Notch Inhibition via GSI Treatment Elevates Protein Synthesis in C2C12 Myotubes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
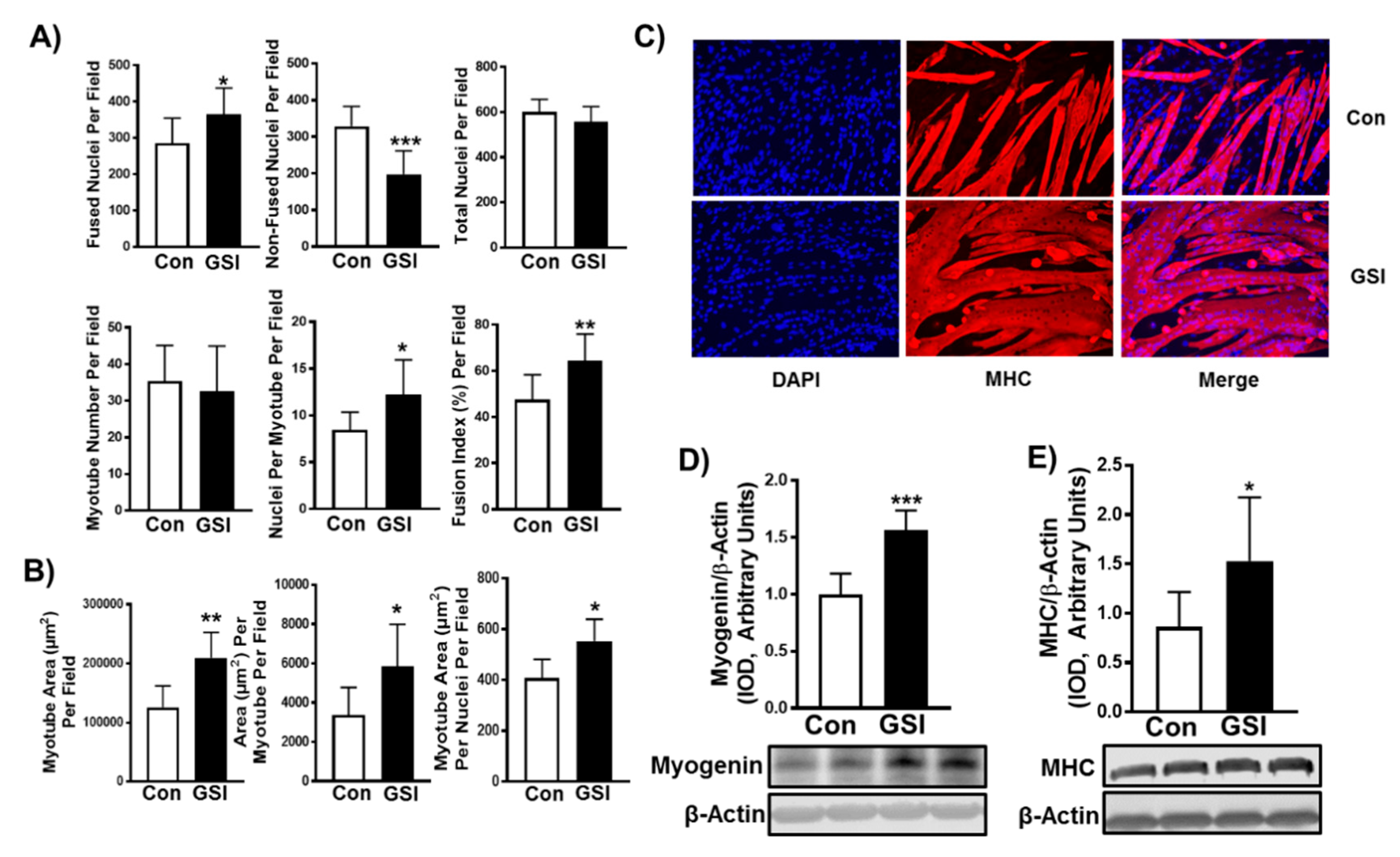
2.2. Myosin Heavy Chain Staining
2.3. Myotube Fusion and Myotube Area
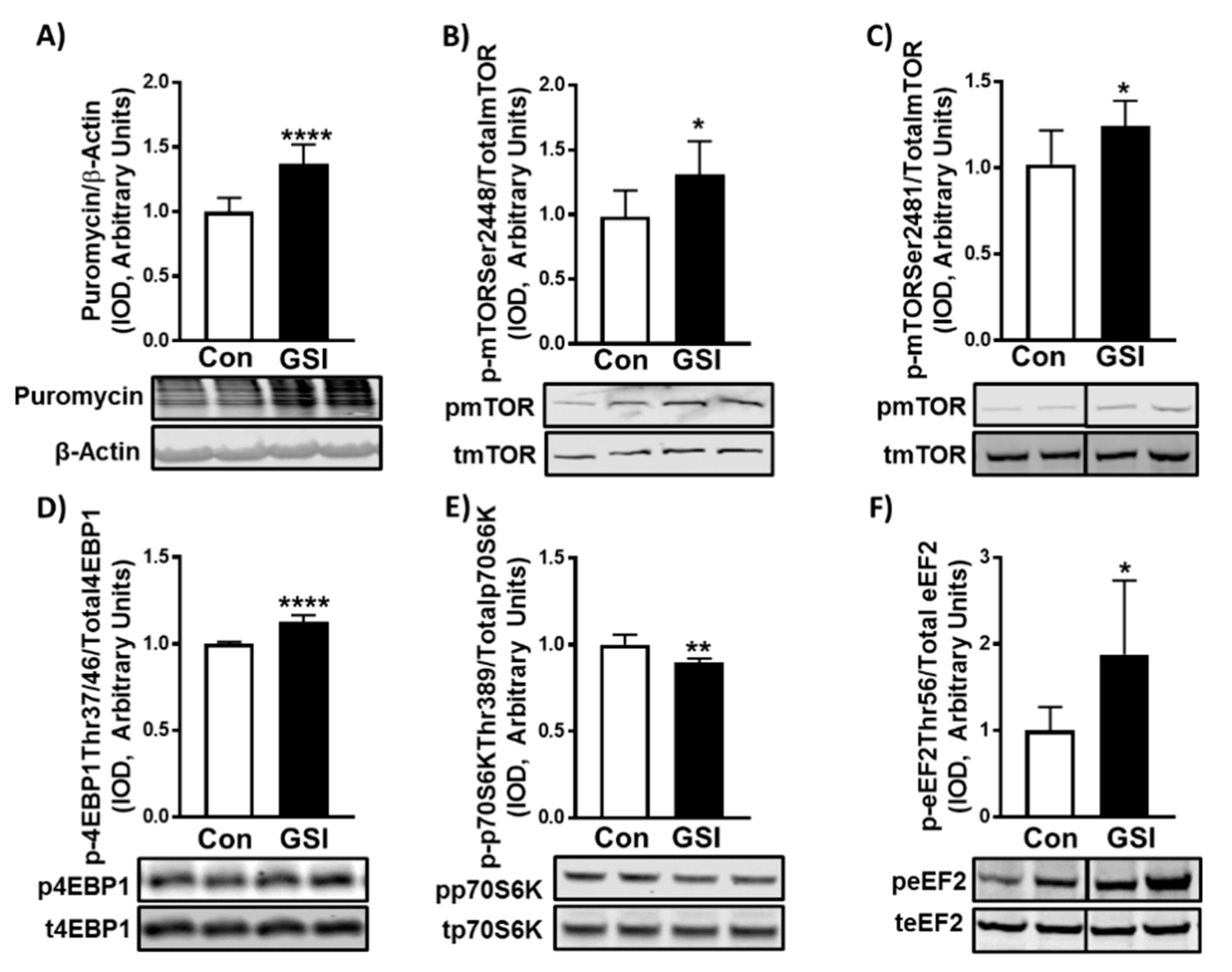
2.4. Protein Synthesis
2.5. Western Blot
2.6. Statistical Analysis
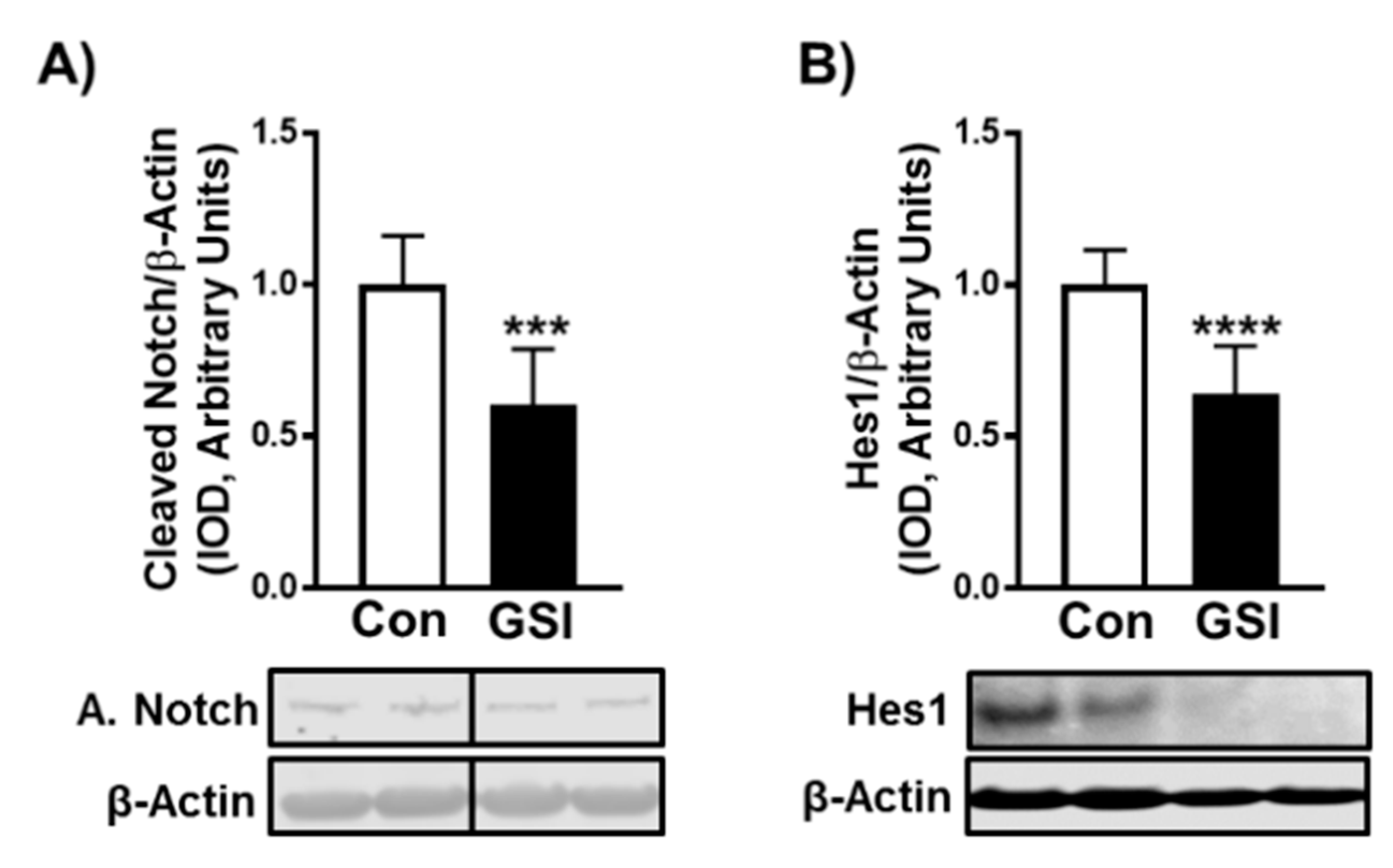
3. Results
3.1. GSI Increases C2C12 Myotube Formation and Hypertrophy
3.2. GSI Increases Protein Synthesis and mTOR Signaling in C2C12 Myotubes
3.3. GSI Increases Signaling Upstream of mTOR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morley, J.E.; Baumgartner, R.N.; Roubenoff, R.; Mayer, J.; Nair, K.S. Sarcopenia. J. Lab. Clin. Med. 2001, 137, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Arthur, S.T.; Cooley, I.D. The effect of physiological stimuli on sarcopenia; Impact of Notch and Wnt signaling on impaired aged skeletal muscle repair. Int. J. Biol. Sci. 2012, 8, 731–760. [Google Scholar] [CrossRef] [PubMed]
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, 11–16. [Google Scholar] [PubMed]
- Brack, A.S.; Conboy, I.M.; Conboy, M.J.; Shen, J.; Rando, T.A. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008, 2, 50–59. [Google Scholar] [CrossRef]
- Conboy, I.M.; Conboy, M.J.; Smythe, G.M.; Rando, T.A. Notch-mediated restoration of regenerative potential to aged muscle. Science 2003, 302, 1575–1577. [Google Scholar] [CrossRef]
- Bi, P.; Kuang, S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol. Metab. 2015, 26, 248–255. [Google Scholar] [CrossRef]
- Mu, X.; Agarwal, R.; March, D.; Rothenberg, A.; Voigt, C.; Tebbets, J.; Huard, J.; Weiss, K. Notch Signaling Mediates Skeletal Muscle Atrophy in Cancer Cachexia Caused by Osteosarcoma. Sarcoma 2016, 2016, 3758162. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Shawber, C.J.; Samuel, V.T.; Birkenfeld, A.L.; Shulman, G.I.; Kitajewski, J.; Accili, D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med. 2011, 17, 961–967. [Google Scholar] [CrossRef]
