Lymphotropic Viruses: Chronic Inflammation and Induction of Cancers


{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
Lymphotropic Viruses and Cancers
2. Inflammation and Oncogenesis
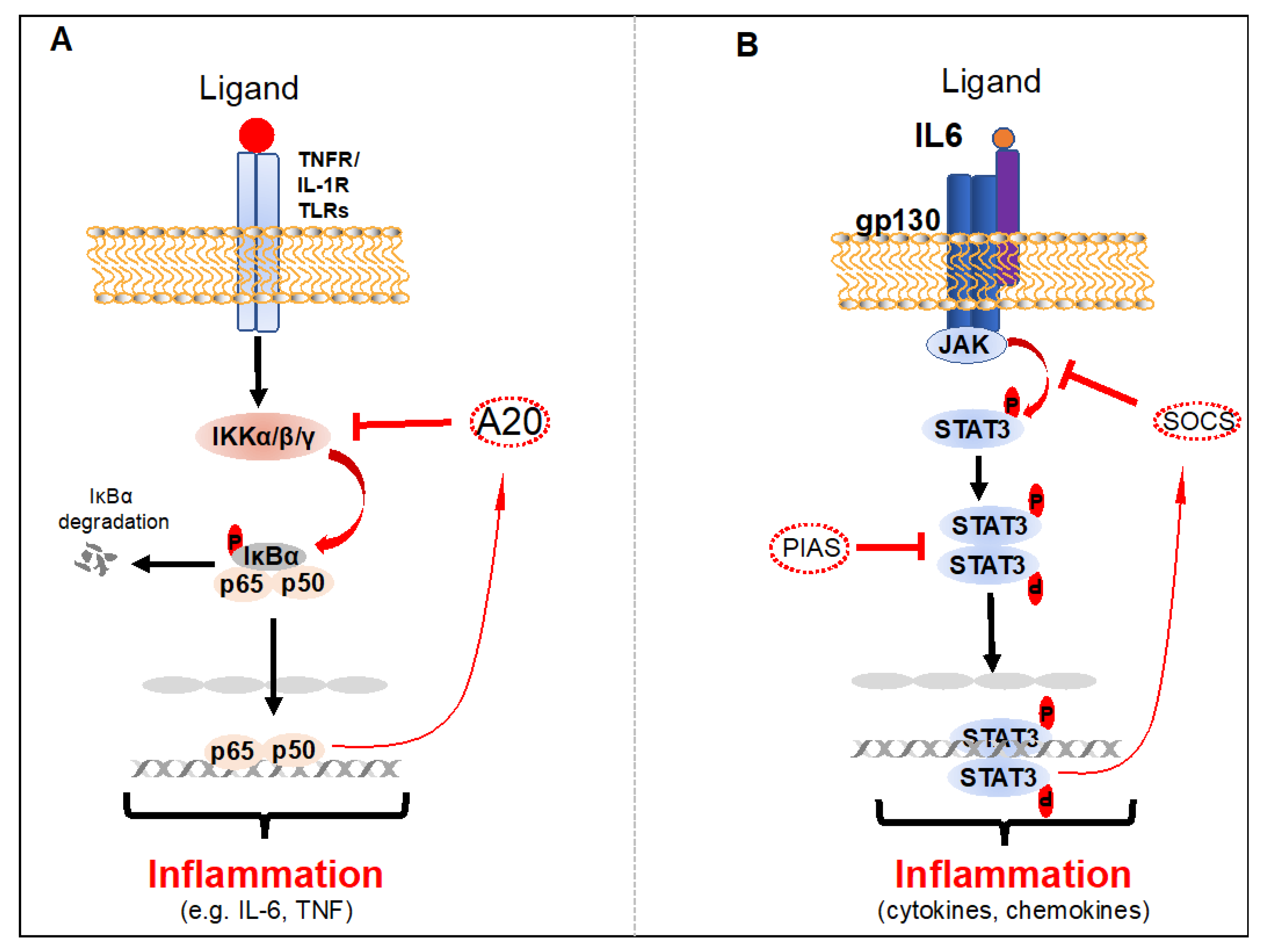
2.1. NF-κB
2.2. STAT3
2.3. Crosstalk between NF-κB and STAT3 in Cancers
3. Role of Cell Surface Molecules (CSMs) in EBV, KSHV or HTLV-1-Induced Inflammation during De Novo Infection
3.1. CSMs in EBV-Induced Inflammation during De Novo Infection
3.2. CSMs in KSHV-Induced Inflammation during De Novo Infection
3.3. CSMs in HTLV-1-Induced Inflammation during De Novo Infection
4. Role of CSMs in the Induction of Inflammation during EBV, KSHV, and HTLV-1 Latency
4.1. CSMs and Chronic Inflammation during EBV Latency
4.2. CSMs and Chronic Inflammation during KSHV Latency
4.3. CSMs and Chronic Inflammation during HTLV-1 Latency
5. Role of CSMs in the Negative Regulation of NF-κB and STAT3 Inhibition
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- Coussens, L. Session 2: Inflammation and Cancer. Toxicol. Pathol. 2004, 32, 732. [Google Scholar] [CrossRef]
- Jha, H.C.; Banerjee, S.; Robertson, E.S. The Role of Gammaherpesviruses in Cancer Pathogenesis. Pathogens 2016, 5, 18. [Google Scholar] [CrossRef]
- Baer, R.; Bankier, A.T.; Biggin, M.D.; Deininger, P.L.; Farrell, P.J.; Gibson, T.J.; Hatfull, G.; Hudson, G.S.; Satchwell, S.C.; Séguin, C.; et al. DNA sequence and expression of the B95-8 Epstein—Barr virus genome. Nat. Cell Biol. 1984, 310, 207–211. [Google Scholar] [CrossRef]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.-C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8]. Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef]
- Vandamme, A.-M. The simian origins of the pathogenic human T-cell lymphotropic virus type I. Trends Microbiol. 1998, 6, 477–483. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Dotti, G. EBV-Associated Lymphoproliferative Disorders: Classification and Treatment. Oncology 2008, 13, 577–585. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Hoeger, B.; Serwas, N.; Boztug, K. Human NF-κB1 Haploinsufficiency and Epstein–Barr Virus-Induced Disease—Molecular Mechanisms and Consequences. Front. Immunol. 2018, 8, 1978. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Velapasamy, S.; Dawson, C.W.; Young, L.S.; Paterson, I.C.; Yap, L.F. The Dynamic Roles of TGF-beta Signalling in EBV-Associated Cancers. Cancers 2018, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.A.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s Sarcoma–Associated Herpesvirus-Like DNA Sequences in AIDS-Related Body-Cavity–Based Lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef]
- Uchiyama, T.; Yodoi, J.; Sagawa, K.; Takatsuki, K.; Uchino, H. Adult T-cell leukemia: Clinical and hematologic features of 16 cases. Blood 1997, 50, 481–492. [Google Scholar] [CrossRef]
