Mitophagy: A New Player in Stem Cell Biology



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
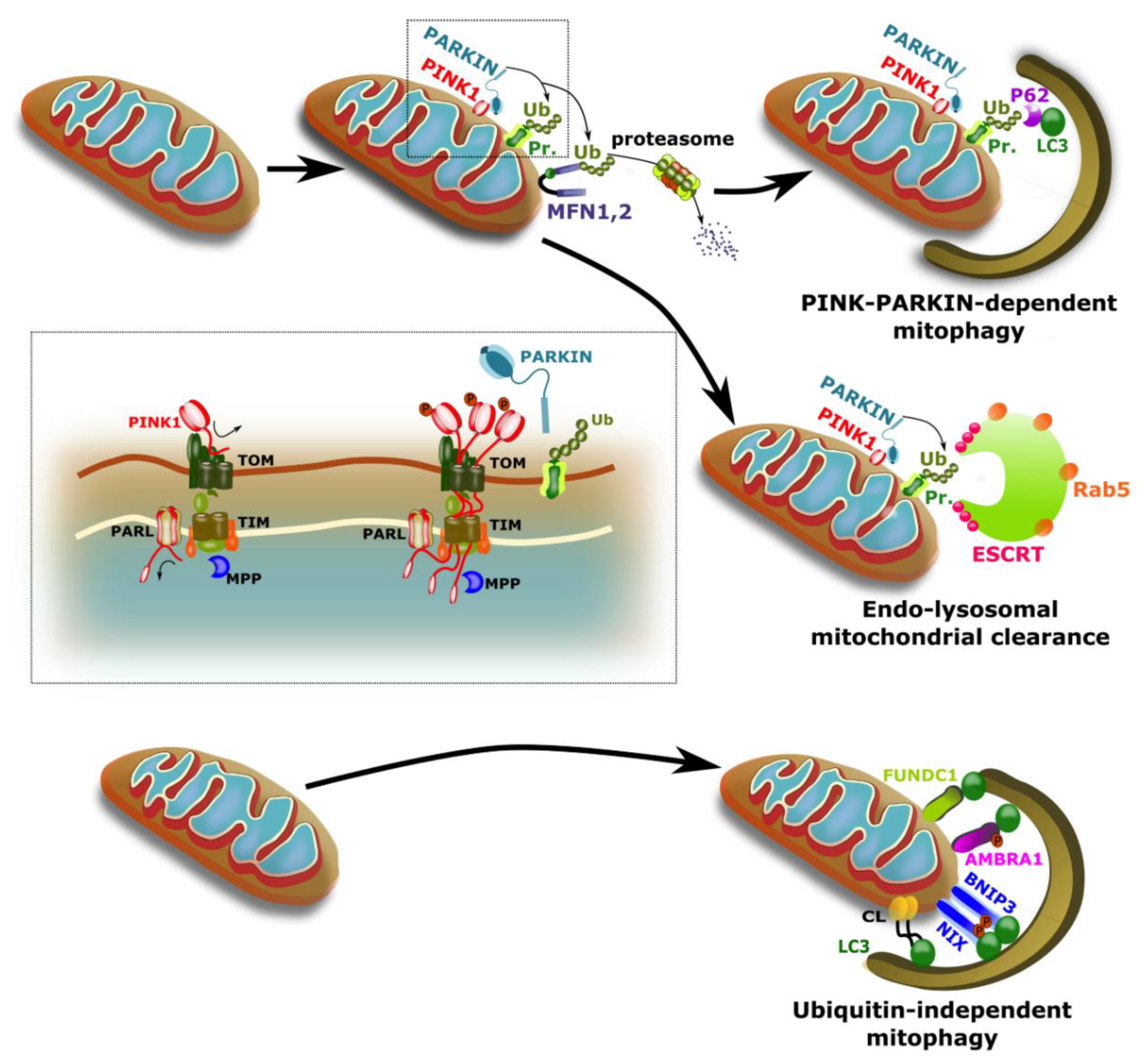
2. Mitophagy
2.1. Important Players in Mitophagy
2.2. Overlap and Cross Talk between Mitophagy Pathways
3. Mitochondria and Stem Cells
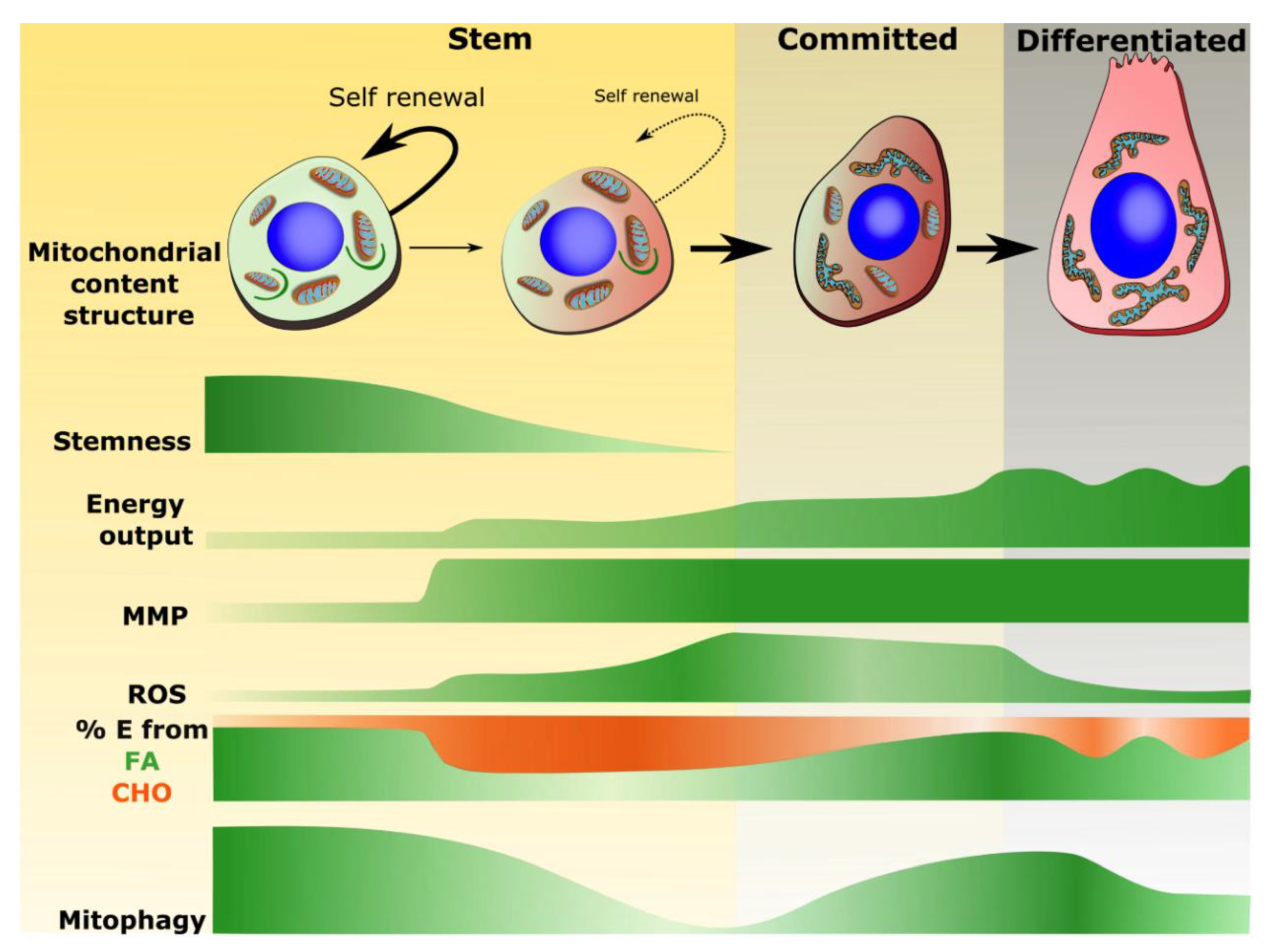
3.1. Mitochondrial Properties Associated with Stemness
3.2. Mitochondrial Phenotype Shifts Associated with Commitment and Differentiation
4. Mitophagy in Stem Cells
4.1. Mitophagy and iPSC Reprogramming
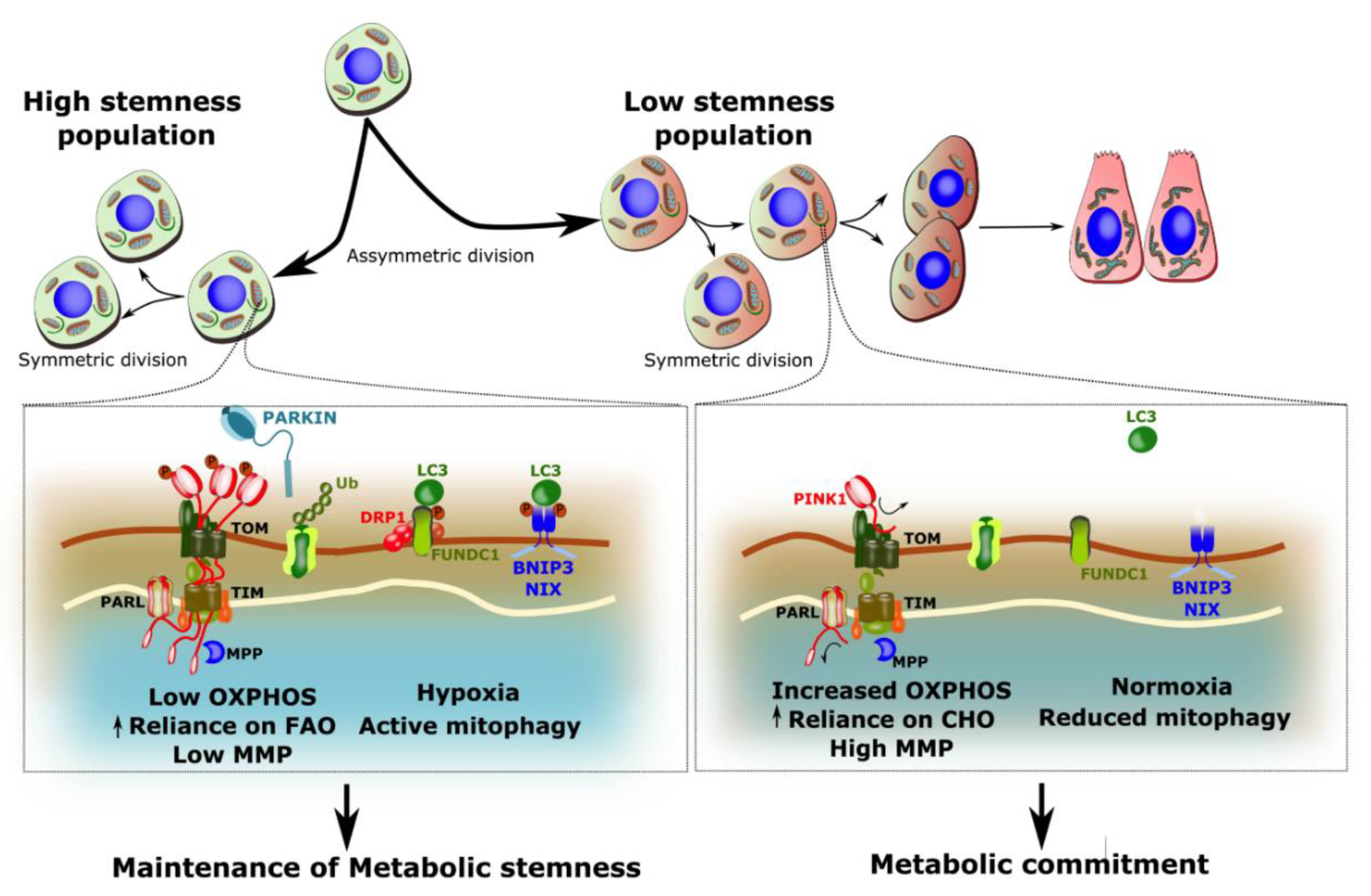
4.2. Mitophagy in Stem Cell Maintenance
4.3. Mitophagy in Stem Cell Differentiation
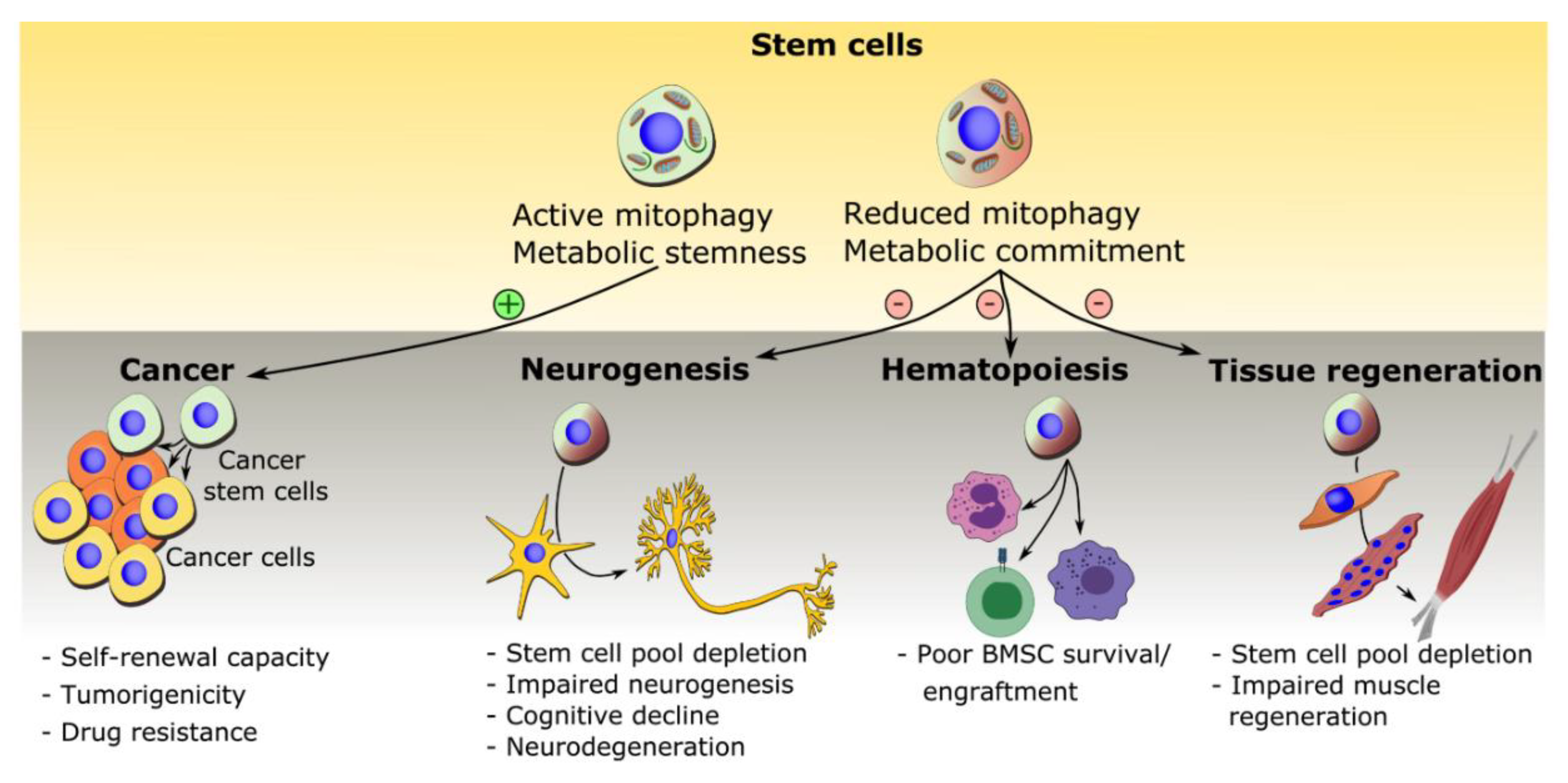
5. Mitophagy in Disease and Treatment
5.1. Mitophagy in Cancer Stem Cells
5.2. Mitophagy in Neural Stem Cell Depletion and Impaired Neurogenesis
5.3. Mitophagy in Bone Marrow Mesenchymal Stem Cells and Bone Diseases
5.4. Mitophagy in Ageing
5.5. Therapeutic Interventions
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Kim, I.; Lemasters, J.J. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am. J. Physiol. Physiol. 2011, 300, C308–C317. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense Against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nat. Cell Biol. 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 2015, 128, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Ghezzi, D. Faculty Opinions recommendation of PINK1 cleavage at position A103 by the mitochondrial protease PARL. J. Cell Biol. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Hoshino, A.; Wang, W.-J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nat. Cell Biol. 2019, 575, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.-K.; Lee, S.H.; Kitada, T.; Kim, J.-M.; Chung, J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 2008, 377, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine. Open Biol. 2012; 2, 120080. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. Faculty Opinions recommendation of PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2016, 205, 143–153. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Fogel, A.I.; Wang, C.; Van Der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Hammerling, B.C.; Najor, R.H.; Cortez, M.Q.; Shires, S.E.; Leon, L.J.; Gonzalez, E.R.; Boassa, D.; Phan, S.; Thor, A.; Jimenez, R.E.; et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat. Commun. 2017, 8, 14050. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef]
- Diwan, A.; Koesters, A.G.; Odley, A.M.; Pushkaran, S.; Baines, C.P.; Spike, B.T.; Daria, D.; Jegga, A.G.; Geiger, H.; Aronow, B.J.; et al. Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6794–6799. [Google Scholar] [CrossRef]