- Bjornson, C.R.R.; Cheung, T.H.; Liu, L.; Tripathi, P.V.; Steeper, K.M.; Rando, T.A. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 2012, 30, 232–242. [Google Scholar] [CrossRef]
- Mourikis, P.; Mourikis, P.; Sambasivan, R.; Castel, D.; Rocheteau, P.; Bizzarro, V.; Tajbakhsh, S. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 2012, 30, 243–252. [Google Scholar] [CrossRef]
- Wen, Y.; Bi, P.; Liu, W.; Asakura, A.; Keller, C.; Kuang, S. Constitutive Notch activation upregulates Pax7 and promotes the self-renewal of skeletal muscle satellite cells. Mol. Cell. Biol. 2012, 32, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Anderson, G.B.; Dooley, M.S.; Hornberger, T.A. The role of mTOR signaling in the regulation of protein synthesis and muscle mass during immobilization in mice. Dis. Model. Mech. 2015, 8, 1059–1069. [Google Scholar] [CrossRef]
- Ge, Y.; Chen, J. Mammalian target of rapamycin (mTOR) signaling network in skeletal myogenesis. J. Biol. Chem. 2012, 287, 43928–43935. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.M.; Pavlath, G.K. Molecular control of mammalian myoblast fusion. Methods Mol. Biol. 2008, 475, 115–133. [Google Scholar] [PubMed]
- Park, I.H.; Chen, J. Mammalian target of rapamycin (mTOR) signaling is required for a late-stage fusion process during skeletal myotube maturation. J. Biol. Chem. 2005, 280, 32009–32017. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- Agley, C.C.; Velloso, C.P.; Lazarus, N.R.; Harridge, S.D.R. An image analysis method for the precise selection and quantitation of fluorescently labeled cellular constituents: Application to the measurement of human muscle cells in culture. J. Histochem. Cytochem. 2012, 60, 428–438. [Google Scholar] [CrossRef]
- Goodman, C.A.; Hornberger, T.A. Measuring protein synthesis with SUnSET: A valid alternative to traditional techniques? Exerc. Sport Sci. Rev. 2013, 41, 107–115. [Google Scholar] [CrossRef]
