A New TGF-β1 Inhibitor, CTI-82, Antagonizes Epithelial–Mesenchymal Transition through Inhibition of Phospho-SMAD2/3 and Phospho-ERK

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Cell Viability Assays
2.3. Reporter Gene Assay
2.4. RNA Extraction and Quantitative Real-Time PCR
2.5. Western Blot Analysis
2.6. Matrigel Invasion Assay and Wound-Healing Assay
2.7. Confocal Microscopy
2.8. Statistical Analysis
3. Results
3.1. Screening of a New TGF-β1 Inhibitor in Chalcone Derivatives
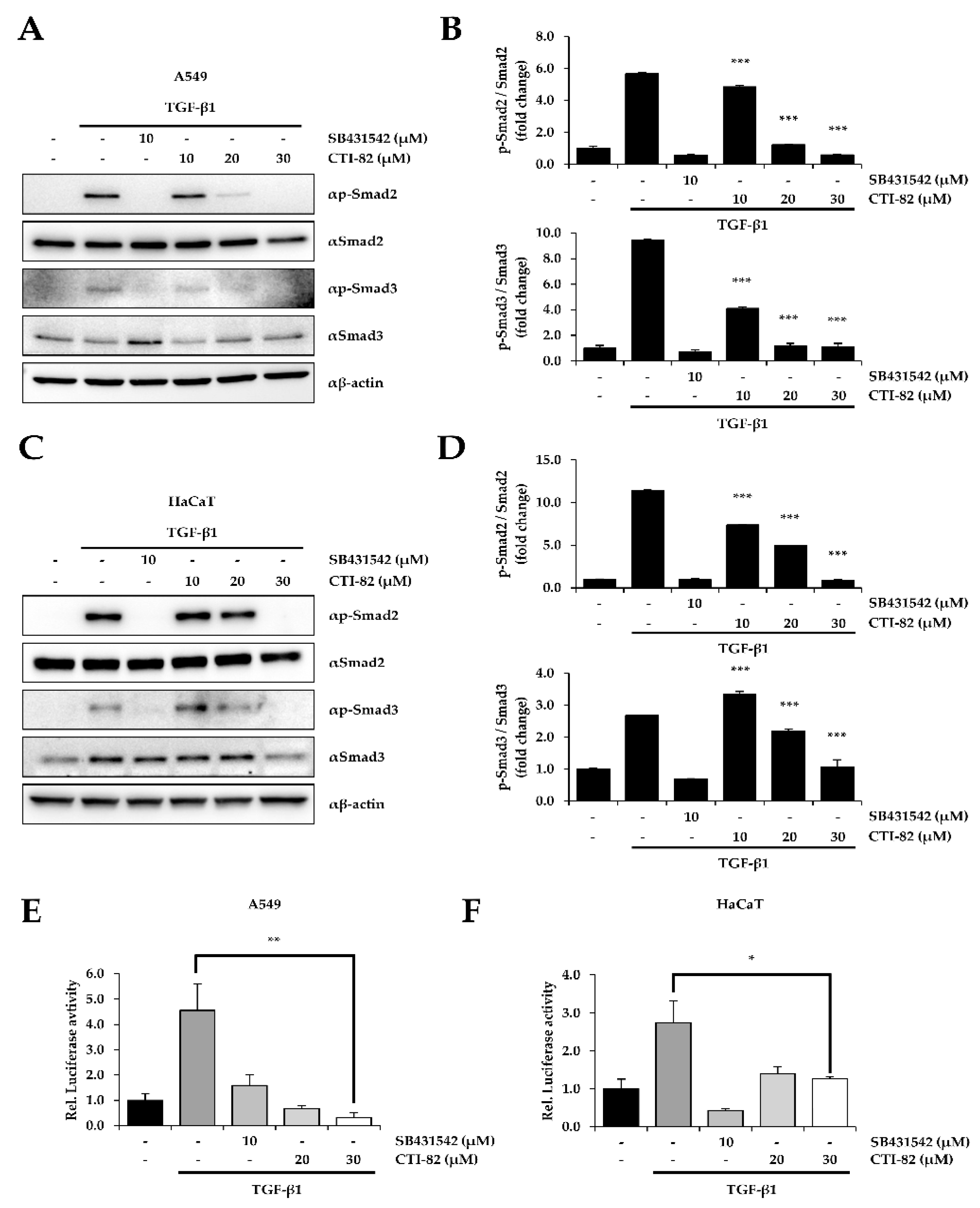
3.2. CTI-82 Inhibits the Phosphorylation of SMAD2/3 Induced by TGF-β1
3.3. CTI-82 Antagonizes TGF-β1-Induced EMT
3.4. CTI-82 Attenuates the Phosphorylation of ERK1/2 Induced by TGF-β1
3.5. CTI-82 Represses TGF-β1-Induced EMT Transcripts
3.6. CTI-82 Inhibits TGF-β1-Induced Cell Migration and Invasion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Sabe, H. Cancer early dissemination: Cancerous epithelial-mesenchymal transdifferentiation and transforming growth factor beta signalling. J. Biochem. 2011, 149, 633–639. [Google Scholar] [CrossRef]
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-beta in cancer. J. Pathol. 2011, 223, 205–218. [Google Scholar] [CrossRef]
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Grunert, S.; Jechlinger, M.; Beug, H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell. Biol. 2003, 4, 657–665. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Xie, D.; Gore, C.; Liu, J.; Pong, R.C.; Mason, R.; Hao, G.; Long, M.; Kabbani, W.; Yu, L.; Zhang, H.; et al. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.H.; Moustakas, A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Tan, E.J.; Peinado, H.; Cano, A.; Heldin, C.H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Patel, K.D. Matrix metalloproteinase-2 (MMP-2) and MMP-9 in pulmonary pathology. Exp. Lung Res. 2005, 31, 599–621. [Google Scholar] [CrossRef]
- Yu, H.; Konigshoff, M.; Jayachandran, A.; Handley, D.; Seeger, W.; Kaminski, N.; Eickelberg, O. Transgelin is a direct target of TGF-beta/Smad3-dependent epithelial cell migration in lung fibrosis. FASEB J. 2008, 22, 1778–1789. [Google Scholar] [CrossRef]
- Fong, Y.C.; Hsu, S.F.; Wu, C.L.; Li, T.M.; Kao, S.T.; Tsai, F.J.; Chen, W.C.; Liu, S.C.; Wu, C.M.; Tang, C.H. Transforming growth factor-beta1 increases cell migration and beta1 integrin up-regulation in human lung cancer cells. Lung Cancer 2009, 64, 13–21. [Google Scholar] [CrossRef]
- Kingsley, D.M. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef]
- Roberts, A.B.; Heine, U.I.; Flanders, K.C.; Sporn, M.B. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann. N. Y. Acad. Sci. 1990, 580, 225–232. [Google Scholar] [CrossRef]
