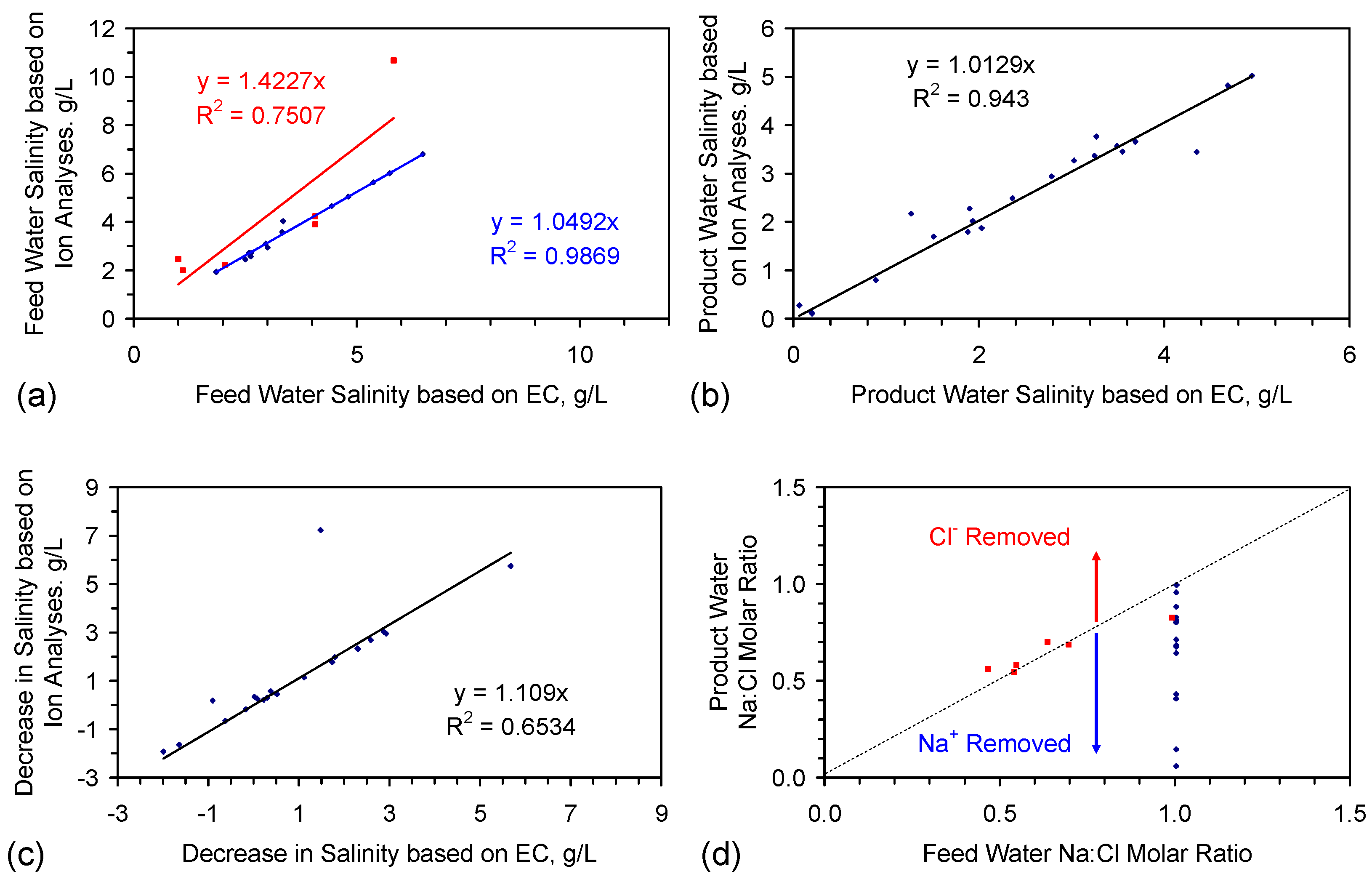
- EC analyses provide a reasonable estimate of feed water salinity when the saline water is constructed from chemically-pure NaCl (Figure 3a), i.e., {F} approximates to 0.54;
- EC analyses can underestimate the salinity of feed water, when the feed water is constructed using halite (E144 Trial Series) (Figure 3a), i.e., {F} is greater than 0.54;
- EC analyses provide a reasonable indication of product water salinity. The regression correlation indicates that the product water salinity has remained unchanged while the samples were held in storage (Figure 3b);
- Decreases in salinity based on EC may underestimate the actual change in salinity by about 10% (Figure 3c);
- The product water molar Na:Cl ratios indicate that preferential removal of Na+ ions (relative to Cl− ions) can occur (and vice versa) during the desalination process (Figure 3d).
4. Discussion
- accelerate the rate of ZVI desalination to allow small compact units to be used to produce the product water;
- produce a product water that is stable when placed in storage;
- adjust the Na:Cl molar ratio in the product water to optimize irrigation water quality and maximize crop yields.
5. Conclusions
Acknowledgments
Conflicts of Interest
Appendix A: Tables of Data and Ion Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | Maximum Temperature (°C) | Duration (h) |
|---|---|---|
| CSD1a | 50 | 15 |
| CSD1b | 44 | 124 |
| CSD1c | 39 | 30 |
| CSD1d | 36 | 11 |
| Trial | Feed Water (g·L−1) | Product Water (g·L−1) | ||||
|---|---|---|---|---|---|---|
| Na+ | Cl− | NaCl | Na+ | Cl− | NaCl | |
| CSD1a | 0.962 | 1.489 | 2.452 | 0.005 | 0.131 | 0.136 |
| CSD1b | 0.962 | 1.489 | 2.452 | 0.004 | 0.104 | 0.108 |
| CSD1c | 1.009 | 1.562 | 2.570 | 0.173 | 0.625 | 0.799 |
| CSD1d | 1.155 | 1.788 | 2.943 | 0.545 | 1.251 | 1.796 |
| Trial | Feed Water Volume (L) | Product Water Volume (L) | Duration (h) | Air Flow Duration (%) |
|---|---|---|---|---|
| E143a | 5.8 | 5.0 | 9.00 | 100.0% |
| E143b | 5.8 | 5.0 | 8.33 | 60.6% |
| E143c | 5.8 | 5.1 | 4.37 | 58.6% |
| E143d | 5.8 | 5.1 | 4.02 | 43.8% |
| E143e | 5.8 | 5.1 | 3.87 | 44.2% |
| E143f | 5.8 | 5.2 | 2.70 | 100.0% |
| E143g | 5.8 | 5.0 | 3.08 | 100.0% |
| E143h | 5.8 | 4.3 | 14.30 | 100.0% |
| E143i | 5.8 | 4.4 | 8.90 | 50.6% |
| E143j | 5.8 | 4.3 | 7.50 | 29.3% |
| E143k | 5.8 | 4.3 | 3.58 | 72.1% |
| Trial | Feed Water (g·L−1) | Product Water (g·L−1) | ||||
|---|---|---|---|---|---|---|
| Na+ | Cl− | NaCl | Na+ | Cl− | NaCl | |
| E143a | 2.363 | 3.658 | 6.021 | 0.024 | 0.255 | 0.279 |
| E143b | 1.829 | 2.831 | 4.660 | 0.497 | 1.201 | 1.698 |
| E143c | 1.982 | 3.067 | 5.049 | 0.617 | 1.403 | 2.020 |
| E143d | 2.210 | 3.422 | 5.632 | 1.001 | 1.940 | 2.942 |
| E143e | 2.668 | 4.130 | 6.799 | 1.837 | 2.985 | 4.821 |
| E143f | 0.761 | 1.178 | 1.939 | 1.294 | 2.277 | 3.570 |
| E143g | 1.068 | 1.653 | 2.722 | 0.784 | 1.709 | 2.494 |
| E143h | 1.068 | 1.653 | 2.722 | 1.171 | 2.199 | 3.370 |
| E143i | 1.219 | 1.886 | 3.105 | 1.961 | 3.064 | 5.026 |
| E143j | 1.405 | 2.175 | 3.580 | 1.123 | 2.147 | 3.270 |
| E143k | 1.585 | 2.454 | 4.039 | 0.784 | 2.985 | 3.769 |
| Trial | Product Water (Adjusted for Water Loss) (g·L−1) | Net Ion Removal (g·L−1) | ||||
|---|---|---|---|---|---|---|
| Na+ | Cl− | NaCl | Na+ | Cl− | NaCl | |
| E143a | 0.0207 | 0.2198 | 0.2405 | 2.342 | 3.438 | 5.780 |
| E143b | 0.4284 | 1.0353 | 1.4638 | 1.401 | 1.796 | 3.196 |
| E143c | 0.5425 | 1.2337 | 1.7762 | 1.439 | 1.833 | 3.273 |
| E143d | 0.8802 | 1.7059 | 2.5869 | 1.330 | 1.716 | 3.045 |
| E143e | 1.6153 | 2.6247 | 4.2392 | 1.053 | 1.505 | 2.560 |
| E143f | 1.1490 | 2.0218 | 3.1699 | −0.388 | −0.844 | −1.231 |
| E143g | 0.6759 | 1.4733 | 2.1500 | 0.392 | 0.180 | 0.572 |
| E143h | 0.8682 | 1.6303 | 2.4984 | 0.200 | 0.023 | 0.224 |
| E143i | 1.4877 | 2.3244 | 3.8128 | −0.269 | −0.438 | −0.708 |
| E143j | 0.8326 | 1.5917 | 2.4243 | 0.572 | 0.583 | 1.156 |
| E143k | 0.5812 | 2.2130 | 2.7943 | 1.004 | 0.241 | 1.245 |
| Item | Na (g·L−1) | Cl (g·L−1) | NaCl (g·L−1) | Na:Cl Molar Ratio |
|---|---|---|---|---|
| Feed Water | 1.585 | 2.454 | 4.039 | 1.000 |
| Product Water | 0.784 | 2.985 | 3.769 | 0.407 |
| Manifold Water | 13.653 | 14.966 | 28.620 | 0.912 |
| ZVI Cartridge Water | 446.678 | 144.185 | 590.862 * | 3.098 |
| Trial | Analyses Based on EC | Direct Ion Analyses | NaCl Removed (g·L−1) | NaCl Removed * (g·L−1) | ||||
|---|---|---|---|---|---|---|---|---|
| Feed Water (g·L−1) | Product Water (g·L−1) | Feed Water (g·L−1) | Product Water (g·L−1) | EC Analyses | Ion Analyses | EC Analyses | Ion Analyses | |
| E143a | 5.74 | 0.06 | 6.02 | 0.28 | 5.68 | 5.74 | 5.688 | 5.780 |
| E143b | 4.44 | 1.51 | 4.66 | 1.70 | 2.93 | 2.96 | 3.138 | 3.196 |
| E143c | 4.81 | 1.93 | 5.05 | 2.02 | 2.88 | 3.03 | 3.113 | 3.273 |
| E143d | 5.37 | 2.79 | 5.63 | 2.94 | 2.58 | 2.69 | 2.917 | 3.045 |
| E143e | 6.48 | 4.69 | 6.8 | 4.82 | 1.79 | 1.98 | 2.356 | 2.560 |
| E143f | 1.85 | 3.49 | 1.94 | 3.57 | −1.64 | −1.63 | −1.249 | −1.231 |
| E143g | 2.59 | 2.36 | 2.72 | 2.49 | 0.23 | 0.23 | 0.556 | 0.572 |
| E143h | 2.63 | 3.25 | 2.72 | 3.37 | −0.62 | −0.65 | 0.221 | 0.224 |
| E143i | 2.96 | 4.95 | 3.11 | 5.03 | −1.99 | −1.92 | −0.795 | −0.708 |
| E143j | 3.33 | 3.03 | 3.58 | 3.27 | 0.30 | 0.31 | 1.084 | 1.156 |
| E143k | 3.35 | 3.27 | 4.04 | 3.77 | 0.08 | 0.27 | 0.926 | 1.245 |
| Trial | Feed Water Volume (L) | Product Water Volume (L) | Duration (h) | Air Flow Duration (%) |
|---|---|---|---|---|
| E144a | 5.8 | 4.4 | 6.48 | 58.64% |
| E144b | 5.8 | 4.4 | 104.30 | 67.59% |
| E144c | 5.8 | 4.9 | 690.00 | 0.00% |
| E144d | 5.8 | 4.6 | 291.00 | 0.00% |
| E144e | 5.8 | 4.4 | 537.00 | 0.00% |
| E144f | 5.8 | 3.9 | 1295.00 | 0.00% |
| Trial | Feed Water (g·L−1) | Product Water (g·L−1) | ||||
|---|---|---|---|---|---|---|
| Na+ | Cl− | NaCl | Na+ | Cl− | NaCl | |
| E144a | 1.134 | 2.770 | 3.904 | 1.074 | 2.381 | 3.455 |
| E144b | 1.312 | 2.925 | 4.237 | 1.123 | 2.537 | 3.660 |
| E144c | 0.636 | 1.826 | 2.462 | 0.593 | 1.684 | 2.276 |
| E144d | 0.522 | 1.480 | 2.002 | 0.593 | 1.582 | 2.174 |
| E144e | 0.512 | 1.707 | 2.219 | 0.497 | 1.378 | 1.875 |
| E144f | 4.161 | 6.519 | 10.680 | 1.196 | 2.251 | 3.446 |
| Trial | Product Water (Adjusted for Water Loss) (g·L−1) | Net Ion Removal (g·L−1) | ||||
|---|---|---|---|---|---|---|
| Na+ | Cl− | NaCl | Na+ | Cl− | NaCl | |
| E144a | 0.8148 | 1.8063 | 2.6210 | 0.319 | 0.964 | 1.283 |
| E144b | 0.8519 | 1.9246 | 2.7766 | 0.460 | 1.000 | 1.460 |
| E144c | 0.5010 | 1.4227 | 1.9228 | 0.135 | 0.403 | 0.539 |
| E144d | 0.4652 | 1.2411 | 1.7055 | 0.057 | 0.239 | 0.297 |
| E144e | 0.3770 | 1.0454 | 1.4224 | 0.135 | 0.662 | 0.797 |
| E144f | 0.8042 | 1.5136 | 2.3171 | 3.357 | 5.005 | 8.363 |
| Trial | Trial Analyses Based on EC | Direct Ion Analyses | NaCl Removed (g·L−1) | |||
|---|---|---|---|---|---|---|
| Feed Water (g·L−1) | Product Water (g·L−1) | Feed Water (g·L−1) | Product Water (g·L−1) | EC Analyses | Ion Analyses | |
| E144a | 4.07 | 3.55 | 3.904 | 3.455 | 0.52 | 0.449 |
| E144b | 4.07 | 3.69 | 4.237 | 3.66 | 0.38 | 0.577 |
| E144c | 1 | 1.9 | 2.462 | 2.276 | −0.9 | 0.186 |
| E144d | 1.1 | 1.27 | 2.002 | 2.174 | −0.17 | −0.172 |
| E144e | 2.05 | 2.03 | 2.219 | 1.875 | 0.02 | 0.344 |
| E144f | 5.83 | 4.35 | 10.68 | 3.446 | 1.48 | 7.234 |
Appendix B: Reaction Route Group A
- direct reaction with ZVI to produce a product;
- catalysed reaction involving ZVI;
- removal in hydration shells;
- adsorption by ZVI.