- Goodman, C.A.; Mabrey, D.M.; Frey, J.W.; Miu, M.H.; Schmidt, E.K.; Pierre, P.; Hornberger, T.A. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011, 25, 1028–1039. [Google Scholar] [CrossRef]
- Hinds, T.D.; Peck, B.; Shek, E.; Stroup, S.; Hinson, J.; Arthur, S.; Marino, J.S. Overexpression of Glucocorticoid Receptor beta Enhances Myogenesis and Reduces Catabolic Gene Expression. Int. J. Mol. Sci. 2016, 17, 232. [Google Scholar] [CrossRef] [PubMed]
- Rosa de Andrade, I.; Corrêa, S.; Fontenele, M.; de Oliveira Teixeira, J.D.; Abdelhay, E.; Costa, M.L.; Mermelstein, C. γ-Secretase Inhibition Induces Muscle Hypertrophy in a Notch-Independent Mechanism. Proteomics 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Mourikis, P.; Tajbakhsh, S. Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev. Biol. 2014, 14, 2. [Google Scholar]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Tani, S.; Tamura, K.; Minoguchi, S.; Kurooka, H.; Honjo, T. Delta-induced Notch signaling mediated by RBP-J. inhibits MyoD expression and myogenesis. J. Biol. Chem. 1999, 274, 7238–7244. [Google Scholar] [PubMed]
- Buas, M.F.; Kabak, S.; Kabak, T. Inhibition of myogenesis by Notch: Evidence for multiple pathways. J. Cell Physiol. 2009, 218, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kamei, C.N.; Layne, M.D.; Jain, M.K.; Liao, J.K.; Lee, M.E.; Chin, M.T. Regulation of myogenic terminal differentiation by the hairy-related transcription factor CHF2. J. Biol. Chem. 2001, 276, 18591–18596. [Google Scholar] [CrossRef]
- Ge, Y.; Wu, A.-L.; Warnes, C.; Liu, J.; Zhang, C.; Kawasome, H.; Terada, N.; Boppart, M.D.; Schoenherr, C.J.; Chen, J. mTOR regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am. J. Physiol. Cell Physiol. 2009, 297, 1434–1444. [Google Scholar] [CrossRef]