- Rogov, V.V.; Suzuki, H.; Marinković, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubašić, M.; Šprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2009, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Linbo, C.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. Faculty Opinions recommendation of Ambra1 regulates autophagy and development of the nervous system. Fac. Opin. Post-Publ. Peer Rev. Biomed. Lit. 2007, 447, 1121–1125. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Di Rita, A.; Peschiaroli, A.; Pasquale, D.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin Interacts with Ambra1 to Induce Mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Désaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Ni, H.-M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W.; Yin, X.-M. Nix Is Critical to Two Distinct Phases of Mitophagy, Reactive Oxygen Species-mediated Autophagy Induction and Parkin-Ubiquitin-p62-mediated Mitochondrial Priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef]
- Naeem, S.; Qi, Y.; Tian, Y.; Zhang, Y. NIX compensates lost role of parkin in cd-induced mitophagy in HeLa cells through phosphorylation. Toxicol. Lett. 2020, 326, 1–10. [Google Scholar] [CrossRef]
- Park, J.-S.; Koentjoro, B.; Sue, C.M. Commentary: Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Front. Mol. Neurosci. 2017, 10, 297. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nat. Cell Biol. 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Von Coelln, R.; Thomas, B.; Savitt, J.M.; Lim, K.L.; Sasaki, M.; Hess, E.J.; Dawson, V.L.; Dawson, T.M. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc. Natl. Acad. Sci. USA 2004, 101, 10744–10749. [Google Scholar] [CrossRef]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) Repression of PGC-1α Contributes to Neurodegeneration in Parkinson’s Disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Peng, W.; Zhang, J.; Dong, W.; Wu, J.; Wang, T.; Xie, Z. P53 and Parkin co-regulate mitophagy in bone marrow mesenchymal stem cells to promote the repair of early steroid-induced osteonecrosis of the femoral head. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Suomalainen, A.; Hämäläinen, R.H. Stem cells, mitochondria and aging. Biochim. Biophys. Acta (BBA) Bioenergy 2015, 1847, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Chacón-Martínez, C.A.; Koester, J.; Wickström, S.A. Signaling in the stem cell niche: Regulating cell fate, function and plasticity. Development 2018, 145, dev165399. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Dzierzak, E. The many faces of hematopoietic stem cell heterogeneity. Development 2016, 143, 4571–4581. [Google Scholar] [CrossRef]
- Wilson, N.K.; Kent, D.G.; Buettner, F.; Shehata, M.; Macaulay, I.C.; Calero-Nieto, F.J.; Castillo, M.S.; Oedekoven, C.A.; Diamanti, E.; Schulte, R.; et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 2015, 16, 712–724. [Google Scholar] [CrossRef]
- Greulich, P.; Simons, B.D. Dynamic heterogeneity as a strategy of stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2016, 113, 7509–7514. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; Maclaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.-E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef]