B.1. Direct Reaction with ZVI
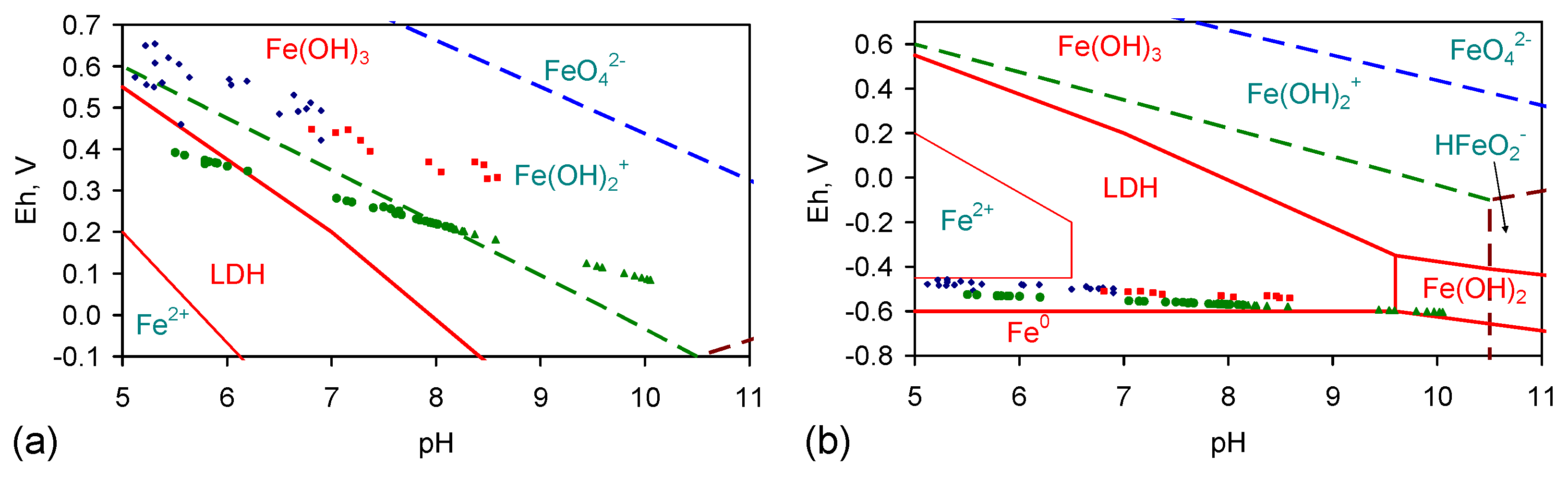
- β-FeOOH (akaganeite), e.g., [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53], where the Na+ and Cl− ions are concentrated in tunnels within the structure and in its hydrated ionic shell [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. β-FeOOH is the dominant FeOOH corrosion species when the water contains significant concentrations of Cl− ions [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53], i.e., Log(Cl−/OH−) is greater than 1.16 and preferably greater than eight [2,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. The general ZVI corrosion route is [2,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]:Fe0 → Fe(OH)2→ GR1(Cl−)→β-FeOOH (akaganeite)—medium Eh route [2]
Fe0→Fe(OH)2→Fe(OH)3→β-FeOOH—high Eh route [2]
Fe0→FeII→FeII + FeIII→FeIII [2] - Green Rust 1 (chloride) ((NaI(a = 1 – c − b)FeII(b = 1 − c − a)FeIIIc(OH)g]x−·[(x/n)Cln−·mH2O]x+))[2] (GR1(Cl−)), ferrous hydroxychloride, e.g., [54,55,56,57,58,59,60,61]. The Na+ and Cl− ions are concentrated within the layered molecular structure [54,55,56,57,58,59,60,61]. The general ZVI corrosion route is [2,54,55,56,57,58,59,60,61]:Fe0→Fe(OH)2→GR1(Cl−)—low Eh route [2]
Fe0→Fe(OH)2→GR1(HCO3−)→GR1(Cl−)—low Eh route coupled with decreasing pH [2]
Fe0→Fe(OH)2→GR1(HCO3−)→GR1(Cl−)→GR1(SO3−)→GR2(SO4−) [2]
Fe0→FeII→FeII + FeIII [2]
- 3.
B.2. Direct Catalytic Reaction with ZVI
- A role as a catalyser [65]:Fe0 + nCl− = Fen+nCl− + ne− (production of intermediate)
Fen+nCl− = Fen+ + nCl− (ionisation of Fe)
Fe0→FeII→FeII + FeIII→FeIII→FeIV
- 2.
- A role as phase distributor [65]: The end corrosion product (rust) at the water ZVI interface is FeOOH (Equation (B1)). The formation of the surface rust (FeOOH) species results in the initial corrosion (-OH) terminal groups at the rust-water interface changing to (-OH2)+ groups [65]. This change both attracts Cl− ions and allows Cl− ions to migrate through the rust to the metal surface [65] (e.g., Appendix E).
B.3. Removal in Hydration Shells
- Negatively-charged terminal surface: which can be defined as FeOH0.5− and takes the general structure: (OH)-(OH)-Fe-O-O-Fe-R (where R = a repeat of the stoichiometric atomic layer sequence or tethering surface. R can include hydrated layers) [2]. A double hydrated terminal surface takes the form: (H2O)-(H2O)-(OH)-(OH)-Fe-O-O-Fe-R [2]. This surface type has a negative charge and can be expected to remove Na+ ions from the water [2].
- Positively-charged interface terminal surface: which can be defined as FeOH20.5+ and takes the general structure: (OH2)-(OH)-Fe-O-O-Fe-R [2]. A double hydrated interface terminal surface takes the form: (H2O)-(H2O)-(OH2)-(OH)-Fe-O-O-Fe-R [2]. This surface type has a positive charge and can be expected to remove Cl− ions from the water [2].
B.4. Adsorption
≡Fe-OH = ≡Fe-O− + H+
≡Fe-OH + L− (e.g., Cl−) = ≡Fe-L (e.g., Fe-Cl) + OH−
Appendix C: Reaction Route Group B
NO3− + H3O+[H2O]2 = NH4+ + 3HO− + 1.5O2 (g,aq)
2NO3− + 12H+ + 10e− = N2 (g, aq) + 6H2O
NO3− + 2H+ = NO2− + H2O
C.1. n-Fe Surface-Based Reaction or Aqueous ZVM Ion-Based Reaction [71]
HzCxCly (corrosion product or pollutant) + e− = HzCxCly−
HzCxCly− + Fen+ = [HzCxCly-Fe](n−1)+
[HzCxCly- Fe](n−1)+ + H2O = [Hz+1CxCly-1] + Fe-Cl(n−1)+ (ion adduct) + OH + e−
xCs(+/−) sites form in the corroding iron when the iron contains carbon [2].
C.2. n-Fe-Hydroxide/Peroxide Surface-Based Reaction or Aqueous Fe-Hydroxide/Peroxide Ion-Based Reaction [71]
FejOkHdn+ + HzCxCly (corrosion product or pollutant) + e− = [HzCxCly-FejOkHd](n−1)+
[HzCxCly-FejOkHd](n−1)+ + H2O = [Hz+1CxCly-1] + FejOk+1Hd+1(n−2)+ + Cl + e−
C.3. n-Fe Surface-Based Reaction or Aqueous ZVM Ion-Based Reaction [71]
HzCx N3Cly− + Fen+ = [HzCx N3Cly-Fe](n−1)+
[HzCx N3Cly-Fe](n−1)+ + H2O = [Hz+1Cx N3Cly-1] + FeCl(n−1)+ + OH + e−
C.4. Implications of Surface-Based Reactions
- the Na+ and Cl− ion concentrations measured in the product water within the reactor to decline as the concentration of radical products increased;
- the desalination reactions to be reversed in the stored water, as the pH changes;
- the relative molar proportion of Na:Cl ions in the product water to be adjusted during reactor operation.
C.5. Pressurization by CO2
C.6. Pressurization by O2
C.7. Generic ZVI Reactions Associated with CO2 and O2 Gas Charging
C.8. Na Removal Associated with Fe Valency Changes
(≡[(OH)6(OH2)3](6 − n)−) + (6-n)Na+ = (≡[(OH)6(OH2)3](6−n)−·(6 − n)Na+)
(≡) = site with a charge of n(+), e.g., ≡(OH2)+
(≡[(OH)6(OH2)3](6 − n)−) can be (≡[(OH)m(OH2)y]·z(OH2)+) (m − n − z)−
In the presence of CO2 the adsorption site may potentially take the form:
(≡[(OH)m(CO2)d(HCO3)e(CO3)f (OH2)y] z(OH2)+) (m + e + 2f − n − z)−
C.9. Catalysed Cl Removal Associated with Fe
HzCxCly-FejOkHd](n − 1)+ = HzCxCly + [FejOkHd]n+ + e−
C.10. UV Absorption Peaks which May Be Associated with Catalysed Cl Removal
C.10.1. UV-Visible Absorbance Peaks Associated with Cl Species
C.10.2. UV-Visible Absorbance Peaks Associated with Nano-Particles Produced during Desalinations
Appendix D: Significance of Higher Residual Salinities in the Manifold and ZVI Cartridge
D.1. Salinity Concentration Associated with ZVI Corrosion Products
D.2. Recovery of NaCl Held in ZVI Corrosion Products
D.3. Significance of Water Volume Reduction
D.3.1. Observed Water Volume Reduction
- Water losses associated with evaporation at the gas: water contact;
- Water losses associated with humidification as a dry gas is bubbled through the water.
D.3.2. Impact of High Water Losses Due to Suboptimal Reactor Design on Desalination Assessment
D.4. Different Na+ and Cl− Ion Concentrations in Different Parts of the Reactor
Appendix E: Location of the Desalination Sites
E.1. Basic Desalination Model: Observations
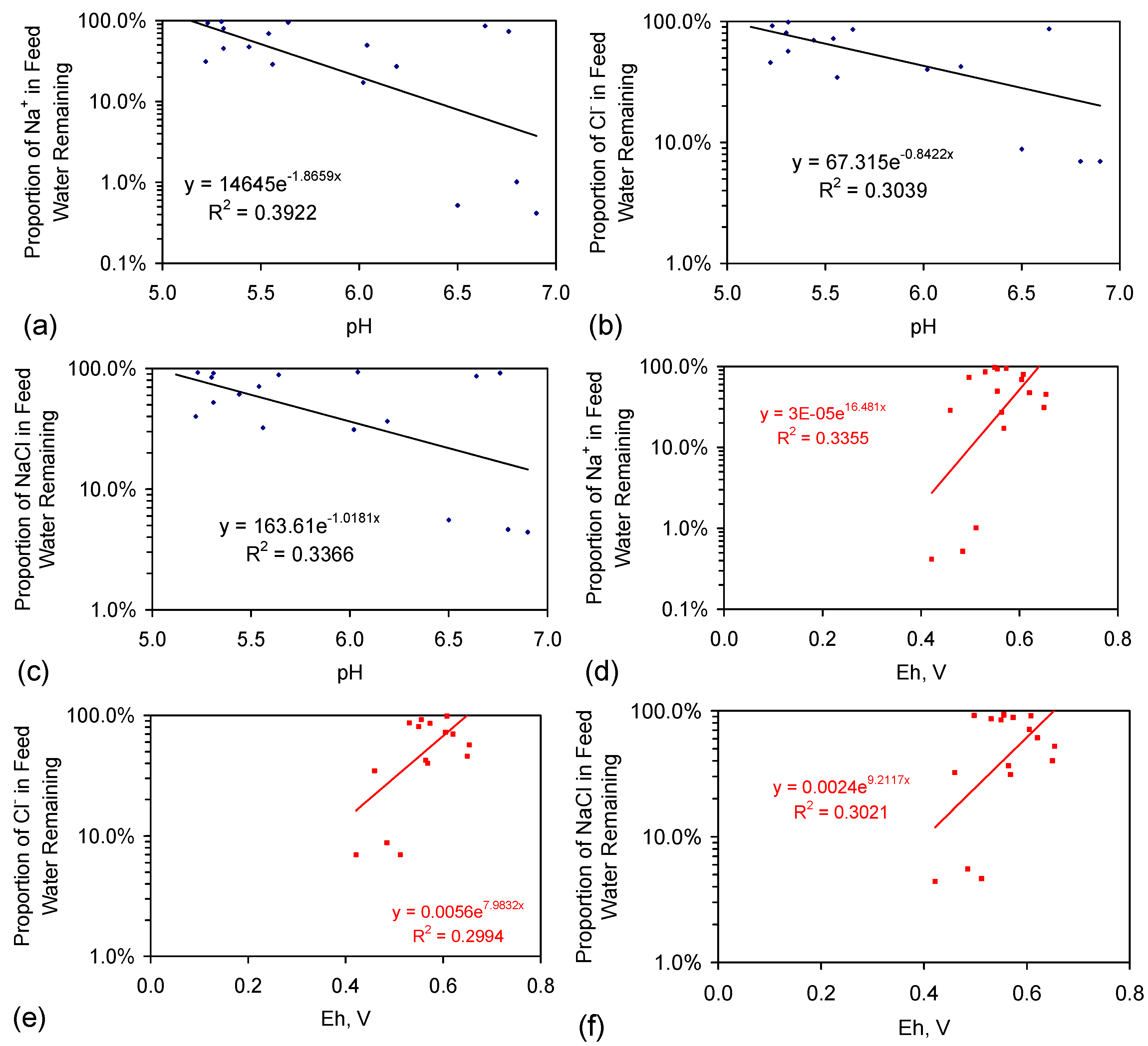
E.2. Mass Balance Considerations
E.2.1. Mass Balance Calculation
Cl− (feed water), g·L−1 = Cl− (product water) + Cl− (adsorbed) + Cl− (manifold + cartridge)
- CSD trial group: total (g) removed: Na+ = 28.23; Cl− = 35.42; NaCl removed = 54.5% of ZVI adsorption potential.
- E143 trial group: total (g) removed: Na+ = 2.52; Cl− = 37.77; NaCl removed = 57.3% of ZVI adsorption potential; total (g) removed from the product water: Na+ = 49.92; Cl− = 55.18;
- E144 trial group: total (g) removed: Na+ = 25.88; Cl− = 47.98; NaCl removed = 277% of ZVI adsorption potential.