- Kaimori, A.; Potter, J.; Kaimori, J.Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Robertson, H.; Marshall, H.L.; Pekalski, M.; Zhao, L.; Booth, T.A.; Jones, D.E.; Burt, A.D.; Kirby, J.A. Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab. Invest. 2008, 88, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Ko, H.; So, Y.; Jeon, H.; Jeong, M.H.; Choi, H.K.; Ryu, S.H.; Lee, S.W.; Yoon, H.G.; Choi, K.C. TGF-beta1-induced epithelial-mesenchymal transition and acetylation of Smad2 and Smad3 are negatively regulated by EGCG in human A549 lung cancer cells. Cancer Lett. 2013, 335, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Sales, E.; Mohaddes, G.; Alipour, M.R. Chalcones as putative hepatoprotective agents: Preclinical evidence and molecular mechanisms. Pharmacol. Res. 2018, 129, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Deb Majumdar, I.; Devanabanda, A.; Fox, B.; Schwartzman, J.; Cong, H.; Porco, J.A., Jr.; Weber, H.C. Synthetic cyclohexenyl chalcone natural products possess cytotoxic activities against prostate cancer cells and inhibit cysteine cathepsins in vitro. Biochem. Biophys. Res. Commun. 2011, 416, 397–402. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, E.; Baek, K.H.; Kwon, H.B.; Woo, H.; Lee, E.S.; Kwon, Y.; Na, Y. Chalcones, inhibitors for topoisomerase I and cathepsin B and L, as potential anti-cancer agents. Bioorg. Med. Chem. Lett. 2013, 23, 3320–3324. [Google Scholar] [CrossRef]
- Matarrese, P.; Ascione, B.; Ciarlo, L.; Vona, R.; Leonetti, C.; Scarsella, M.; Mileo, A.M.; Catricala, C.; Paggi, M.G.; Malorni, W. Cathepsin B inhibition interferes with metastatic potential of human melanoma: An in vitro and in vivo study. Mol. Cancer 2010, 9, 207. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, M.; Wang, W.; Song, Y.; Chen, G.; Wang, Z.; Liang, Z. Downregulation of cathepsin L suppresses cancer invasion and migration by inhibiting transforming growth factorbetamediated epithelialmesenchymal transition. Oncol. Rep. 2015, 33, 1851–1859. [Google Scholar] [CrossRef]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef]
- Rasanen, K.; Vaheri, A. TGF-beta1 causes epithelial-mesenchymal transition in HaCaT derivatives, but induces expression of COX-2 and migration only in benign, not in malignant keratinocytes. J. Dermatol. Sci. 2010, 58, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Jeon, H.; Lee, D.; Choi, H.K.; Kang, K.S.; Choi, K.C. Sanguiin H6 suppresses TGF-beta induction of the epithelial-mesenchymal transition and inhibits migration and invasion in A549 lung cancer. Bioorg. Med. Chem. Lett. 2015, 25, 5508–5513. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jeon, Y.; Kim, T.; Lim, W.C.; Ham, J.; Park, Y.H.N.; Kim, T.J.; Ko, H. Combined treatment with zingerone and its novel derivative synergistically inhibits TGF-beta1 induced epithelial-mesenchymal transition, migration and invasion of human hepatocellular carcinoma cells. Bioorg. Med. Chem. Lett. 2017, 27, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, K.; Nakatsuka, K.; Matsuo, N.; Yoshioka, H. p38 MAPK mediates the expression of type I collagen induced by TGF-beta 2 in human retinal pigment epithelial cells ARPE-19. Invest. Ophthalmol. Vis. Sci. 2004, 45, 2431–2437. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Ko, H.; Kim, S.J.; Shim, S.H.; Chang, H.; Ha, C.H. Shikonin Induces Apoptotic Cell Death via Regulation of p53 and Nrf2 in AGS Human Stomach Carcinoma Cells. Biomol. Ther. 2016, 24, 501–509. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kang, K.S.; Choi, K.C.; Ko, H. Cardamonin induces autophagy and an antiproliferative effect through JNK activation in human colorectal carcinoma HCT116 cells. Bioorg. Med. Chem. Lett. 2015, 25, 2559–2564. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Kim, J.M.; Kim, S.J.; Shim, S.H.; Ha, C.H.; Chang, H.I. Induction of apoptosis by genipin inhibits cell proliferation in AGS human gastric cancer cells via Egr1/p21 signaling pathway. Bioorg. Med. Chem. Lett. 2015, 25, 4191–4196. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.H.; Ko, H.; Jeon, H.; Sung, G.J.; Park, S.Y.; Jun, W.J.; Lee, Y.H.; Lee, J.; Lee, S.W.; Yoon, H.G.; et al. Delphinidin induces apoptosis via cleaved HDAC3-mediated p53 acetylation and oligomerization in prostate cancer cells. Oncotarget 2016, 7, 56767–56780. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Choi, W.I.; Ko, H.; So, Y.; Kang, K.S.; Kim, I.; Kim, K.; Yoon, H.G.; Kim, T.J.; Choi, K.C. Neobavaisoflavone sensitizes apoptosis via the inhibition of metastasis in TRAIL-resistant human glioma U373MG cells. Life Sci. 2014, 95, 101–107. [Google Scholar] [CrossRef]
- Kim, J.M.; Ko, H.; Kim, S.J.; Shim, S.H.; Ha, C.H.; Chang, H.I. Chemopreventive Properties of Genipin on AGS Cell Line via Induction of JNK/Nrf2/ARE Signaling Pathway. J. Biochem. Mol. Toxicol. 2016, 30, 45–54. [Google Scholar] [CrossRef]
- Lee, D.; Ko, H.; Kim, Y.J.; Kim, S.N.; Choi, K.C.; Yamabe, N.; Kim, K.H.; Kang, K.S.; Kim, H.Y.; Shibamoto, T. Inhibition of A2780 Human Ovarian Carcinoma Cell Proliferation by a Rubus Component, Sanguiin H-6. J. Agric. Food Chem. 2016, 64, 801–805. [Google Scholar] [CrossRef]
- Ko, H.; Jeong, M.H.; Jeon, H.; Sung, G.J.; So, Y.; Kim, I.; Son, J.; Lee, S.W.; Yoon, H.G.; Choi, K.C. Delphinidin sensitizes prostate cancer cells to TRAIL-induced apoptosis, by inducing DR5 and causing caspase-mediated HDAC3 cleavage. Oncotarget 2015, 6, 9970–9984. [Google Scholar] [CrossRef]
- Li, P.H.; Jiang, H.; Zhang, W.J.; Li, Y.L.; Zhao, M.C.; Zhou, W.; Zhang, L.Y.; Tang, Y.D.; Dong, C.Z.; Huang, Z.S.; et al. Synthesis of carbazole derivatives containing chalcone analogs as non-intercalative topoisomerase II catalytic inhibitors and apoptosis inducers. Eur. J. Med. Chem. 2018, 145, 498–510. [Google Scholar] [CrossRef]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef]
- Pohlers, D.; Brenmoehl, J.; Loffler, I.; Muller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-beta and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef]
- Shah, S.; Fourgeaud, C.; Derieux, S.; Mirshahi, S.; Contant, G.; Pimpie, C.; Lo Dico, R.; Soria, J.; Pocard, M.; Mirshahi, M. The close relationship between heparanase and epithelial mesenchymal transition in gastric signet-ring cell adenocarcinoma. Oncotarget 2018, 9, 33778–33787. [Google Scholar] [CrossRef][Green Version]