- Papa, L.; Djedaini, M.; Hoffman, R. Mitochondrial Role in Stemness and Differentiation of Hematopoietic Stem Cells. Stem Cells Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Slack, R.S. Mitochondrial and Reactive Oxygen Species Signaling Coordinate Stem Cell Fate Decisions and Life Long Maintenance. Antioxid. Redox Signal. 2018, 28, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, P.; Kannan, P.; Mlody, B.G.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef] [PubMed]
- Forni, M.F.; Peloggia, J.; Trudeau, K.; Shirihai, O.S.; Kowaltowski, A.J. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem Cells 2016, 34, 743–755. [Google Scholar] [CrossRef]
- Beckervordersandforth, R.; Ebert, B.; Schäffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron 2017, 93, 560–573.e6. [Google Scholar] [CrossRef]
- Luchsinger, L.L.; De Almeida, M.J.; Corrigan, D.J.; Mumau, M.; Snoeck, H.-W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nat. Cell Biol. 2016, 529, 528–531. [Google Scholar] [CrossRef]
- Liang, R.; Arif, T.; Kalmykova, S.; Kasianov, A.; Lin, M.; Menon, V.; Qiu, J.; Bernitz, J.M.; Moore, K.; Lin, F.; et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020, 26, 359–376.e7. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-Oxoacid Dehydrogenase Complexes in Mitochondria Can Produce Superoxide/Hydrogen Peroxide at Much Higher Rates Than Complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef]
- Seo, B.J.; Yoon, S.H.; Do, J.T. Mitochondrial Dynamics in Stem Cells and Differentiation. Int. J. Mol. Sci. 2018, 19, 3893. [Google Scholar] [CrossRef]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, T.; Wang, L.; Cai, Y.; Zhong, X.; He, X.; Hu, L.; Tian, S.; Wu, M.; Hui, L.; et al. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 2017, 36, 1330–1347. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Pilz, G.-A.; Ghesquière, B.; Kovacs, W.J.; Wegleiter, T.; Moore, D.L.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Slack, R.S. Mitochondrial activity in the regulation of stem cell self-renewal and differentiation. Curr. Opin. Cell Biol. 2017, 49, 1–8. [Google Scholar] [CrossRef]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Y.Z.Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Braun, S.M.G.; Zurkirchen, L.; Von Schoultz, C.; Zamboni, N.; Araúzo-Bravo, M.J.; Kovacs, W.J.; Karalay, Ö.; Suter, U.; Machado, R.A.C.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nat. Cell Biol. 2013, 493, 226–230. [Google Scholar] [CrossRef]
- Ryall, J.G.; Cliff, T.; Dalton, S.; Sartorelli, V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 2015, 17, 651–662. [Google Scholar] [CrossRef]
- Repele, A.; Lupi, R.; Eaton, S.; Urbani, L.; De Coppi, P.; Campanella, M. Cell metabolism sets the differences between subpopulations of satellite cells (SCs). BMC Cell Biol. 2013, 14, 1–7. [Google Scholar] [CrossRef][Green Version]
- Chakkalakal, J.V.; Christensen, J.; Xiang, W.; Tierney, M.T.; Boscolo, F.S.; Sacco, A.; Brack, A.S. Early forming label-retaining muscle stem cells require p27kip1 for maintenance of the primitive state. Development 2014, 141, 1649–1659. [Google Scholar] [CrossRef]