Cl− + S2↔ClS2→C(Cl) + S2
NaO− + S2↔NaOS2→C(NaO) + S2
ClO− + S2↔ClOS2→C(ClO) + S2
HzCxOnCly−+ S2↔HzCxOnCly S2→C(HzCxOnCly) + S2
Initial State↔Adsorbed State (via Step 1)→Product (via Step 2)
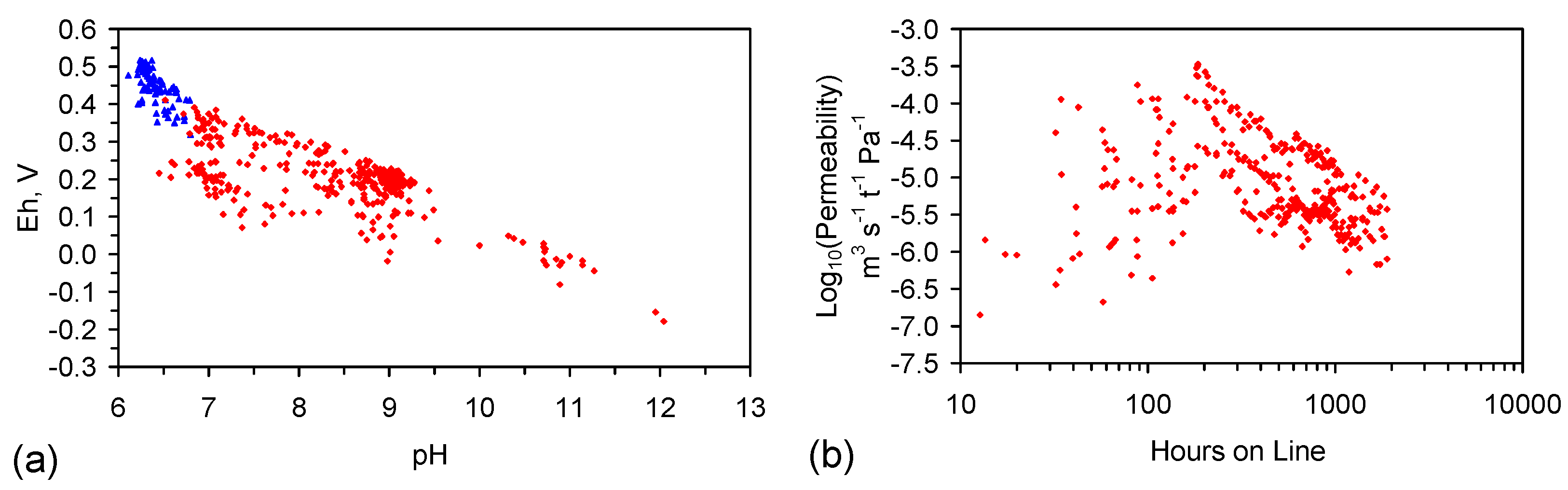
E.3. pH Considerations
E.4. Loss of Reactivity with Time
E.4.1. Porosity Reduction
E.4.2. Impact of ZVI Ageing on Reactivity
E.4.3. Impact of ZVI Particle Aggregation on Permeability
E.4.4. Interpretation of the Relationship between Permeability Decline and Reactivity
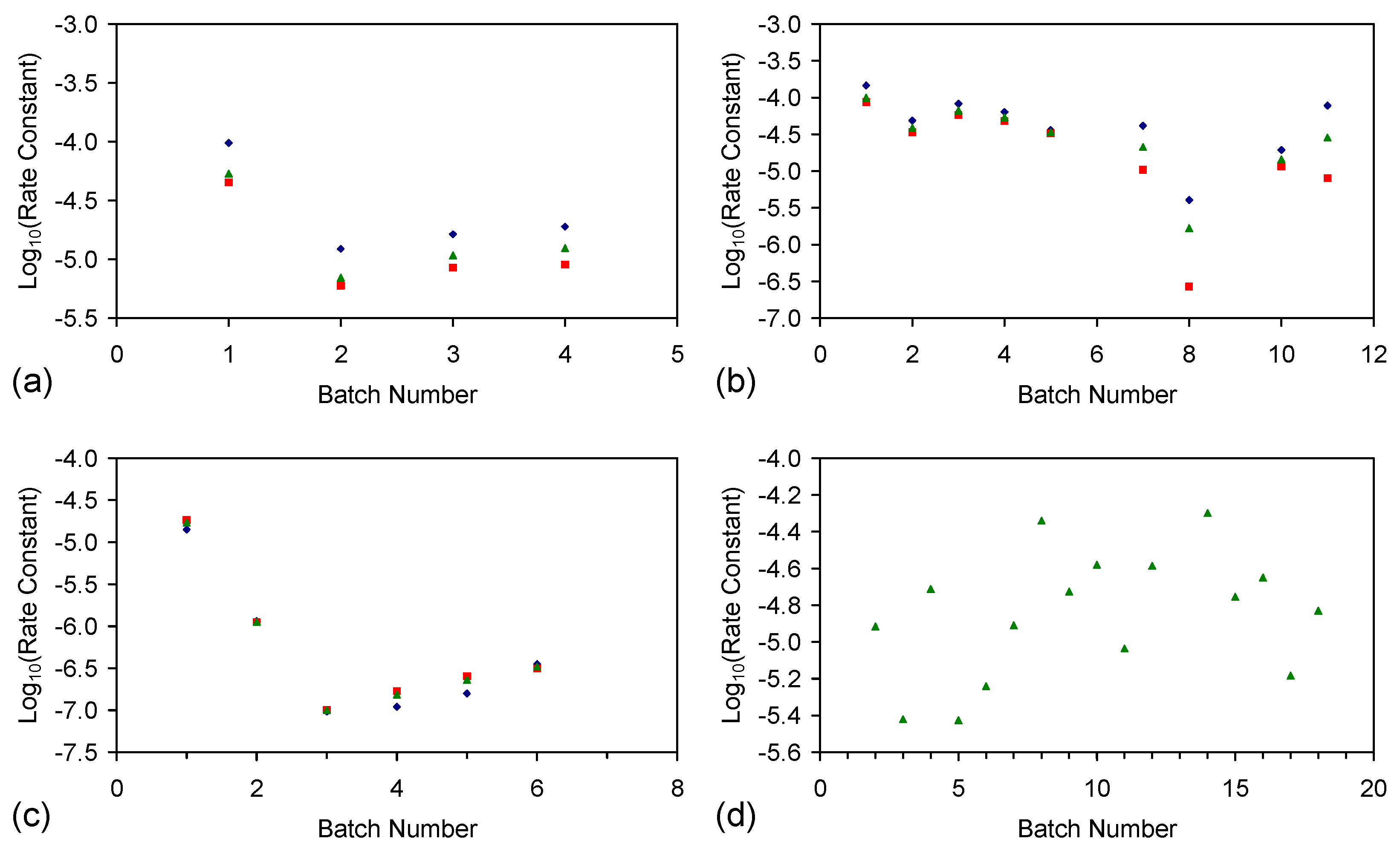
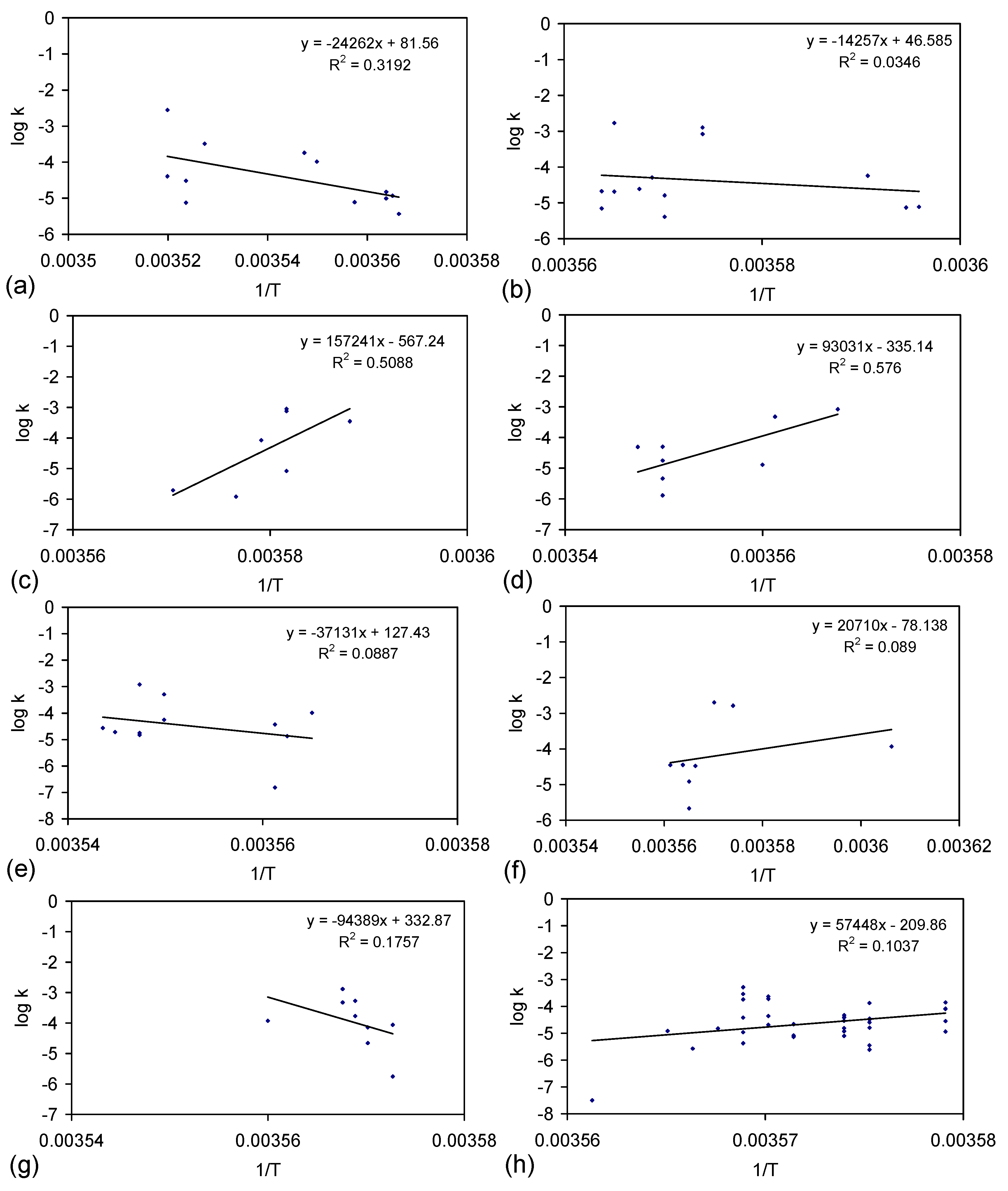
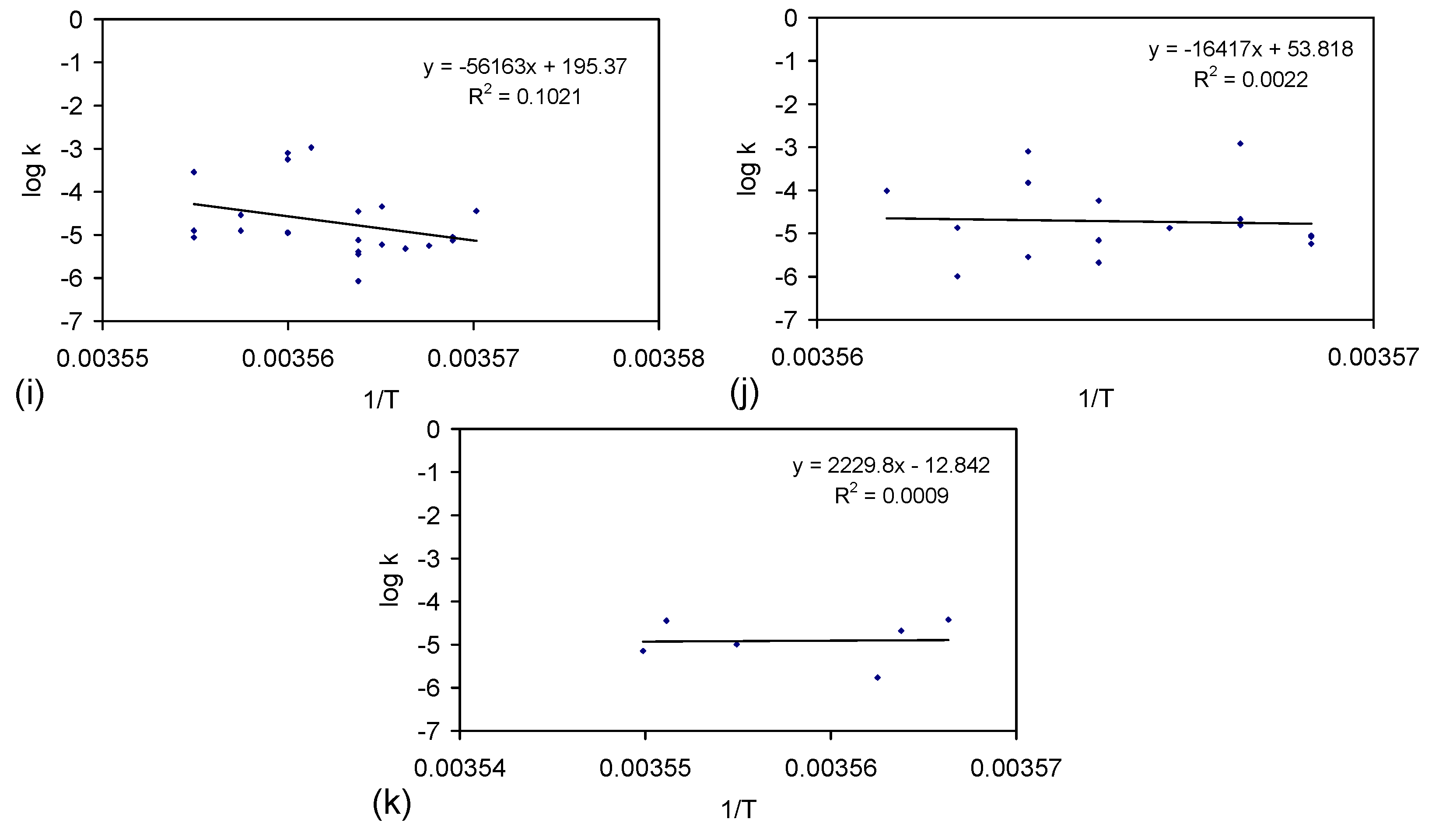
E.4.5. The Rate Constant, k
kCl = ln(Cl−t = 0/Cl−t = n)/t
kNa = ln(Na−t = 0/Na−t = n)/t
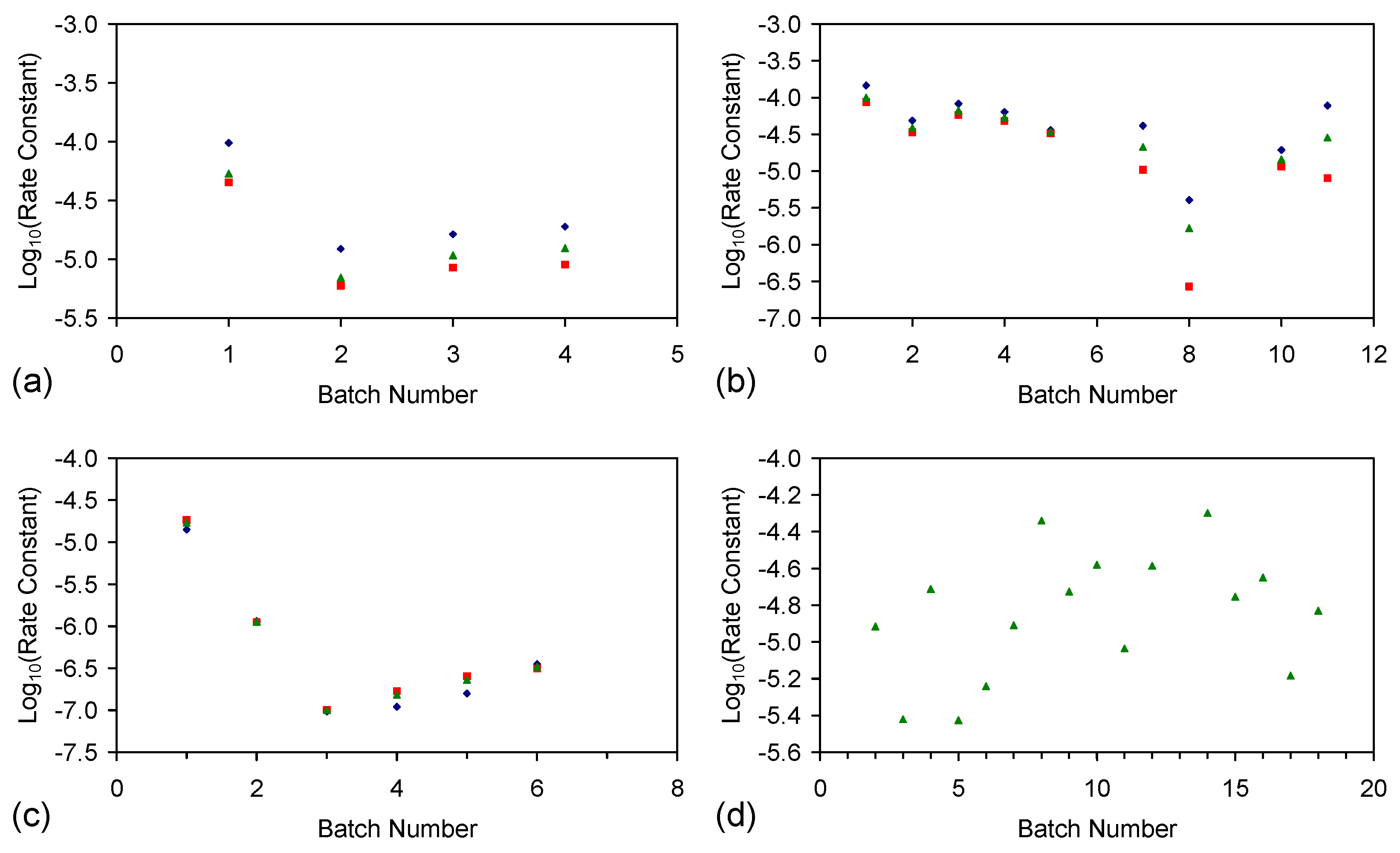
E.4.6. Control Analysis of the Rate of ZVI Permeability Decline with Time
kp = Qflow rate/Φ
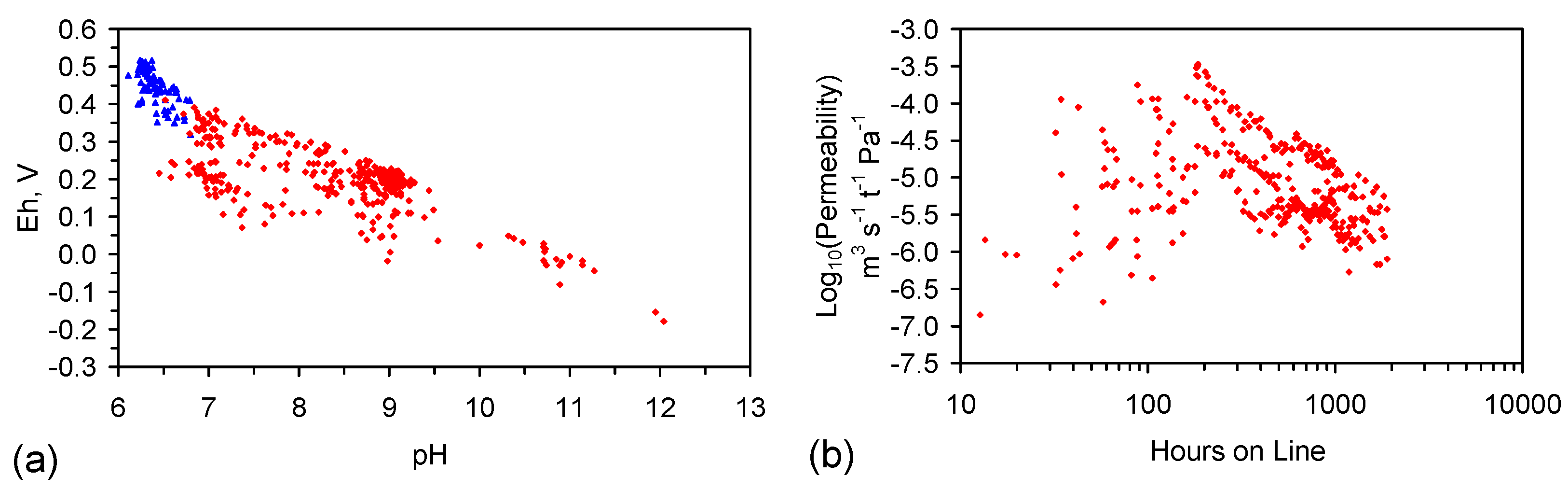
E.4.7. Intra-Particle or Inter-Particle Location of the ZVI Desalination Sites
E.4.8. Intra-Particle Location of the ZVI Desalination Sites
E143 ZVI Cartridge Reuse
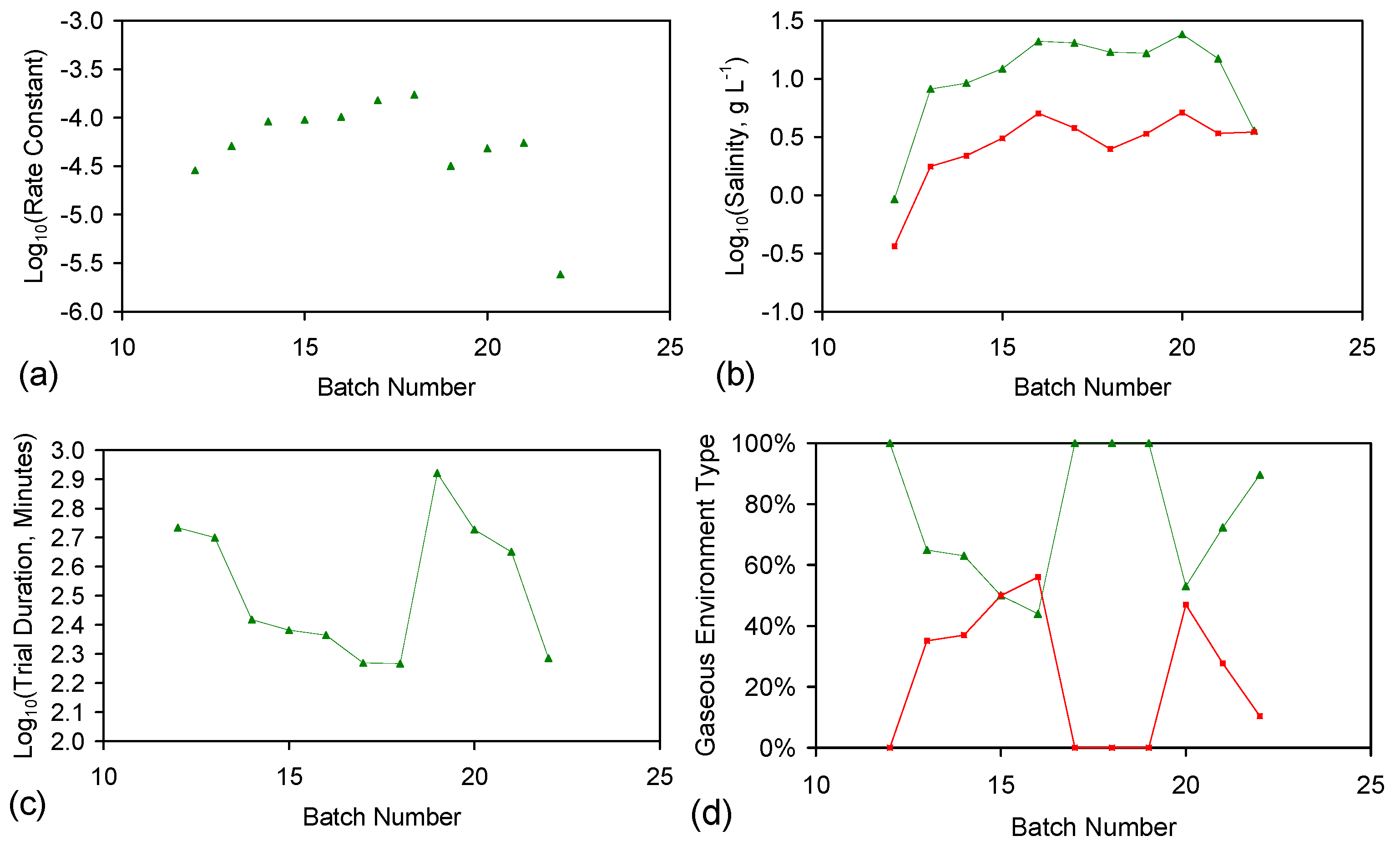
E146 ZVI Cartridge Reuse
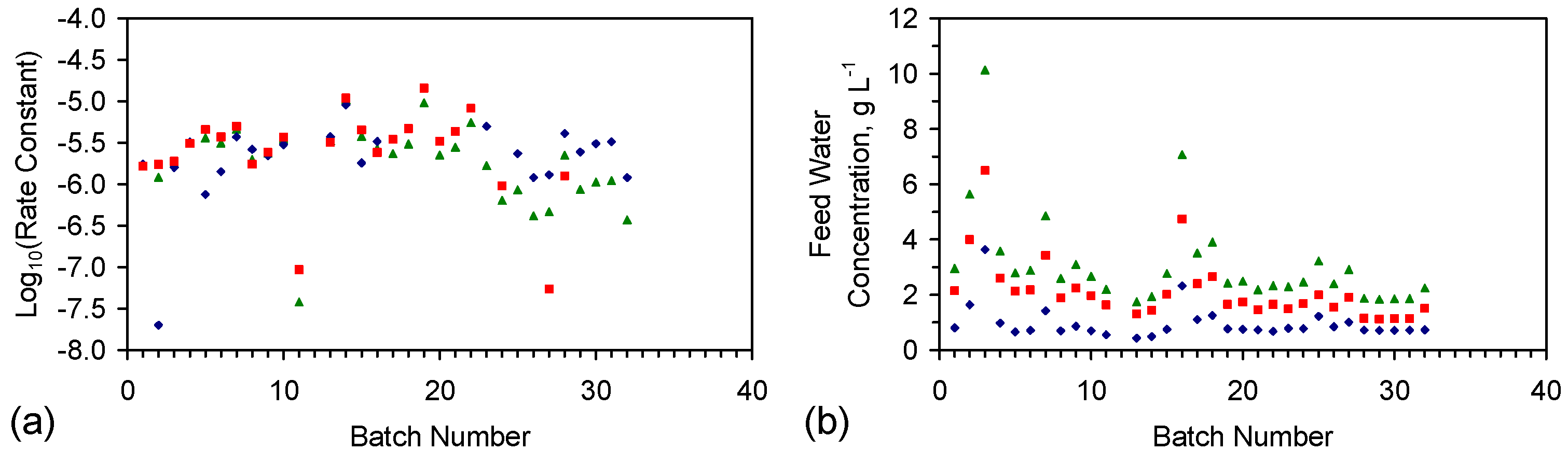
Appendix F: Nature of the Reacted (Catalytic) ZVI Material
F.1. Catalyst
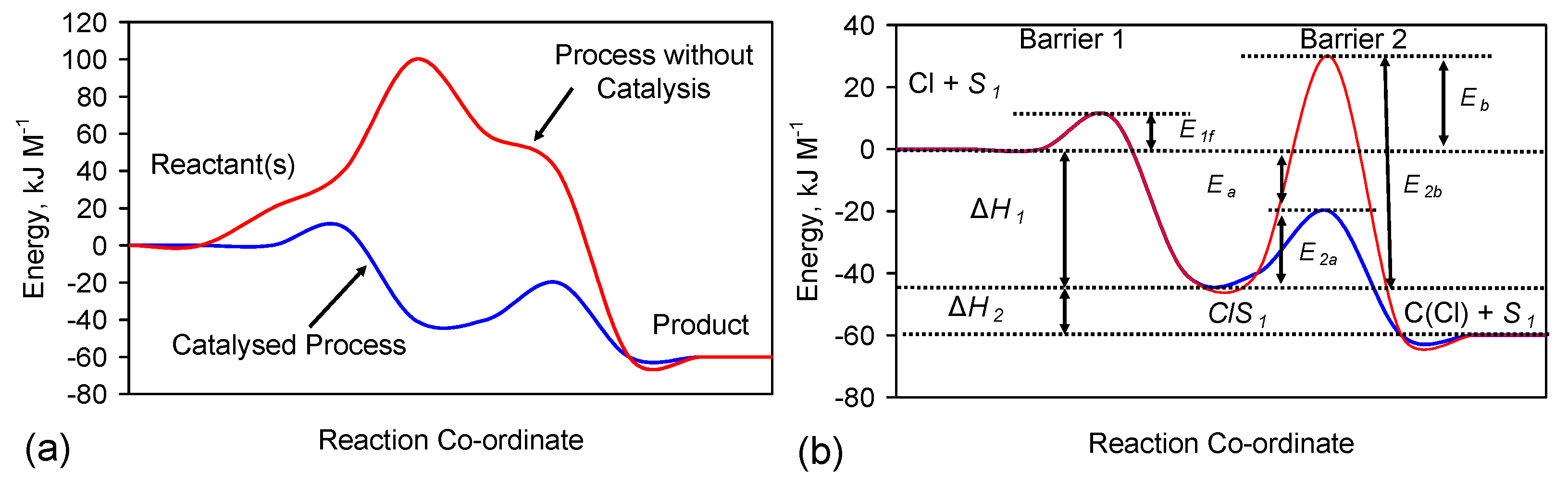
Reverse Step 1 = ClS1→Cl− + S1 Rate, v1r = k1r [ClS1] k1r = A1r exp(−E1r/RT)