- Gessain, A.; Vernant, J.; Maurs, L.; Barin, F.; Gout, O.; Calender, A.; De Thé, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 326, 407–410. [Google Scholar] [CrossRef]
- Morrison, B.J.; Labo, N.; Miley, W.J.; Whitby, D. Serodiagnosis for Tumor Viruses. Semin. Oncol. 2015, 42, 191–206. [Google Scholar] [CrossRef]
- Myoung, J.; Ganem, D. Active lytic infection of human primary tonsillar B cells by KSHV and its noncytolytic control by activated CD4+ T cells. J. Clin. Investig. 2011, 121, 1130–1140. [Google Scholar] [CrossRef]
- Pique, C.; Jones, K.S. Pathways of cell-cell transmission of HTLV-1. Front. Microbiol. 2012, 3, 378. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.A.; Hernandez-Hopkins, D.; Vider, J.; Ponomarev, V.; Hyjek, E.; Schattner, E.J.; Cesarman, E. NF-κB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood 2006, 107, 3295–3302. [Google Scholar] [CrossRef] [PubMed]
- Read, S.A.; Douglas, M.W. Virus induced inflammation and cancer development. Cancer Lett. 2014, 345, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.T.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E. The alternative NF-κB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF- B, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef]
- Pujari, R.; Hunte, R.; Khan, W.N.; Shembade, N. A20-mediated negative regulation of canonical NF-kappaB signaling pathway. Immunol. Res. 2013, 57, 166–171. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, E.W. Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cell. Mol. Immunol. 2012, 9, 123–130. [Google Scholar] [CrossRef]
- Lütticken, C.; Wegenka, U.; Yuan, J.; Buschmann, J.; Schindler, C.; Ziemiecki, A.; Harpur, A.G.; Wilks, A.F.; Yasukawa, K.; Taga, T.; et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 1994, 263, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Standke, G.J.; Meier, V.S.; Groner, B. Mammary gland factor activated by prolactin on mammary epithelial cells and acute-phase response factor activated by interleukin-6 in liver cells share DNA binding and transactivation potential. Mol Endocrinol. 1994, 8, 469–477. [Google Scholar] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.M.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Lai, R. STAT3 in Cancer—Friend or Foe? Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef]
- Prasad, S.; Pandey, M.K.; Yadav, V.R.; Aggarwal, B.B. Gambogic acid inhibits STAT3 phosphorylation through activation of protein tyrosine phosphatase SHP-1: Potential role in proliferation and apoptosis. Cancer Prev. Res. 2011, 4, 1084–1094. [Google Scholar] [CrossRef]