- Mohammad, K.; Dakik, P.; Medkour, Y.; Mitrofanova, D.; Titorenko, V.I. Quiescence Entry, Maintenance, and Exit in Adult Stem Cells. Int. J. Mol. Sci. 2019, 20, 2158. [Google Scholar] [CrossRef]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Novotova, M.; Fortin, D.; Garnier, A.; Ventura-Clapier, R.; Veksler, V.; Joubert, F. Postnatal development of mouse heart: Formation of energetic microdomains. J. Physiol. 2010, 588, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Seyer, P.; Grandemange, S.; Busson, M.; Carazo, Á.; Gamaléri, F.; Pessemesse, L.; Casas, F.; Cabello, G.; Wrutniak-Cabello, C. Mitochondrial activity regulates myoblast differentiation by control of c-Myc expression. J. Cell. Physiol. 2006, 207, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Rochard, P.; Rodier, A.; Casas, F.; Cassar-Malek, I.; Marchal-Victorion, S.; Daury, L.; Wrutniak, C.; Cabello, G. Mitochondrial Activity Is Involved in the Regulation of Myoblast Differentiation through Myogenin Expression and Activity of Myogenic Factors. J. Biol. Chem. 2000, 275, 2733–2744. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef]
- Wei, P.; Dove, K.K.; Bensard, C.; Schell, J.C.; Rutter, J. The Force Is Strong with This One: Metabolism (Over)powers Stem Cell Fate. Trends Cell Biol. 2018, 28, 551–559. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; A Teitell, M. Metabolic Regulation in Pluripotent Stem Cells during Reprogramming and Self-Renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic Oxidative Bioenergetics Transitions into Pluripotency-Dependent Glycolysis to Facilitate Nuclear Reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Iv, C.A.E.; Ramalho-Santos, J.; Van Houten, B.; Schatten, G. Energy Metabolism in Human Pluripotent Stem Cells and Their Differentiated Counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef]
- Flores, A.; Schell, J.; Krall, A.S.; Jelinek, D.; Miranda, M.; Grigorian, M.; Braas, D.; White, A.C.; Zhou, J.L.; Graham, N.A.; et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol. 2017, 19, 1017–1026. [Google Scholar] [CrossRef]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef]
- Ren, R.; Ocampo, A.; Liu, G.-H.; Belmonte, J.C.I. Regulation of Stem Cell Aging by Metabolism and Epigenetics. Cell Metab. 2017, 26, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Rando, T.A. Interaction between epigenetic and metabolism in aging stem cells. Curr. Opin. Cell Biol. 2017, 45, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.L.; Timoskainen, S.; West, F.D.; Vekterud, K.; Boquest, A.C.; Ährlund-Richter, L.; Stice, S.L.; Collas, P. Lineage-Specific Promoter DNA Methylation Patterns Segregate Adult Progenitor Cell Types. Stem Cells Dev. 2010, 19, 1257–1266. [Google Scholar] [CrossRef]