Forward Step 2 = NaS1→C(Cl) + S1 Rate, v2 = k2 [ClS1] k2 = A2 exp(-E2/RT)
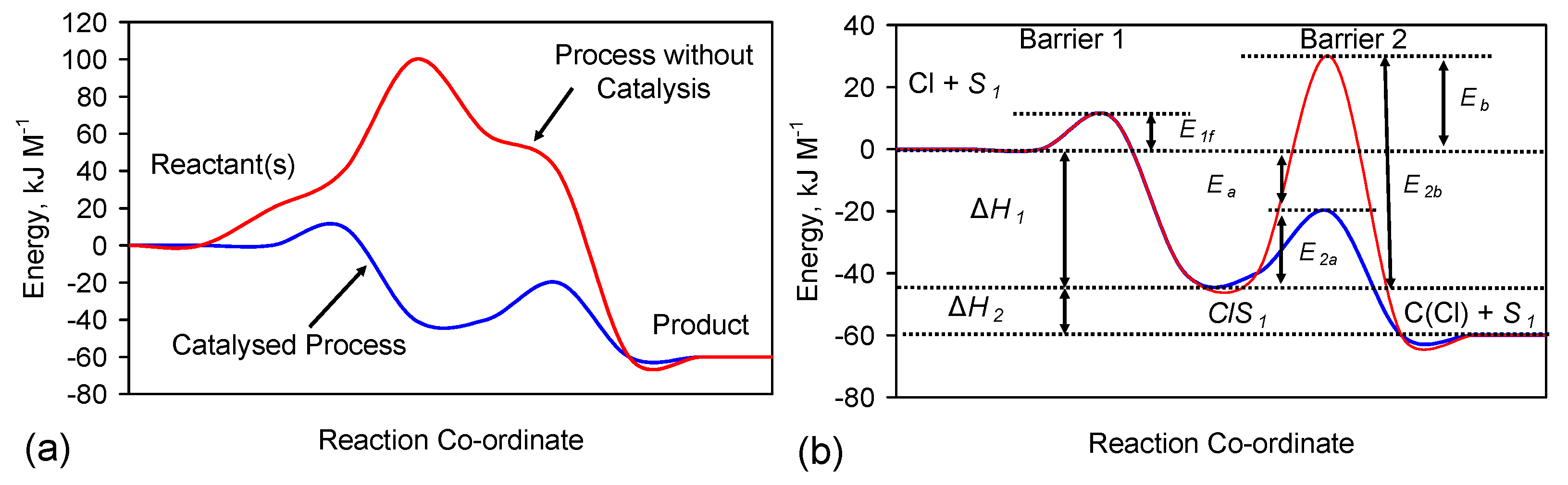
F.2. Fe Catalyst
F.3. Krasilshchikov Pathway
NaOH + OH− = NaO− + H2O
NaO− = NaO + e−
Me-OH + OH− = Me-O− + H2O
Me-O− = Me-O + e−
2Me-O = 2Me + O2
F.4. ZVI Desalination Catalyst
F.4.1. Initial ZVI Desalination Catalyst Operation
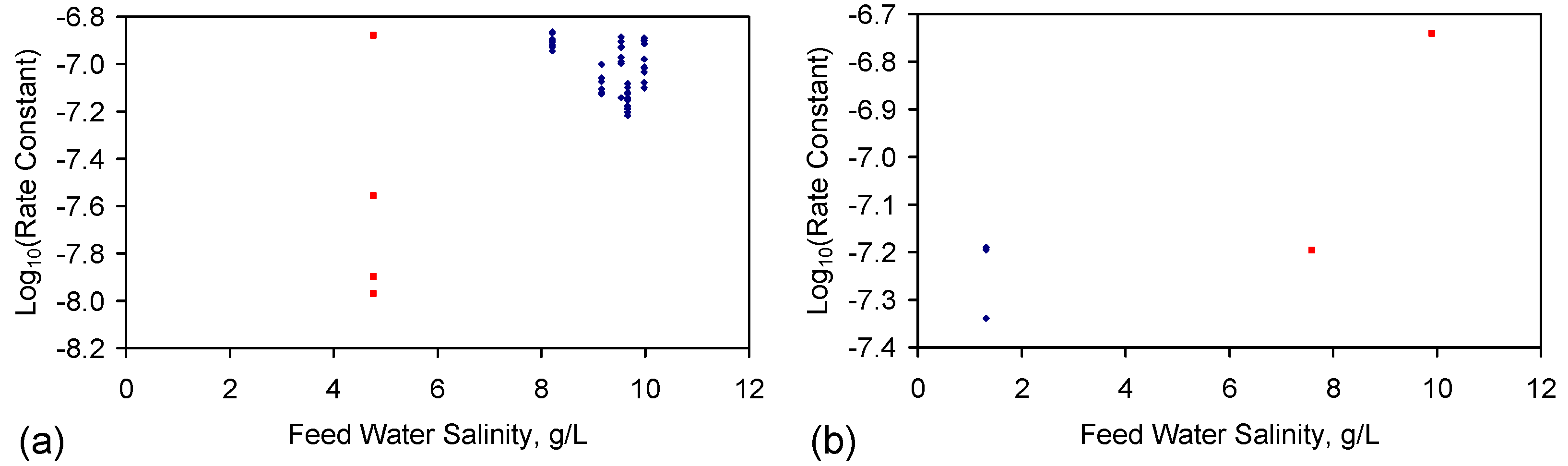

F.4.2. Gas-Pressured ZVI Desalination Catalyst Operation
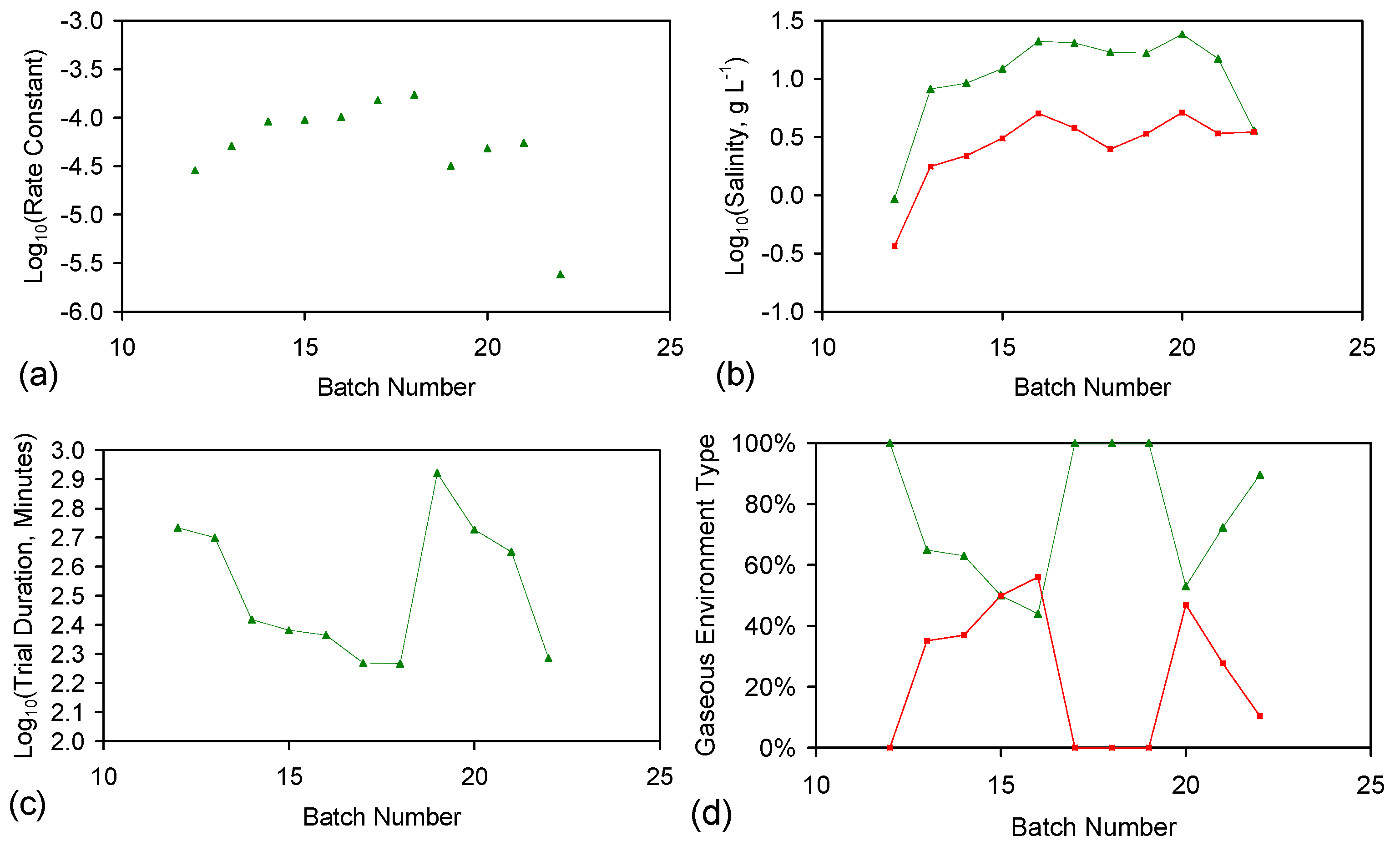
- Processed ZVI constructed [2] using the “incipient wetness technique and impregnation by soaking technique” [141] provides (Figure F2a) a single-use application for the desalination of water tanks (Figure 1). Reuse of the ZVM TP without recharge (ST6 trials (Figure F2a) [2]) results in a lower rate constant.
- The desalination process is catalytic;
- The desalination catalytic sites are located in the intra-layer porosity of LDH and related species;
- The catalyst can be recharged during the desalination process by providing an appropriate gas charge at an appropriate pressure.
Appendix G: Composition Characteristics of Suitable Saline Water
- (i)
- Total dissolved solids (TDS). This is normally measured using EC (electrical conductivity) where TDS = fEC where f and EC vary with temperature [142]. A value for f of 0.55 is commonly used to determine the TDS of water containing NaCl and 0.75 for water dominated by calcium carbonate [142]. The EC of water produced during ZVI desalination can be higher than the EC of the feed water [2,3].