- Sgrignani, J.; Garofalo, M.; Matkovic, M.; Merulla, J.; Catapano, C.V.; Cavalli, A. Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. Int. J. Mol. Sci. 2018, 19, 1591. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D.M. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.D.; Jang, I.; Cho, Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef]
- Kim, D.J.; Tremblay, M.L.; DiGiovanni, J. Protein Tyrosine Phosphatases, TC-PTP, SHP1, and SHP2, Cooperate in Rapid Dephosphorylation of Stat3 in Keratinocytes Following UVB Irradiation. PLoS ONE 2010, 5, e10290. [Google Scholar] [CrossRef]
- Peyser, N.D.; Du, Y.; Li, H.; Lui, V.W.Y.; Xiao, X.; Chan, T.A.; Grandis, J.R. Loss-of-Function PTPRD Mutations Lead to Increased STAT3 Activation and Sensitivity to STAT3 Inhibition in Head and Neck Cancer. PLoS ONE 2015, 10, e0135750. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef]
- Peyser, N.D.; Freilino, M.; Wang, L.; Zeng, Y.; Li, H.; Johnson, D.E.; Grandis, J.R. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 2015, 35, 1163–1169. [Google Scholar] [CrossRef]
- Lui, V.W.Y.; Peyser, N.D.; Ng, P.K.-S.; Hritz, J.; Zeng, Y.; Lu, Y.; Li, H.; Wang, L.; Gilbert, B.R.; General, I.J.; et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1114–1119. [Google Scholar] [CrossRef]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef]
- Saydmohammed, M.; Joseph, D.; Syed, V. Curcumin suppresses constitutive activation of STAT-3 by up-regulating protein inhibitor of activated STAT-3 (PIAS-3] in ovarian and endometrial cancer cells. J. Cell. Biochem. 2010, 110, 447–456. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific Inhibition of Stat3 Signal Transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Franceschi, S. Infections and cancer: Established associations and new hypotheses. Crit. Rev. Oncol. 2009, 70, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef]
- Chen, L.-F.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of Nuclear NF-kappa B Action Regulated by Reversible Acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- D’Addario, M.; Libermann, T.A.; Xu, J.; Ahmad, A.; Menezes, J. Epstein-Barr virus and its glycoprotein-350 upregulate IL-6 in human B-lymphocytes via CD21, involving activation of NF-κB and different signaling pathways. J. Mol. Biol. 2001, 308, 501–514. [Google Scholar] [CrossRef]
- Fingeroth, J.D.; Weis, J.J.; Tedder, T.F.; Strominger, J.L.; Biro, P.A.; Fearon, D.T. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. USA 1984, 81, 4510–4514. [Google Scholar] [CrossRef]
- D’Addario, M.; Ahmad, A.; Morgan, A.; Menezes, J. Binding of the Epstein-Barr virus major envelope glycoprotein gp350 results in the upregulation of the TNF-alpha gene expression in monocytic cells via NF-kappaB involving PKC, PI3-K and tyrosine kinases. J. Mol. Biol. 2000, 298, 765–778. [Google Scholar] [CrossRef]