- Hwang, I.-Y.; Kwak, S.; Lee, S.; Kim, H.; Lee, S.E.; Kim, J.-H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H.; Jin, X.; et al. Psat1-Dependent Fluctuations in α-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab. 2016, 24, 494–501. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nat. Cell Biol. 2015, 518, 413–416. [Google Scholar] [CrossRef]
- Mortensen, M.; Ferguson, D.J.P.; Simon, A.K. Mitochondrial clearance by autophagy in developing erythrocytes: Clearly important, but just how much so? Cell Cycle 2010, 9, 1901–1906. [Google Scholar] [CrossRef]
- Simon, H.-U. Regulation of eosinophil and neutrophil apoptosis--similarities and differences. Immunol. Rev. 2001, 179, 156–162. [Google Scholar] [CrossRef]
- García-Prat, L.; Sousa-Victor, P.; Muñoz-Cánoves, P. Proteostatic and Metabolic Control of Stemness. Cell Stem Cell 2017, 20, 593–608. [Google Scholar] [CrossRef]
- Oliver, L.; Hue, E.; Priault, M.; Vallette, F.M. Basal Autophagy Decreased During the Differentiation of Human Adult Mesenchymal Stem Cells. Stem Cells Dev. 2012, 21, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Salemi, S.; Yousefi, S.; A Constantinescu, M.; Fey, M.F.; Simon, H.-U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2011, 22, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nat. Cell Biol. 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.-L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef]
- Mortensen, M.; Ferguson, D.J.P.; Edelmann, M.J.; Kessler, B.; Morten, K.J.; Komatsu, M.; Simon, A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2009, 107, 832–837. [Google Scholar] [CrossRef]
- Mortensen, M.; Watson, A.S.; Simon, A.K. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011, 7, 1069–1070. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, E.R.A.M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nat. Cell Biol. 2017, 543, 205–210. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Feixiang, B.; Yang, L.; Long, Q.; Chen, K.; Tang, H.; Wu, Y.; Liu, Z.; Zhou, Y.; Qi, J.; Zheng, L.; et al. BNIP3L-dependent mitophagy accounts for mitochondrial clearance during 3 factors-induced somatic cell reprogramming. Autophagy 2017, 13, 1543–1555. [Google Scholar] [CrossRef]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef]
- Jin, G.; Xu, C.; Zhang, X.; Long, J.; Rezaeian, A.H.; Liu, C.; Furth, M.E.; Kridel, S.; Pasche, B.; Bian, X.-W.; et al. Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol. 2018, 19, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Cadete, V.J.; Vasam, G.; Menzies, K.J.; Burelle, Y. Mitochondrial quality control in the cardiac system: An integrative view. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 1991, 294, 158–162. [Google Scholar] [CrossRef]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, Å.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef]
- Da Dawson, T.M.; Pirooznia, S. Faculty Opinions recommendation of Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Fac. Opin. Post-Publ. Peer Review Biomed. Lit. 2015, 348, 340–343. [Google Scholar] [CrossRef]