- (ii)
- The major ions. The major ions fall into two groups (anions and cations). Common practice is to determine the cation and anion concentrations and an ionic balance error [142], e.g.:PC = Principal Cations (meq·L−1) = Na+/22.99 + 2Ca2+/40.08 + 2Mg2+/24.31 + K+/39
PA = Principal Anions (meq·L−1) = Cl−/35.45 + 2(SO42−)/96.06 + NO3−/62 + Alkalinity
Ion Balance Error (IBE) = (PC − PA)/(PC + PA) × 100%
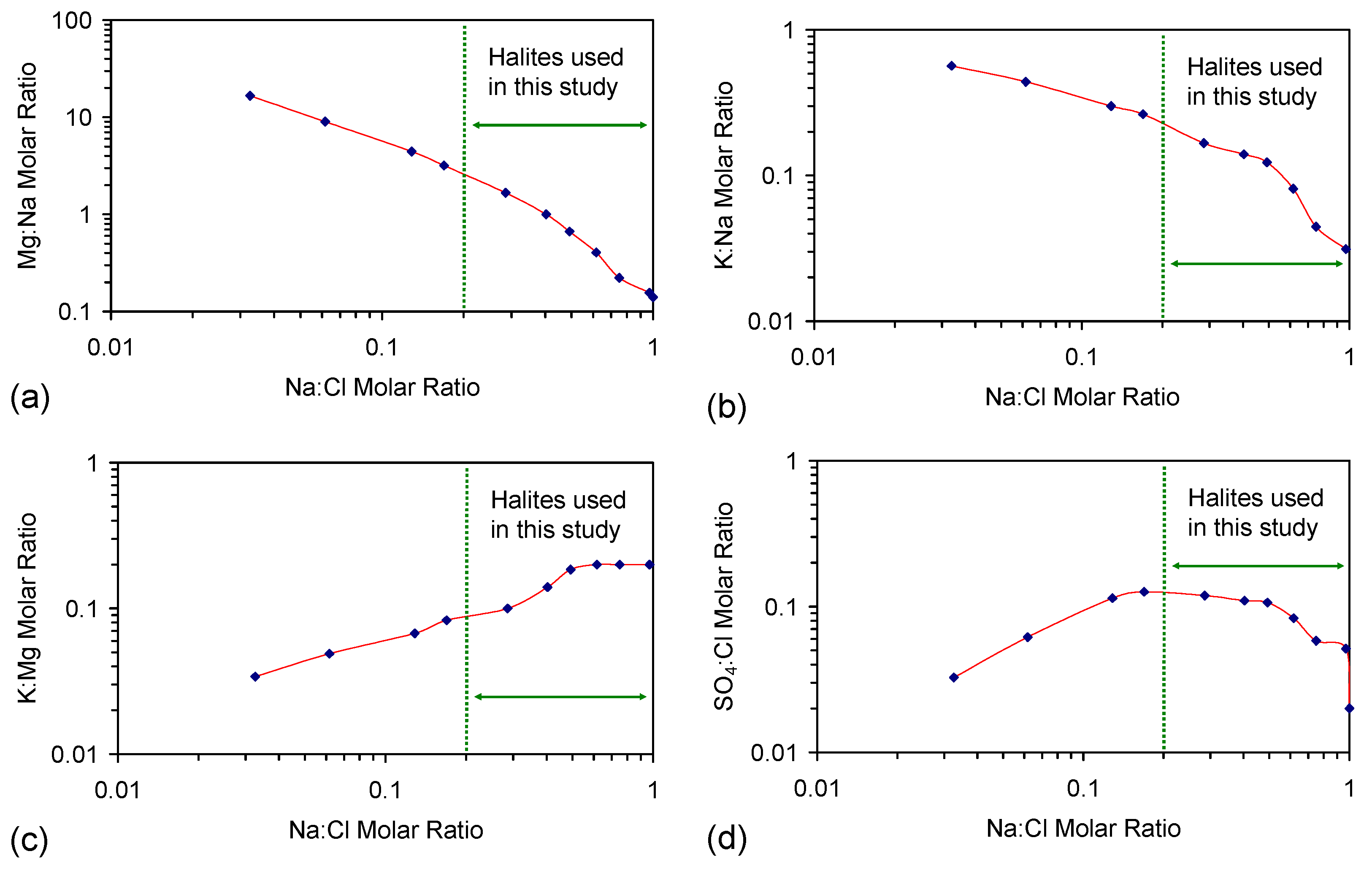
G.1. Synthetic Water Constructed from Halite
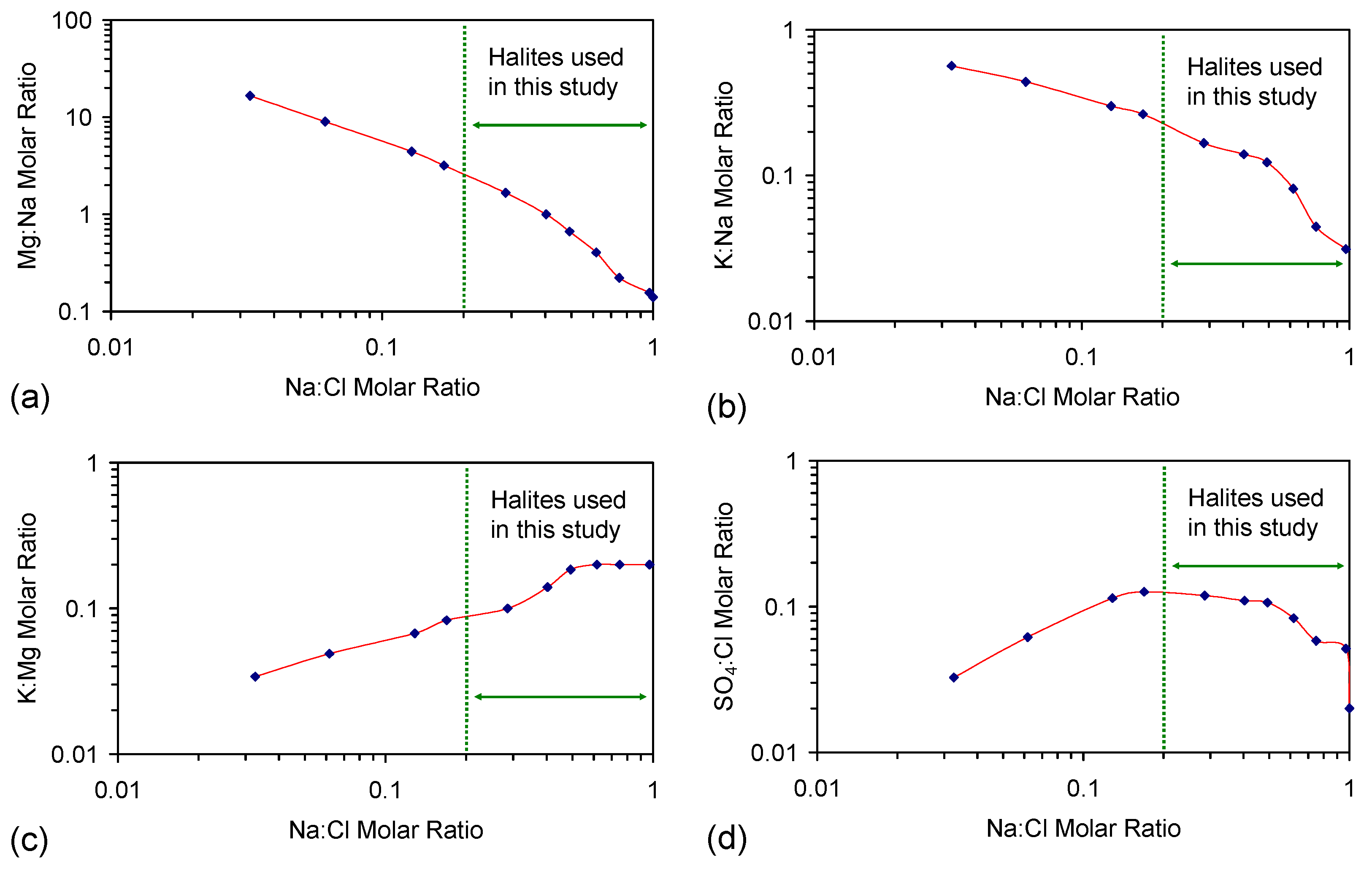
G.2. Interaction of Other Cations and Anions with ZVI
SAR = Na+/((Ca2+ + Mg2+)0.5) when cation concentrations are in mmol·L−1
SARadj = Na+/(0.5(Caeq2+ + Mg2+)0.5) when units are meq·L−1
SARadj = Na+/((Caeq2+ + Mg2+)0.5) when units are mmol·L−1
Caeq2+ = X (PCO2)1/3= 2 × 10Log(X) × (PCO2)1/3
Is = (1.3477SC + 0.5355)/1000
Log (X) = (1/3)[4.6629+0.6103 Log(Is) + 0.844 [Log (Is)]2 + 2Log(Ca2+/2HCO3−)]
G.3. Desalination of More Complex Water
Appendix H: Desalination Pathway
- The NaCl is removed with the gas when it is bubbled through the water. This has been investigated by passing the product gas through a series of downstream traps. No increases in salinity have been observed in the downstream traps (e.g., [1]).