- Bouillie, S.; Barel, M.; Frade, R. Signaling through the EBV/C3d receptor (CR2, CD21] in human B lymphocytes: Activation of phosphatidylinositol 3-kinase via a CD19-independent pathway. J. Immunol. 1999, 162, 136–143. [Google Scholar]
- Arredouani, M.S.; Bhasin, M.K.; Sage, D.R.; Dunn, L.K.; Gill, M.B.; Agnani, D.; Libermann, T.A.; Fingeroth, J.D. Analysis of Host Gene Expression Changes Reveals Distinct Roles for the Cytoplasmic Domain of the Epstein-Barr Virus Receptor/CD21 in B-Cell Maturation, Activation, and Initiation of Virus Infection. J. Virol. 2014, 88, 5559–5577. [Google Scholar] [CrossRef] [PubMed]
- D’Addario, M.; Ahmad, A.; Xu, J.W.; Menezes, J. Epstein-Barr virus envelope glycoprotein gp350 induces NF-kappaB activation and IL-1beta synthesis in human monocytes-macrophages involving PKC and PI3-K. FASEB J. 1999, 13, 2203–2213. [Google Scholar]
- Sun, S.C.; Cesarman, E. NF-kappaB as a target for oncogenic viruses. Curr. Top. Microbial. Immunol. 2011, 349, 197–244. [Google Scholar]
- Koganti, S.; De La Paz, A.; Freeman, A.F.; Bhaduri-McIntosh, S. B Lymphocytes from Patients with a Hypomorphic Mutation in STAT3 Resist Epstein-Barr Virus-Driven Cell Proliferation. J. Virol. 2013, 88, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bhaduri-McIntosh, S. A Central Role for STAT3 in Gammaherpesvirus-Life Cycle and -Diseases. Front. Microbiol. 2016, 7, 1052. [Google Scholar] [CrossRef]
- De Oliveira, D.E.; Ballon, G.; Cesarman, E. NF-κB signaling modulation by EBV and KSHV. Trends Microbiol. 2010, 18, 248–257. [Google Scholar] [CrossRef]
- Takada, H.; Imadome, K.I.; Shibayama, H.; Yoshimori, M.; Wang, L.; Saitoh, Y.; Uota, S.; Yamaoka, S.; Koyama, T.; Shimizu, N.; et al. EBV induces persistent NF-kappaB activation and contributes to survival of EBV-positive neoplastic T- or NK-cells. PLoS ONE 2017, 12, e0174136. [Google Scholar]
- Kumar, B.; Chandran, B. KSHV Entry and Trafficking in Target Cells—Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics. Viruses 2016, 8, 305. [Google Scholar] [CrossRef]
- Sadagopan, S.; Sharma-Walia, N.; Veettil, M.V.; Raghu, H.; Sivakumar, R.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 2007, 81, 3949–3968. [Google Scholar] [CrossRef]
- He, Z.; Zhao, J.; Zhang, J.; Jung, J.U.; Feng, P. NF-kappaB activation coordinated by IKKbeta and IKKepsilon enables latent infection of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014, 88, 444–455. [Google Scholar] [CrossRef][Green Version]
- Hunte, R.; Alonso, P.; Thomas, R.; Bazile, C.A.; Ramos, J.C.; van der Weyden, L.; Dominguez-Bendala, J.; Khan, W.N.; Shembade, N. CADM1 is essential for KSHV-encoded vGPCR-and vFLIP-mediated chronic NF-kappaB activation. PLoS Pathog. 2018, 14, e1006968. [Google Scholar] [CrossRef] [PubMed]