- Adams, W.C.; Chen, Y.-H.; Kratchmarov, R.; Yen, B.; Nish, S.A.; Lin, W.-H.W.; Rothman, N.J.; Luchsinger, L.L.; Klein, U.; Busslinger, M.; et al. Anabolism-associated Mitochondrial Stasis Driving Lymphocyte Differentiation over Self-renewal. Cell Rep. 2016, 17, 3142–3152. [Google Scholar] [CrossRef]
- Esteban-Martínez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef]
- Sin, J.; Andres, A.M.; Taylor, D.J.R.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef]
- Tang, A.H.; Rando, T.A. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nat. Cell Biol. 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Veeraraghavalu, K.; Choi, S.H.; Zhang, X.; Sisodia, S.S. Endogenous expression of FAD-linked PS1 impairs proliferation, neuronal differentiation and survival of adult hippocampal progenitors. Mol. Neurodegener. 2013, 8, 41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Agnihotri, S.K.; Shen, R.; Li, J.; Gao, X.; Büeler, H. Loss of PINK1 leads to metabolic deficits in adult neural stem cells and impedes differentiation of newborn neurons in the mouse hippocampus. FASEB J. 2017, 31, 2839–2853. [Google Scholar] [CrossRef] [PubMed]
- Wiatr, K.; Szlachcic, W.J.; Trzeciak, M.; Figlerowicz, M.; Figiel, M. Huntington Disease as a Neurodevelopmental Disorder and Early Signs of the Disease in Stem Cells. Mol. Neurobiol. 2018, 55, 3351–3371. [Google Scholar] [CrossRef]
- Orosco, L.A.; Ross, A.P.; Cates, S.L.; Scott, S.E.; Wu, D.; Sohn, J.; Pleasure, D.; Pleasure, S.J.; Adamopoulos, I.E.; Zarbalis, K.S. Loss of Wdfy3 in mice alters cerebral cortical neurogenesis reflecting aspects of the autism pathology. Nat. Commun. 2014, 5, 4692. [Google Scholar] [CrossRef]
- E Terrillion, C.; Abazyan, B.; Yang, Z.; Crawford, J.; Shevelkin, A.V.; Jouroukhin, Y.; Yoo, K.H.; Cho, C.H.; Roychaudhuri, R.; Snyder, S.H.; et al. DISC1 in Astrocytes Influences Adult Neurogenesis and Hippocampus-Dependent Behaviors in Mice. Neuropsychopharmacol. 2017, 42, 2242–2251. [Google Scholar] [CrossRef]
- Napoli, E.; Song, G.; Panoutsopoulos, A.A.; Riyadh, M.A.; Kaushik, G.; Halmai, J.; Levenson, R.; Zarbalis, K.S.; Giulivi, C. Beyond autophagy: A novel role for autism-linked Wdfy3 in brain mitophagy. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef]
- Wang, Z.T.; Lu, M.H.; Zhang, Y.; Ji, W.L.; Lei, L.; Wang, W.; Fang, L.P.; Wang, L.W.; Yu, F.; Wang, J.; et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell 2019, 18, e12860. [Google Scholar] [CrossRef]
- Piñero-Martos, E.; Ortega-Vila, B.; Pol-Fuster, J.; Cisneros-Barroso, E.; Ruiz-Guerra, L.; Medina-Dols, A.; Heine-Suñer, D.; Lladó, J.; Olmos, G.; Vives-Bauzà, C. Disrupted in schizophrenia 1 (DISC1) is a constituent of the mammalian mitochondrial contact site and cristae organizing system (MICOS) complex, and is essential for oxidative phosphorylation. Hum. Mol. Genet. 2016, 25, 4157–4169. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Sproul, A.; Martinez, H.; Paquet, D.; Gerges, M.; Noggle, S.; Starkov, A.A. Autophagy Induction by Bexarotene Promotes Mitophagy in Presenilin 1 Familial Alzheimer’s Disease iPSC-Derived Neural Stem Cells. Mol. Neurobiol. 2019, 56, 8220–8236. [Google Scholar] [CrossRef]