- The NaCl is removed as gaseous components. The product gas composition has been monitored (SRI 8610C thermal conductivity detector (manufactured by SRI Instruments, Torrance, CA, USA)), and only H2 [1], N2, CO2, O2 and H2O have been identified. Mass balance analyses of the feed gases and product gases have indicated that no additional species are present in the product gas. Consequently, flue gas analyses for chlorinated gaseous products using ECD (electron capture detectors) or DELCD (dry electrolytic conductivity detectors) have not been undertaken.
H.1. Nature of the Desalination Pathway


H.2. Gas Saturation and Gas Pressure Implications
CK = d/3τ Inter-particle
CF = ϕ/τ Intra-Particle
Appendix I: Mineralogical Issues
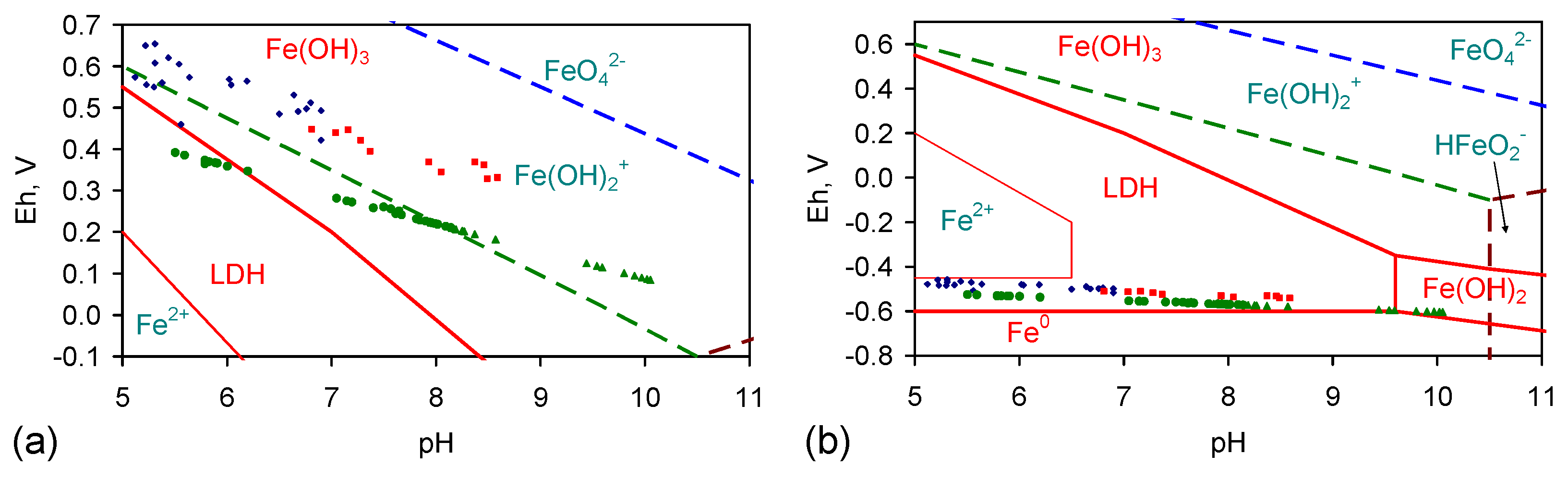
I.1. Desalination Mechanism: Step 1

I.2. Desalination Mechanism: Amorphous Iron Hydroxides
I.3. Humic Acids and Microbiota
References
- Antia, D.D.J. Desalination of groundwater and impoundments using nano-zero valent iron, Fe0. Meteor. Hydrol. Water Manag. 2015, 3, 21–38. [Google Scholar]
- Antia, D.D.J. Desalination of water using ZVI, Fe0. Water 2015, 7, 3671–3831. [Google Scholar] [CrossRef]
- Fronczyk, J.; Pawluk, K.; Michniak, M. Application of permeable reactive barriers near roads for chloride ions removal. Ann. Warsaw Univ. Life Sci. SGGW Land Reclaim. 2010, 42, 249–259. [Google Scholar] [CrossRef]
- Fronczyk, J.; Pawluk, K.; Garbulewski, K. Multilayer PRBs—Effective technology for protection of the groundwater environment in traffic infrastructures. Chem. Eng. Trans. 2012, 28, 67–72. [Google Scholar]
- Hwang, Y.; Kim, D.; Shin, H.-S. Inhibition of nitrate reduction by NaCl adsorption on a nano-zero valent iron surface during concentrate treatment for water reuse. Environ. Technol. 2015, 36, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, R.T.; Puls, R.W.; Sewell, G.W. Long-term performance of permeable reactive barriers using zero valent iron: Geochemical and microbiological effects. Ground Water 2003, 41, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Puls, R.W.; Blowes, D.W.; Gilham, R.W. Long-term performance monitoring for a permeable reactive barrier at the US Coast guard support center, Elizabeth City, North Carolina. J. Hazard. Mater. 1999, 68, 109–124. [Google Scholar] [CrossRef]
- Wilkin, R.T.; Acree, S.D.; Ross, R.R.; Puls, R.W.; Lee, T.R.; Woods, L.L. Fifteen-year assessment of a permeable reactive barrier for treatment of chromate and trichloroethylene in groundwater. Sci. Total Environ. 2014, 468–469, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Moline, G.R.; Kamolpornwijit, W.; West, O.R. Influence of hydrochemical processes on zero valent iron reactive barrier performance: A field investigation. J. Contam. Hydrol. 2005, 78, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Savoie, J.G.; Kent, D.B.; Smith, R.L.; le Blanc, D.R.; Hubble, D.W. Changes in Ground-Water Quality Near Two Granular Iron Permeable Reactive Barriers in a Sand and Gravel Aquifer, Cape Cod, Massachusetts, 1997–2000; Water Resources Investigation Report 03-4309; US Geological Survey: Reston, VA, USA, 2004; p. 77. [Google Scholar]
- Wada, Y.; Bierkens, M.F.P. Sustainability of global water use: Past reconstruction and future projections. Environ. Res. Lett. 2014, 9. [Google Scholar] [CrossRef]
- Amarasinghe, U.A.; Smakhtin, V. Global Water Demand Projections: Past, Present and Future; Report 156; International Water Management Institute (IWMI): Columbo, Sri Lanka, 2014. [Google Scholar]
- Knapp, K.C.; Baerenklau, K.A. Ground water quantity and quality management: Agricultural production and aquifer salinization over long time scales. J. Agric. Resour. Econ. 2006, 31, 616–641. [Google Scholar]
- Panta, S.; Flowers, T.; Lane, P.; Doyle, R.; Haros, G.; Shaala, S. Halophyte agriculture: Success stories. Environ. Exp. Bot. 2014, 107, 71–83. [Google Scholar] [CrossRef]
- Food and Agricultural Organization (FAO). The State of the World’s Land and Water Resources for Food and Agriculture (SOLAW)—Managing Systems at Risk; Food and Agricultural Organization of the United Nations and Earth Scan: Abingdon, UK, 2011. [Google Scholar]
- Payen, S.; Basset-Mens, C.; Follain, S.; Grunberger, O.; Marlet, S.; Nunez, M.; Perret, S. Pass the salt please! From a review to a theoretical framework for integrating salinization impacts in food LCA. In Proceedings of the 9th International Conference on Life Cycle Assessment in the Agri-Foods Sector, San Francisco, CA, USA, 8–10 October 2014.
- Schyns, J.F.; Hamaideh, A.; Hoekstra, A.Y.; Mekonnen, M.M.; Schyns, M. Mitigating the risk of extreme water scarcity and dependency: The case of Jordan. Water 2015, 7, 5705–5730. [Google Scholar] [CrossRef]
- Warsinger, D.M.; Mistry, K.H.; Nayar, K.G.; Chung, H.W.; Lienhard, V.J.H. Entropy generation of desalination powered by varied temperature waste heat. Entropy 2015, 17, 7530–7566. [Google Scholar] [CrossRef]
- Eshoul, N.M.; Agnew, B.; Al-Weshahi, M.A.; Atab, M.S. Energy analysis of a two pass reverse osmosis (RO) desalination unit with and without an energy recovery turbine (ERT) and pressure exchanger (PX). Energies 2015, 8, 6910–6925. [Google Scholar] [CrossRef]
- Al Hashemi, R.; Zarreen, S.; Al Raisi, A.; Al Marzooqi, F.A.; Hasan, S.W. A review of desalination trends in Gulf Cooperation council countries. Int. Interdiscip. J. Sci. Res. 2014, 1, 72–96. [Google Scholar]
- Sorour, M.; Hani, H.A.; Shaalan, H.F.; Al-Bazedi, G.A. Preliminary techno-economics assessment of developed desalination/salt recovery facility based on membrane and thermal techniques. Desalin. Water Treat. 2014. [Google Scholar] [CrossRef]
- Venkatesan, R. Comparison between LTTD and RO process of sea-water desalination: An integrated economic, environmental and ecological framework. Curr. Sci. 2014, 106, 378–386. [Google Scholar]
- Hofges, A.; Hansen, K.; McLeod, D. The economics of bulk water transport in Southern California. Resources 2014, 3, 703–720. [Google Scholar]
- Kjellsson, J.B.; Webber, M.E. The energy-water nexus: Spatially resolved analysis of the potential for desalinating brackish groundwater by use of solar energy. Resources 2015, 4, 476–489. [Google Scholar] [CrossRef]
- Chang, H.; Chang, C.-L.; Hung, C.-Y.; Chen, T.-W.; Ho, C.-D. Optimization study of small scale solar membrane distillation desalination systems (s-SMDDS). Int. J. Environ. Res. Public Health 2014, 11, 12064–12087. [Google Scholar] [CrossRef] [PubMed]
- Antia, D.D.J. Sustainable zero-valent metal (ZVM) water treatment associated with diffusion, infiltration, abstraction and recirculation. Sustainability 2010, 2, 2988–3073. [Google Scholar] [CrossRef]
- Ebbing, D.D.; Gammon, S.D. General Chemistry, 6th ed.; Houghton Mifflin Co.: Boston, MA, USA, 1999. [Google Scholar]
- British Standards Institution. Specification for Salt for Spreading on Highways for Winter Maintenance; BS 3247:2011; British Standards Institution: London, UK, 2013. [Google Scholar]
- Garcia-Veigas, J.; Cendon, D.I.; Pueyo, J.J.; Peryt, T.D. Zechstein saline brines in Poland, evidence of overturned anoxic ocean during the Late Permian mass extinction event. Chem. Geol. 2011, 290, 189–201. [Google Scholar] [CrossRef]
- Dalard, F.; Gourbeyre, Y.; Degrigny, C. Chloride removal from archeological cast iron by pulsating current. Stud. Conserv. 2002, 47, 117–121. [Google Scholar] [CrossRef]
- Radkova, L.; Fojtkova, P.; Kozakova, Z.; Krcma, F.; Sazavska, V.; Kujawa, A. Sample temperature during corrosion removal by low pressure low temperature hydrogen RF plasma. Romanian Rep. Phys. 2015, 67, 586–599. [Google Scholar]
- Reguer, S.; Dillmann, P.; Mirambet, F. Buried iron archeological artefacts: Corrosion mechanisms related to the presence of Cl− containing phases. Corros. Sci. 2007, 49, 2726–2744. [Google Scholar] [CrossRef]
- Reguer, S.; Neff, D.; Remazeilles, C.; Guilminot, E.; Nicot, F.; Pele, C.; Meguelati, M.; Mirambet, F.; Dillmann, P.; Refait, P.; et al. Desalinisation of iron archaeological artifacts: Understanding of chlorine removal mechanisms of the corrosion layers supported by characterization techniques. Innov. Investig. Metal Artifacts 2007, 2, 60–68. [Google Scholar]
- Rimmer, M.; Wang, Q. Assessing the effects of alkaline desalination treatments for archaeological iron using scanning electron microscopy. Br. Mus. Tech. Res. Bull. 2010, 4, 79–86. [Google Scholar]
- Rimmer, M.; Watkinson, D.; Wang, Q. The efficiency of chloride extraction from archaeological objects using deoxygenated alkaline solutions. Stud. Conserv. 2012, 57, 29–41. [Google Scholar] [CrossRef]
- Bayle, M.; de Vivies, P.; Memet, J-B.; Foy, E.; Dillmann, P.; Neff, D. Corrosion product transformations in alkaline baths under pressure and high temperature: The sub-critical stabilization of marine iron artifacts stored under atmospheric conditions. Mater. Corros. 2015. [Google Scholar]
- Asenath-Smith, E.; Estroff, L.A. Role of akaganeite (β-FeOOH) in the growth of hematite (α-Fe2O3) in an inorganic silica hydrogel. Cryst. Growth Des. 2015, 15, 3388–3398. [Google Scholar] [CrossRef]
- Tadic, M.; Milosevic, I.; Kraji, S.; Saboungi, M-L.; Motte, L. Ferromagnetic behaviour and exchange bias effect in akaganeite nanorods. Appl. Phys. Lett. 2015, 116. [Google Scholar] [CrossRef]
- Remazeilles, C.; Refait, P.H. On the formation of β-FeOOH (akaganeite) in chloride containing environments. Corros. Sci. 2007, 49, 844–857. [Google Scholar] [CrossRef]
- Mackay, A.L. Beta-ferric oxyhydroxide. Mineral. Mag. 1960, 32, 545–557. [Google Scholar] [CrossRef]
- Mackay, A.L. Beta-ferric oxyhydroxide—Akaganeite. Mineral. Mag. 1962, 33, 270–280. [Google Scholar] [CrossRef]
- Bottero, J.-Y.; Manceau, A.; Villieras, F.; Tchoubar, D. Structure and mechanisms of formation of FeOOH (Cl) polymer. Langmuir 1994, 10, 316–319. [Google Scholar] [CrossRef]
- Dante, S.; Hou, Z.; Risbud, S.; Stroeve, P. Nucleation of iron oxy-hydroxide nanoparticles by layer-by-layer polyionic assemblies. Langmuir 1999, 15, 2176–2182. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, K. Crystal splitting in the growth of β-FeO(OH). J. Cryst. Growth 2007, 308, 185–188. [Google Scholar] [CrossRef]
- Song, H.-J.; Liu, L.; Jia, X.-H. Synthesis of multiwalled carbon nanotubes/β-FeOOH nanocomposites with high adsorption capacity. J. Nanopart. Res. 2012, 14. [Google Scholar] [CrossRef]
- Parameshwari, R.; Priyadarshini, P.; Chandrasekaran, G. Optimization, structural, spectroscopic and magnetic studies on stable akaganeite nanoparticles via co-precipitation method. Am. J. Mater. Sci. 2011, 1, 18–25. [Google Scholar]
- Konishi, H.; Yamashita, M.; Uchida, H.; Mizuki, J. Characterization of rust layer formed on Fe, Fe-Ni and Fe-Cr alloys exposed to Cl− rich environment by Cl and Fe K-edge XANES Measurements. Mater. Trans. 2005, 46, 329–336. [Google Scholar] [CrossRef]
- Post, J.E.; Heany, P.J.; von Dreele, R.B.; Hanson, J.C. Neutron and temperature resolved synchrotron X-ray powder diffraction study of akaganeite. Am. Mineral. 2003, 88, 782–788. [Google Scholar] [CrossRef]
- Yue, J.; Jiang, X.; Yu, A. Experimental and theoretical study on the β-FeOOH nanorods: Growth and conversion. J. Nanopart. Res. 2011, 13, 3961–3974. [Google Scholar] [CrossRef]
- Xu, Z.; Liang, J.; Zhou, L. Template—Free hydrothermal synthesis of β-FeOOH nanorods and their catalytic activity in the degradation of methyl orange by a photo-Fenton-like process. Open J. Inorg. Non Met. Mater. 2013, 3, 58–65. [Google Scholar] [CrossRef]
- Garcia, K.E.; Barrero, C.A.; Morales, A.L.; Greneche, J.M. Characterization of akaganeite synthesed in the presence of Al3+, Cr3+ and Cu2+ ions and urea. Mater. Chem. Phys. 2008, 112, 120–126. [Google Scholar] [CrossRef]