- Zhang, P.; Liang, X.; Shan, T.; Jiang, Q.; Deng, C.; Zheng, R.; Kuang, S. mTOR is necessary for proper satellite cell activity and skeletal muscle regeneration. Biochem. Biophys. Res. Commun. 2015, 463, 102–108. [Google Scholar] [CrossRef]
- Ciciliot, S.; Schiaffino, S. Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 2010, 16, 906–914. [Google Scholar] [CrossRef]
- White, R.B.; Biérinx, A.S.; Gnocchi, V.F.; Zammit, P.S. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 2010, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Jaam, B.A.; Heu, K.; Pennarubia, F.; Segelle, A.; Magnol, L.; Germot, A.; Legardinier, S.; Blanquet, V.; Maftah, A. Reduced Notch signalling leads to postnatal skeletal muscle hypertrophy in Pofut1cax/cax mice. Open Biol. 2016, 6. [Google Scholar] [CrossRef]
- Der Vartanian, A.; Audfray, A.; Jaam, B.A.; Janot, M.; Legardinier, S.; Maftah, A.; Germot, A. Protein O-fucosyltransferase 1 expression impacts myogenic C2C12 cell commitment via the Notch signaling pathway. Mol. Cell. Biol. 2015, 35, 391–405. [Google Scholar] [CrossRef]
- Schuster-Gossler, K.; Cordes, R.; Gossler, A. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc. Natl. Acad. Sci. USA 2007, 104, 537–542. [Google Scholar] [CrossRef]
- Vasyutina, E.; Lenhard, D.C.; Wende, H.; Erdmann, B.; Epstein, J.A.; Birchmeier, C. RBP-J. (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4443–4448. [Google Scholar] [CrossRef]
- Chan, S.M.; Weng, A.P.; Tibshirani, R.; Aster, J.C.; Utz, P.J. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood 2007, 110, 278–286. [Google Scholar] [CrossRef]
- Ciofani, M.; Zuniga-Pflucker, J.C. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat. Immunol. 2005, 6, 881–888. [Google Scholar] [CrossRef]
- Sade, H.; Krishna, S.; Sarin, A. The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J. Biol. Chem. 2004, 279, 2937–2944. [Google Scholar] [CrossRef]
- Ravitz, M.J.; Chen, L.; Lynch, M.; Schmidt, E.V. c-myc Repression of TSC2 contributes to control of translation initiation and Myc-induced transformation. Cancer Res. 2007, 67, 11209–11217. [Google Scholar] [CrossRef]