- Walter, J.; Bolognin, S.; Antony, P.M.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.; Thu, D.; Waldvogel, H.J.; Faull, R.L.; Luthi-Carter, R. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat. Methods 2011, 8, 945–947. [Google Scholar] [CrossRef]
- Yan, C.; Luo, L.; Guo, C.-Y.; Goto, S.; Urata, Y.; Shao, J.-H.; Li, T.-S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef]
- Takeda, M.; Koseki, J.; Takahashi, H.; Miyoshi, N.; Nishida, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Yamamoto, H.; et al. Disruption of Endolysosomal RAB5/7 Efficiently Eliminates Colorectal Cancer Stem Cells. Cancer Res. 2019, 79, 1426–1437. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.-H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef]
- Rosen, J.M.; Jordan, C.T. The Increasing Complexity of the Cancer Stem Cell Paradigm. Science 2009, 324, 1670–1673. [Google Scholar] [CrossRef]
- E Visvader, J.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.-Q.; Li, Q.; Wang, G.-H.; Sun, F.-F.; Huang, G.-J.; Bian, X.-W.; Yu, S.-C.; Qian, G.-S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-A.; Wang, C.-Y.; Hsieh, Y.-T.; Chen, Y.-J.; Wei, Y.-H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2014, 14, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef]
- Lee, J.K.; Mathews, K.; Schlaggar, B.; Perlmutter, J.; Paulsen, J.S.; Epping, E.; Burmeister, L.; Nopoulos, P. Measures of growth in children at risk for Huntington disease. Neurology 2012, 79, 668–674. [Google Scholar] [CrossRef]
- Woda, J.M.; Calzonetti, T.; Hilditch-Maguire, P.; Duyao, M.P.; A Conlon, R.; Macdonald, M.E. Inactivation of the Huntington’s disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMC Dev. Biol. 2005, 5, 17. [Google Scholar] [CrossRef]
- Sardo, V.L.; Zuccato, C.; Gaudenzi, G.; Vitali, B.; Ramos, C.; Tartari, M.; A Myre, M.; A Walker, J.; Pistocchi, A.; Conti, L.; et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat. Neurosci. 2012, 15, 713–721. [Google Scholar] [CrossRef]
- Molero, A.E.; Gokhan, S.; Gonzalez, S.; Feig, J.L.; Alexandre, L.C.; Mehler, M.F. Impairment of developmental stem cell-mediated striatal neurogenesis and pluripotency genes in a knock-in model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 21900–21905. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastián, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Heurtier, V.; Owens, N.; Gonzalez, I.; Mueller, F.; Proux, C.; Mornico, D.; Clerc, P.; Dubois, A.; Navarro, P. The molecular logic of Nanog-induced self-renewal in mouse embryonic stem cells. Nat. Commun. 2019, 10, 1109. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, N.J.; Porteous, D.J. DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacol. 2012, 62, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.L.; Song, H. DISC1 partners with GSK3beta in neurogenesis. Cell 2009, 136, 990–992. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Wang, W.E.; Zeng, C. How to Improve the Survival of Transplanted Mesenchymal Stem Cell in Ischemic Heart? Stem Cells Int. 2015, 2016, 1–14. [Google Scholar] [CrossRef]