- Ishikawa, T.; Katoh, R.; Yasukawa, A.; Kandori, K.; Nakayama, T.; Yuse, F. Influence of metals on the formation of β-FeOOH particles. Corros. Sci. 2001, 43, 1727–1738. [Google Scholar] [CrossRef]
- Kyzas, G.; Peleka, E.N.; Deliyanni, E.A. Nanocrystalline akaganeite as adsorbent for surfactant removal from aqueous solutions. Materials 2013, 6, 184–197. [Google Scholar] [CrossRef]
- Remazeilles, C.; Refait, P. Formation, fast oxidation and thermodynamic data of Fe(II) hydroxychlorides. Corros. Sci. 2008, 50, 856–864. [Google Scholar] [CrossRef]
- Refait, P.; Genin, J.M.R. The mechanism of oxidation of ferrous hydroxychloride beta-Fe2(OH)3Cl in chloride containing aqueous solution: The formation of beta-FeOOH akaganeite, an X-ray diffraction, Mossbauer spectroscopy and electrochemical study. Corros. Sci. 1997, 39, 539–553. [Google Scholar] [CrossRef]
- Refait, P.; Drissi, S.H.; Pytkiewicz, J.; Genin, J.M.R. The anodic species competition in iron aqueous corrosion: Role of various green rust compounds. Corros. Sci. 1997, 39, 1699–1710. [Google Scholar] [CrossRef]
- Refait, P.; Abdelmoula, M.; Genin, J.M.R. Mechanisms of formation and structure of green rust one in aqueous corrosion of iron in the presence of chloride ions. Corros. Sci. 1998, 40, 1547–1560. [Google Scholar] [CrossRef]
- Al-Moubaraki, A.H.; Al-Judaibi, A.; Asiri, M. Corrosion of C-steel in the Red Sea: Effect of immersion time and inhibitor concentration. Int. J. Electrochem. Sci. 2015, 10, 4252–4278. [Google Scholar]
- Ruby, C.; Aissa, R.; Gehin, A.; Cortot, J.; Adelmoula, M.; Genin, J.M.R. Green rusts synthesis by coprecipitation of FeII-FeIII ions and mass balance diagram. C. R. Geosci. 2006, 338, 420–432. [Google Scholar] [CrossRef]
- Taylor, R.M. 1984 Influence of chloride on the formation of iron oxides from Fe(II) chloride. I. Effect of [Cl]/[Fe] on the formation of magnetite. Clays Clay Miner. 1984, 32, 167–174. [Google Scholar] [CrossRef]
- Refait, P.; Genin, J.M.R. The transformation of chloride containing green rust one into sulphated green rust two by oxidation in mixed Cl− and SO42− aqueous media. Corros. Sci. 1994, 36, 55–65. [Google Scholar] [CrossRef]
- Ockermann, L.T.; Schreyer, J.M. Preparation of sodium ferrate. J. Am. Chem. Soc. 1951, 73, 5478. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 1st ed.; NACE International: Cebelcor, Houston, TX, USA, 1974. [Google Scholar]
- Nesic, S. Key issues related to the internal corrosion of oil and gas pipelines—A review. Corros. Sci. 2007, 49, 4308–4338. [Google Scholar] [CrossRef]
- Perez, F.R.; Barrero, C.A.; Walker, A.R.H.; Garcia, K.E.; Nomura, K. Effects of chloride concentration, immersion time and steel composition on the spinel phase formation. Mater. Chem. Phys. 2009, 117, 214–223. [Google Scholar] [CrossRef]
- Ning, J.; Zheng, Y.; Young, D.; Brown, B.; Nesic, S. A Thermodynamic study of hydrogen sulphide corrosion of mild steel. In Proceedings of the NACE Corrosion 2013 Conference, Orlando, FL, USA, 17–21 March 2013; p. 2462.
- Rahmanto, W.H.; Gumawan, R.N. Corrosion rate of copper and iron in seawater based on resistance measurement. J. Coast. Dev. 2002, 5, 67–74. [Google Scholar]
- Sherif, E.-S.S.; Abdo, H.S.; Almajid, A.A. Corrosion behavior of cast iron in freely aerated stagnant Arabian Gulf seawater. Materials 2015, 8, 2127–2138. [Google Scholar] [CrossRef]
- Zakowski, K.; Narozony, M.; Szocinski, M.; Darowicki, K. Influence of water salinity on corrosion risk-the case of the southern Baltic Sea coast. Environ. Monit. Assess. 2014, 186, 4871–4879. [Google Scholar] [CrossRef] [PubMed]
- Antia, D.D.J. Groundwater water remediation by static diffusion using nano-zero valent metals [ZVM](Fe0, Cu0, Al0), n-FeHn+, n-Fe(OH)x, n-FeOOH, n-Fe-[OxHy](n+/−). In Nanomaterials for Environmental Protection, 1st ed.; Kharisov, B.I., Kharissova, O.V., Dias, H.V.R., Eds.; Wiley Inc.: Hoboken, NJ, USA, 2014; Chapter 1; pp. 3–25. [Google Scholar]
- Antia, D.D.J. Water remediation—Water remediation using nano-zero-valent metals (n-ZVM). In CRC Concise Encyclopedia of Nanotechnology, 1st ed.; Kharisov, B.I., Kharissova, O.V., Ortiz-Mendez, U., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2016; Chapter 84; pp. 1103–1120. [Google Scholar]
- Rustad, J.R.; Felmy, A.R.; Hay, B.P. Molecular statics calculations for iron oxide and oxyhydroxide minerals: Towards a flexible model of the reactive mineral-water interface. Geochim. Cosmochim. Acta 1996, 60, 1553–1562. [Google Scholar] [CrossRef]
- Hiemstra, T.; van Riemsdijk, W.H. Adsorption and surface oxidation of Fe(II) on metal (hydr)oxides. Geochim. Cosmochim. Acta 2007, 71, 5913–5933. [Google Scholar] [CrossRef]
- Ghose, S.K.; Waychunas, G.A.; Trainor, T.P.; Eng, P.J. Hydrated goethite (α-FeOOH) (100) interface structure: Ordered water and surface functional groups. Geochim. Cosmochim. Acta 2010, 74, 1943–1953. [Google Scholar] [CrossRef]
- Majzian, J.; Mazeina, L.; Navrotsky, A. Enthalpy of water adsorption and surface enthalpy of lepidocrocite (γ-FeOOH). Geochim. Cosmochim. Acta 2007, 71, 615–623. [Google Scholar] [CrossRef]
- Kozin, P.A.; Boily, J.-F. Mineral surface charge development in mixed electrolyte solutions. J. Colloid Interface Sci. 2014, 418, 256–253. [Google Scholar] [CrossRef] [PubMed]
- Sten, P.; Olin, M.; Lehikoinen, J. Surface Complexation on Iron Oxides with Reference to the Oxide Films Formed on Material Surfaces in Nuclear Power Plants; VTT Tiedotteita-Meddelanden-Research Notes 2055; VTT Technical Research Centre of Finland: Espoo, Finland, 2000. [Google Scholar]
- Rives, V. Layered Double Hydroxides: Present and Future; Nova Science Publication: New York, NY, USA, 2001. [Google Scholar]
- Waseda, Y.; Suzuki, S. Characterization of Corrosion Products on Steel Surfaces; Springer: Berlin, Germany, 2006. [Google Scholar]
- Krivovichev, S.V. Structural Crystallography of Inorganic Oxysalts; IUCr Monographs on Crystallography; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Antia, D.D.J. 2008 Oil polymerisation and fluid expulsion from low temperature, low maturity, over pressured sediments. J. Petrol. Geol. 2008, 31, 263–282. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, H.; Wang, H.; Liu, L.; Xiao, C.; Ma, D. The effects of ionic additives on the aqueous phase Fischer-Tropsch synthesis with ruthenium nanoparticle catalyst. Catal. Today 2012, 183, 143–153. [Google Scholar] [CrossRef]
- Sander, S.P.; Friedl, R.R.; Abbatt, J.P.D.; Barker, J.R.; Burkholder, J.B.; Golden, D.M.; Kolb, C.E.; Kurylo, M.J.; Mortgat, G.K.; Wine, P.H.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies; Evaluation Number 17, JPL (Jet Propulsion Laboratory) Publication 10–6; National Aeronautic and Space Administration (NASA): Pasadena, CA, USA, 2011. [Google Scholar]
- Maric, D.; Burrows, J.P.; Meller, R.; Moortgat, G.K. A study of the UV-visible absorption spectrum of molecular chlorine. J. Photochem. Photobiol. A Chem. 1993, 70, 205–214. [Google Scholar] [CrossRef]
- Catoire, V.; Lesclaux, R.; Lightfoot, P.D.; Rayez, M.T. Kinetic study of the reactions of CH2ClO2 with itself and with HO2, and theoretical study of the reactions of CH2ClO, between 251 and 600 K. J. Hys. Chem. 1994, 98, 2889–2898. [Google Scholar] [CrossRef]
- Pye, K. An occurrence of akaganeite (β-FeOOH·Cl) in Recent oxidized carbonate concretions, Norfolk, England. Min. Mag. 1988, 52, 125–126. [Google Scholar] [CrossRef]
- Varnado, C.D.; Rosen, E.L.; Collins, M.S.; Lynch, V.M.; Bielawski, C.W. Synthesis and study of olephin metathesis catalysts supported by redox switchable diaminocarbene[3]ferocenophanes. Dalton Trans. 2013, 42, 13251–13264. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, M.; Gillett, S.L.; Bell, T.W. Redox-Switchable Materials. US Patent application US2005/0227071 A1, 28 December 2001. [Google Scholar]
- Arumugam, K.; Varnado, C.D.; Sproules, S.; Lynch, V.M.; Bielawski, C.W. Redox-switchable ring closing metathesis: Catalyst design, synthesis and study. Chemistry 2013, 19, 10866–10875. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 89th ed.; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]
- Antia, D.D.J. Modification of aquifer pore water by static diffusion using nano-zero-valent metals. Water 2011, 3, 79–112. [Google Scholar] [CrossRef]
- Gavaskar, A.; Tatar, L.; Condit, W. Cost and Performance Report: Nanoscale Zero-Valent Iron Technologies for Source Remediation; Contract Report CR-05–007-ENV; NAVFAC Naval Facilities Engineering Command, Engineering Service Center: Port Hueneme, CA, USA, 2005; p. 44. [Google Scholar]
- Henderson, A.D.; Desmond, A.H. Long-term performance of zero valent iron permeable reactive barriers: A critical review. Environ. Sci. Eng. 2007, 24, 401–423. [Google Scholar] [CrossRef]
- Obiri-Nyarko, F.; Grajales-Mesa, S.J.; Malina, G. An overview of permeable reactive barriers for in situ sustainable groundwater remediation. Chemosphere 2014, 111, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Dionysiou, D.D.; Liu, H. The use of zero valent iron for groundwater remediation and wastewater treatment: A review. J. Hazard. Mater. 2014, 267, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Sun, Y.; Qin, H.; Li, J.; Lo, I.M.C.; He, D.; Dong, H. The limitations of applying zero valent iron technology in contaminants sequestration and the corresponding countermeasures: The development in zero-valent iron technology in the last two decades (1994–2014). Water Res. 2015, 75, 224–248. [Google Scholar] [CrossRef] [PubMed]
- Noubactep, C. Metallic iron for environmental remediation: A review of reviews. Water Res. 2016, 85, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Stefaniuk, M.; Oleszczuk, P.; Ok, Y.S. Review on nano zerovalent iron (nZVI): From synthesis to environmental applications. Chem. Eng. J. 2016, 287, 618–632. [Google Scholar] [CrossRef]
- Lai, K.C.K. Field evaluation of the performance and heterogeneity of a zero valent iron based permeable reactive barrier for groundwater remediation. HKIE Trans. Hong Kong Inst. Eng. 2007, 14, 2–12. [Google Scholar]
- Cundy, A.B.; Hopkinson, L.; Whitby, R.L.D. Use of iron-based technologies in contaminated land and groundwater remediation: A review. Sci. Total Environ. 2008, 400, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Kobbe-Dama, N.; Noubactep, C.; Tchatchueng, J.-B. Metallic iron for water treatment: Prevailing paradigm hinders progress. Fresenius Environ. Bull. 2013, 22, 2953–2957. [Google Scholar]
- Muchitsch, N.; van Nooten, T.; Bastiaens, L.; Kjeldsen, P. Integrated evaluation of the performance of a more than seven year old permeable reactive barrier at a site contaminated with chlorinated aliphatic hydrocarbons (CAHs). J. Contam. Hydrol. 2011, 126, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Gatcha-Bandjun, N.; Noubactep, C.; Benoit, L.M. Water treatment with Fe0/H2O systems: Learning from internal electrolysis. Fresenius Environ. Bull. 2014, 23, 2663–2669. [Google Scholar]
- Tsinde, R.T.; Phukan, M.; Nassi, A.; Noubactep, C.; Ruppert, H. Validating the efficiency of the MB discoloration method for the characterization of Fe0/H2O systems using accelerated corrosion by chloride ions. Chem. Eng. J. 2015, 279, 353–362. [Google Scholar] [CrossRef]
- Mackenzie, P.D.; Horney, D.P.; Sivavec, T.M. Mineral precipitation and porosity losses in granular iron columns. J. Hazard. Mater. 1999, 68, 1–17. [Google Scholar] [CrossRef]
- Luo, P.; Bailey, E.H.; Mooney, S.J. Quantification of changes in zero valent iron morphology using X-ray computed tomography. J. Environ. Sci. 2013, 25, 2344–2351. [Google Scholar] [CrossRef]
- Domga, R.; Togue-Kamga, F.; Noubactep, C.; Tchatchueng, J.-B. Discussing porosity loss of Fe0 packed water filters at ground level. Chem. Eng. J. 2015, 263, 127–134. [Google Scholar] [CrossRef]