- Jo, M.; Lester, R.D.; Montel, V.; Eastman, B.; Takimoto, S.; Gonias, S.L. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J. Biol. Chem. 2009, 284, 22825–22833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The roles of ZEB1 in tumorigenic progression and epigenetic modifications. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Naber, H.P.; Drabsch, Y.; Snaar-Jagalska, B.E.; ten Dijke, P.; van Laar, T. Snail and Slug, key regulators of TGF-beta-induced EMT, are sufficient for the induction of single-cell invasion. Biochem. Biophys. Res. Commun. 2013, 435, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Eischeid, A.N.; Chen, X.M. Col1A1 production and apoptotic resistance in TGF-beta1-induced epithelial-to-mesenchymal transition-like phenotype of 603B cells. PLoS ONE 2012, 7, e51371. [Google Scholar] [CrossRef]
- Czuwara-Ladykowska, J.; Sementchenko, V.I.; Watson, D.K.; Trojanowska, M. Ets1 is an effector of the transforming growth factor beta (TGF-beta) signaling pathway and an antagonist of the profibrotic effects of TGF-beta. J. Biol. Chem. 2002, 277, 20399–20408. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Maeda, M.; Chaika, N.; Johnson, K.R.; Wheelock, M.J. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am. J. Respir. Cell Mol. Biol. 2008, 38, 95–104. [Google Scholar] [CrossRef]
- Bai, X.; Li, Y.Y.; Zhang, H.Y.; Wang, F.; He, H.L.; Yao, J.C.; Liu, L.; Li, S.S. Role of matrix metalloproteinase-9 in transforming growth factor-beta1-induced epithelial-mesenchymal transition in esophageal squamous cell carcinoma. Onco Targets Ther. 2017, 10, 2837–2847. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, J.-H.; Kim, H.; Park, S.-H.; Park, H.; Jeong, M.; Kwak, S.; Sung, G.-J.; Song, J.-H.; Na, Y.; Choi, K.-C. A New TGF-β1 Inhibitor, CTI-82, Antagonizes Epithelial–Mesenchymal Transition through Inhibition of Phospho-SMAD2/3 and Phospho-ERK. Biology 2020, 9, 143. https://doi.org/10.3390/biology9070143
Jeong J-H, Kim H, Park S-H, Park H, Jeong M, Kwak S, Sung G-J, Song J-H, Na Y, Choi K-C. A New TGF-β1 Inhibitor, CTI-82, Antagonizes Epithelial–Mesenchymal Transition through Inhibition of Phospho-SMAD2/3 and Phospho-ERK. Biology. 2020; 9(7):143. https://doi.org/10.3390/biology9070143
Chicago/Turabian StyleJeong, Ji-Hoon, Hyunhee Kim, Seung-Ho Park, Hayeon Park, Minseok Jeong, Sungmin Kwak, Gi-Jun Sung, Ji-Hye Song, Younghwa Na, and Kyung-Chul Choi. 2020. "A New TGF-β1 Inhibitor, CTI-82, Antagonizes Epithelial–Mesenchymal Transition through Inhibition of Phospho-SMAD2/3 and Phospho-ERK" Biology 9, no. 7: 143. https://doi.org/10.3390/biology9070143
APA StyleJeong, J.-H., Kim, H., Park, S.-H., Park, H., Jeong, M., Kwak, S., Sung, G.-J., Song, J.-H., Na, Y., & Choi, K.-C. (2020). A New TGF-β1 Inhibitor, CTI-82, Antagonizes Epithelial–Mesenchymal Transition through Inhibition of Phospho-SMAD2/3 and Phospho-ERK. Biology, 9(7), 143. https://doi.org/10.3390/biology9070143