- Hu, C.; Zhao, L.; Peng, C.; Li, L. Regulation of the mitochondrial reactive oxygen species: Strategies to control mesenchymal stem cell fates ex vivo and in vivo. J. Cell. Mol. Med. 2018, 22, 5196–5207. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef]
- Sart, S.; Song, L.; Li, Y. Controlling Redox Status for Stem Cell Survival, Expansion, and Differentiation. Oxidative Med. Cell. Longev. 2015, 2015, 1–14. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Song, K.; Noh, H.; Lee, S.H. Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L–mitophagy pathway. Aging Cell 2020, 19, e13111. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, H.; Han, D.; Ma, S.; Fan, W.; Wang, Y.; Zhang, R.; Fan, M.; Huang, Y.; Fu, X.; et al. A Novel Mechanism of Mesenchymal Stromal Cell-Mediated Protection against Sepsis: Restricting Inflammasome Activation in Macrophages by Increasing Mitophagy and Decreasing Mitochondrial ROS. Oxidative Med. Cell. Longev. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Koehler, C.L.; Perkins, G.A.; Ellisman, M.H.; Jones, D.L. Pink1 and Parkin regulate Drosophila intestinal stem cell proliferation during stress and aging. J. Cell Biol. 2017, 216, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; Sengupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef]
- Johnson, S.C.; Kaeberlein, M. Rapamycin in aging and disease: Maximizing efficacy while minimizing side effects. Oncotarget 2016, 7, 44876–44878. [Google Scholar] [CrossRef]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A DeltaPsim independent pharmacological regulator of mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-dit-Félix, A.A.; Williams, E.G.; Jha, P.; Sasso, G.L.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, F.; Guo, Z.; Zhao, J.; Song, X.; Yang, H. Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells. Mol. Carcinog. 2018, 57, 193–200. [Google Scholar] [CrossRef]
- Ehartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; McQuibban, G.A. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cairns, G.; Thumiah-Mootoo, M.; Burelle, Y.; Khacho, M. Mitophagy: A New Player in Stem Cell Biology. Biology 2020, 9, 481. https://doi.org/10.3390/biology9120481
Cairns G, Thumiah-Mootoo M, Burelle Y, Khacho M. Mitophagy: A New Player in Stem Cell Biology. Biology. 2020; 9(12):481. https://doi.org/10.3390/biology9120481
Chicago/Turabian StyleCairns, George, Madhavee Thumiah-Mootoo, Yan Burelle, and Mireille Khacho. 2020. "Mitophagy: A New Player in Stem Cell Biology" Biology 9, no. 12: 481. https://doi.org/10.3390/biology9120481
APA StyleCairns, G., Thumiah-Mootoo, M., Burelle, Y., & Khacho, M. (2020). Mitophagy: A New Player in Stem Cell Biology. Biology, 9(12), 481. https://doi.org/10.3390/biology9120481