- Li, L.; Benson, C.H.; Lawson, E.M. Impact of mineral fouling on hydraulic behavior of permeable reactive barriers. Groundwater 2005, 43, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Benson, C.H.; Lawson, E.M. Modeling porosity reductions caused by mineral fouling in continuous wall permeable reactive barriers. J. Contam. Hydrol. 2006, 83, 89–121. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsova, I.; Bayer, P.; Ebert, M.; Finkel, M. Modeling the long term performance of zero valent iron using a spatio-temporal approach for iron aging. J. Contam. Hydrol. 2007, 90, 58–80. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Ruhr, A.S.; Amos, R.T. Investigating dominant processes in ZVI permeable reactive barriers using reactive transport modelling. J. Contam. Hydrol. 2013, 151, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, K.; Noubactep, C. Characterizing the impact of sand addition on the efficiency of granular iron for contaminant removal in batch system. Chem. Eng. J. 2015, 262, 891–896. [Google Scholar] [CrossRef]
- Liu, T.; Li, X.; Waite, T.D. Depassivation of aged Fe0 by ferrous ions: Implications to contaminant degradation. Environ. Sci. Technol. 2013, 47, 13712–13720. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, H.-J.; Kim, H.-E.; Kweon, J.; Lee, B.-D.; Lee, C. Oxidant production from corrosion of nano- and microparticulate zero valent iron in the presence of oxygen: A comparative study. J. Hazard. Mater. 2014, 265, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Noubactep, C. Flaws in the design of Fe(0)-based filtration systems? Chemosphere 2014, 117, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Lai, B.; Yang, P.; Zhou, Y.; Wang, J.; Fang, S. Comparative study on the reactivity of Fe/Cu bimetallic particles and zero valent iron (ZVI) under different conditions of N2, air or without aeration. J. Hazard. Mater. 2015, 297, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yuan, Y.; Lai, B.; Zhou, Y.; Wang, J. Treatment of reverse osmosis (RO) concentrate by the combined Fe/Cu/air and Fenton process (1st Fe/Cu/air—Fenton-2ndFe/Cu/air). J. Hazard. Mater. 2016, 302, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Benson, C.H. Evaluation of five strategies to limit the impact of fouling in permeable reactive barriers. J. Hazard. Mater. 2010, 181, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Kocur, C.M.; O’Carroll, D.M.; Sleep, B.E. Impact of nZVI stability on mobility in porous media. J. Contam. Hydrol. 2013, 145, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Mulder, M. Basic Principles of Membrane Technology; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996. [Google Scholar]
- Ye, G.; van Breugel, P.L.K. Modeling of water permeability in cementitious materials. Mater. Struct. 2006, 39, 877–885. [Google Scholar] [CrossRef]
- Pereira, J.-M.; Arson, C. Retention and permeability properties of damaged porous rocks. Comp. Geotech. 2013, 48, 272–282. [Google Scholar] [CrossRef]
- Berg, C.F. Permeability description by characteristic length, tortuosity, constriction and porosity. Trans. Porous Media 2014, 103, 381–400. [Google Scholar] [CrossRef]
- Brusaert, W. The permeability of a porous medium determined from certain probability laws for pore size distribution. Water Resour. Res. 1968, 4, 425–434. [Google Scholar] [CrossRef]
- Brusaert, W. A concise parameterization of the hydraulic conductivity of unsaturated soils. Adv. Water Resour. 2000, 23, 811–815. [Google Scholar] [CrossRef]
- Brusaert, W. Hydrology an Introduction; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Kaplun, K.; Li, J.; Kawashima, N.; Gerson, A.R. Cu and Fe chalcopyrite leach activation energies and the effect of added Fe3+. Geochim. Cosmochim. Acta 2011, 75, 5865–5878. [Google Scholar] [CrossRef]
- Twigg, M.V. Catalyst Handbook, 2nd ed.; Wolfe Publishing Ltd.: London, UK, 1989. [Google Scholar]
- Revell, L.E.; Williamson, B.E. Why are some reactions slower at higher temperatures? J. Chem. Educ. 2013, 90, 1024–1027. [Google Scholar] [CrossRef]
- Bolobajev, J.; Trapido, M.; Goi, A. Interaction of tannic acid with ferric iron to assist 2,4,6-trichlorophenol catalytic decomposition and reuse of ferric sludge as a source of iron catalyst in Fenton-based treatment. Appl. Catal. B Environ. 2016, 187, 75–82. [Google Scholar] [CrossRef]
- Sheng-tao, J.; Jiang-Zhong, Z.; Shu-li, B.; Yu-jiang, G. Research on Fe-loaded ZSM-5 molecular sieve catalyst in high concentration aniline wastewater treatment. Desalt. Water Treat. 2016, 57, 791–798. [Google Scholar] [CrossRef]
- Xiong, H.; Motchelaho, M.A.; Moyo, M.; Jewell, L.L.; Coville, N.J. Effect of Group 1 alkali metal promoters on Fe/CNT catalysts in Fischer-Tropsch synthesis. Fuel 2015, 150, 687–696. [Google Scholar] [CrossRef]
- Vlamidis, Y.; Scavetta, E.; Gazzano, M.; Tonelli, D. Iron vs. aluminium based layered double hydroxides as water splitting catalysts. Electrochim. Acta 2016, 188, 653–660. [Google Scholar] [CrossRef]
- Bouniol, P. Influence of iron on water radiolysis in cement based materials. J. Nucl. Mater. 2010, 403, 167–183. [Google Scholar]
- Chen, K.-F.; Li, S.; Zhang, W.-X. Renewable hydrogen generation by bimetallic zero valent iron nanoparticles. Chem. Eng. J. 2011, 170, 562–567. [Google Scholar] [CrossRef]
- Reardon, E.J. Capture and storage of hydrogen gas by zero valent iron. J. Contam. Hydrol. 2014, 157, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, A.S.; Jekel, M. Degassing, gas retention and release in Fe(0) permeable reactive barriers. J. Contam. Hydrol. 2014, 159, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Reardon, E.J. Anaerobic corrosion of granular iron: Measurement and interpretation of hydrogen evolution rates. Environ. Sci. Technol. 1995, 29, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.L.; Rochealeau, R.E. Electrochemical behaviour of reactively spluttered iron-doped nickel oxide. J. Electrochem. Soc. 1997, 144, 3072–3077. [Google Scholar] [CrossRef]
- Fominykh, K.; Chernev, P.; Zaharieva, I.; Sicklinger, J.; Stefanic, G.; Doblinger, M.; Muller, A.; Pokharel, A.; Bocklein, S.; Scheu, C.; et al. Iron-doped nickel oxide as highly efficient catalysts for alkaline water splitting. ACS Nano 2015, 9, 5180–5188. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.; Block, J.H.; Delmon, B. Manual of methods and procedures for catalyst characterization (IUPAC Subcommittee on catalyst characterization). Pure Appl. Chem. 1995, 67, 1257–1306. [Google Scholar] [CrossRef]
- Misstear, B.; Banks, D.; Clark, L. Water Wells and Boreholes; John Wiley & Sons Ltd.: Chichester, UK, 2006. [Google Scholar]
- Lesch, S.M.; Suarez, D.L. A short note on calculating the adjusted SAR index. Trans. Am. Soc. Agric. Biol. Eng. 2009, 52, 493–496. [Google Scholar]
- Scholl, S.E.; Mersmann, A.B. On intraparticle total pressure change during gas phase adsorption. Gas. Sep. Purif. 1991, 5, 77–82. [Google Scholar] [CrossRef]
- Kozin, P.A.; Boily, J.F. Proton binding and ion exchange at the akaganeite/water interface. J. Phys. Chem. C 2013, 117, 6409–6419. [Google Scholar] [CrossRef]
- Naftz, D.L.; Morrison, S.J.; Davis, J.A.; Fuller, C.C. Handbook of Groundwater Remediation Using Permeable Reactive Barriers; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Zhao, J.; Lin, W.; Chang, Q.; Li, W.; Lai, Y. Adsoptive characteristics of akaganeite and its environmental applications: A review. Environ. Technol. Rev. 2012, 1, 114–126. [Google Scholar] [CrossRef]
- Scheck, J.; Lemke, T.; Gebauer, D. The role of chloride ions during the formation of akaganeite revisited. Minerals 2015, 5, 778–787. [Google Scholar] [CrossRef]
- Wang, W.; Howe, J.Y.; Gu, B. Structure and morphology evolution of hematite (α-Fe2O3) nanoparticles in forced hydrolysis of ferric chloride. J. Phys. Chem. C 2008, 112, 9203–9208. [Google Scholar] [CrossRef]
- Zhou, P.; Yan, H.; Gu, B. Competitive complexation of metal ions with humic substances. Chemosphere 2014, 58, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Schmitt, J.; Chen, Z.; Liang, L.; McCarthy, J.F. Adsorption and desorption of natural organic matter on iron oxide: Mechanisms and models. Environ. Sci. Technol. 1994, 28, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Schmitt, J.; Chen, Z.; Liang, L.; McCarthy, J.F. Adsorption and desorption of different organic matter fractions on iron oxide. Geochim. Cosmochim. Acta 1995, 59, 219–229. [Google Scholar] [CrossRef]
- Gu, B.; Mehlorn, T.L.; Liang, L.; McCarthy, J.F. Competitive adsorption, displacement, and transport of organic matter on iron oxide: I. Competitive adsorption. Geochim. Cosmochim. Acta 1996, 60, 1943–1950. [Google Scholar] [CrossRef]
- Gu, B.; Mehlorn, T.L.; Liang, L.; McCarthy, J.F. Competitive adsorption, displacement, and transport of organic matter on iron oxide: II displacement and transport. Geochim. Cosmochim. Acta 1996, 60, 2977–2992. [Google Scholar] [CrossRef]
- Gu, B.; Phelps, T.J.; Liang, L.; Dickey, M.J.; Roh, Y.; Kinsall, B.L.; Palumbo, A.V.; Jacobs, G.K. Biogeochemical dynamics in zero valent iron columns: Implications for permeable reactive barriers. Environ. Sci. Technol. 1999, 33, 2170–2177. [Google Scholar] [CrossRef]
- Liang, L.; Hofmann, A.; Gu, B. Ligand-induced dissolution and release of ferrihydrite colloids. Geochim. Cosmochim. Acta 2000, 64, 2027–2037. [Google Scholar] [CrossRef]
- Lee, J.-H.; Roh, Y.; Kim, K.-W.; Hur, H.-G. Organic acid dependent iron mineral formation by a newly isolated iron reducing bacterium, Shewanella sp. HN-41. Geomicrobiol. J. 2012, 24, 31–41. [Google Scholar] [CrossRef]
- Chen, J.; Gu, B.; Royer, R.A.; Burgos, W.D. The roles of natural organic matter in chemical and microbial reduction of ferric iron. Sci. Total Environ. 2003, 307, 167–178. [Google Scholar] [CrossRef]
- Peng, Q.-A.; Shaaban, M.; Wu, Y.; Hu, R.; Wang, B.; Wang, J. The diversity of iron reducing bacteria communities in subtropical paddy soils of China. Appl. Soil Ecol. 2016, 101, 20–27. [Google Scholar] [CrossRef]
- Ko, M.-S.; Cho, K.; Jeong, D.; Lee, S. Identification of the microbes mediating Fe reduction in a deep saline aquifer and their influence during managed aquifer recharge. Sci. Total Environ. 2016, 545–546, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.E.G.; Gonsalves, M.J.B.D.; Nazareth, D.R.; Nagarchi, L. Microbial iron reduction and methane oxidation in subsurface sediments of the Arabian Sea. Mar. Petrol. Geol. 2015, 67, 327–335. [Google Scholar] [CrossRef]

| Trial | Feed Water (g·L−1) | ||
|---|---|---|---|
| Na+ | Cl− | NaCl | |
| CSD1a | 0.962 | 1.489 | 2.452 |
| CSD1b | 0.962 | 1.489 | 2.452 |
| CSD1c | 1.009 | 1.562 | 2.570 |
| CSD1d | 1.155 | 1.788 | 2.943 |
| E143a | 2.363 | 3.658 | 6.021 |
| E143b | 1.829 | 2.831 | 4.660 |
| E143c | 1.982 | 3.067 | 5.049 |
| E143d | 2.210 | 3.422 | 5.632 |
| E143e | 2.668 | 4.130 | 6.799 |
| E143f | 0.761 | 1.178 | 1.939 |
| E143g | 1.068 | 1.653 | 2.722 |
| E143h | 1.068 | 1.653 | 2.722 |
| E143i | 1.219 | 1.886 | 3.105 |
| E143j | 1.405 | 2.175 | 3.580 |
| E143k | 1.585 | 2.454 | 4.039 |
| E144a | 1.134 | 2.770 | 3.904 |
| E144b | 1.312 | 2.925 | 4.237 |
| E144c | 0.636 | 1.826 | 2.462 |
| E144d | 0.522 | 1.480 | 2.002 |
| E144e | 0.512 | 1.707 | 2.219 |
| E144f | 4.161 | 6.519 | 10.680 |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antia, D.D.J. ZVI (Fe0) Desalination: Stability of Product Water. Resources 2016, 5, 15. https://doi.org/10.3390/resources5010015
Antia DDJ. ZVI (Fe0) Desalination: Stability of Product Water. Resources. 2016; 5(1):15. https://doi.org/10.3390/resources5010015
Chicago/Turabian StyleAntia, David D. J. 2016. "ZVI (Fe0) Desalination: Stability of Product Water" Resources 5, no. 1: 15. https://doi.org/10.3390/resources5010015
APA StyleAntia, D. D. J. (2016). ZVI (Fe0) Desalination: Stability of Product Water. Resources, 5(1), 15. https://doi.org/10.3390/resources5010015