- Copp, J.; Manning, G.; Hunter, T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): Phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009, 69, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Perumalsamy, L.R.; Nagala, M.; Banerjee, P.; Sarin, A. A hierarchical cascade activated by non-canonical Notch signaling and the mTOR-Rictor complex regulates neglect-induced death in mammalian cells. Cell Death Differ. 2009, 16, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, L.S.; Fabian, J.R.; Kimball, S.R. Glycogen synthase kinase-3 is the predominant insulin-regulated eukaryotic initiation factor 2B kinase in skeletal muscle. Int. J. Biochem. Cell Biol. 1999, 31, 191–200. [Google Scholar] [CrossRef]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Gordon, B.S.; Kelleher, A.R.; Kimball, S.R. Regulation of muscle protein synthesis and the effects of catabolic states. Int. J. Biochem. Cell Biol. 2013, 45, 2147–2157. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huot, J.R.; Marino, J.S.; Turner, M.J.; Arthur, S.T. Notch Inhibition via GSI Treatment Elevates Protein Synthesis in C2C12 Myotubes. Biology 2020, 9, 115. https://doi.org/10.3390/biology9060115
Huot JR, Marino JS, Turner MJ, Arthur ST. Notch Inhibition via GSI Treatment Elevates Protein Synthesis in C2C12 Myotubes. Biology. 2020; 9(6):115. https://doi.org/10.3390/biology9060115
Chicago/Turabian StyleHuot, Joshua R., Joseph S. Marino, Michael J. Turner, and Susan T. Arthur. 2020. "Notch Inhibition via GSI Treatment Elevates Protein Synthesis in C2C12 Myotubes" Biology 9, no. 6: 115. https://doi.org/10.3390/biology9060115
APA StyleHuot, J. R., Marino, J. S., Turner, M. J., & Arthur, S. T. (2020). Notch Inhibition via GSI Treatment Elevates Protein Synthesis in C2C12 Myotubes. Biology, 9(6), 115. https://doi.org/10.3390/biology9060115