- Dutartre, H.; Clavière, M.; Journo, C.; Mahieux, R. Cell-Free versus Cell-to-Cell Infection by Human Immunodeficiency Virus Type 1 and Human T-Lymphotropic Virus Type 1: Exploring the Link among Viral Source, Viral Trafficking, and Viral Replication. J. Virol. 2016, 90, 7607–7617. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, A.; Alais, S.; Roux, L.; Thoulouze, M.-I.; Alvarez, K.; Journo, C.; Dutartre, H.; Mahieux, R. How to Control HTLV-1-Associated Diseases: Preventing de Novo Cellular Infection Using Antiviral Therapy. Front. Microbiol. 2018, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.C.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R.M. Spread of HTLV-I Between Lymphocytes by Virus-Induced Polarization of the Cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Thoulouze, M.-I.; Alcover, A. Can viruses form biofilms? Trends Microbiol. 2011, 19, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. USA 2010, 107, 20738–20743. [Google Scholar] [CrossRef] [PubMed]
- Pais-Correia, A.-M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.-I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2009, 16, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.L.; Gelmann, E.P.; Cossman, J.; Young, R.A.; Gallo, R.C.; O’Brien, S.; Matis, L.A. Isolation of HTLV-transformed B-lymphocyte clone from a patient with HTLV-associated adult T-cell leukaemia. Nat. Cell Biol. 1984, 310, 505–506. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Itoyama, Y.; Nakamura, N.; Takamatsu, K.; Kira, J.-I.; Iwamasa, T.; Goto, I.; Yamamoto, N. In Vivo Infection of Human T-Cell Leukemia Virus Type I in Non-T Cells. Virology 1993, 196, 25–33. [Google Scholar] [CrossRef]
- Futsch, N.; Prates, G.; Mahieux, R.; Casseb, J.; Dutartre, H. Cytokine Networks Dysregulation during HTLV-1 Infection and Associated Diseases. Viruses 2018, 10, 691. [Google Scholar] [CrossRef]
- Olière, S.; Hernandez, E.; Lézin, A.; Arguello, M.; Douville, R.; Nguyên, T.L.-A.; Olindo, S.; Panelatti, G.; Kazanji, M.; Wilkinson, P.; et al. HTLV-1 Evades Type I Interferon Antiviral Signaling by Inducing the Suppressor of Cytokine Signaling 1 (SOCS1]. PLoS Pathog. 2010, 6, e1001177. [Google Scholar] [CrossRef] [PubMed]
- Charoenthongtrakul, S.; Zhou, Q.; Shembade, N.; Harhaj, N.S.; Harhaj, E.W. Human T cell leukemia virus type 1 Tax inhibits innate antiviral signaling via NF-kappaB-dependent induction of SOCS1. J. Virol. 2011, 85, 6955–6962. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, D.; Zhang, Y.; Zhang, H.; Cheng, H. The autophagy molecule Beclin 1 maintains persistent activity of NF-kappaB and Stat3 in HTLV-1-transformed T lymphocytes. Biochem. Biophys. Res. Commun. 2015, 465, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Arai, A. Chronic active Epstein–Barr virus infection: A bi-faceted disease with inflammatory and neoplastic elements. Immunol. Med. 2018, 41, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Imadome, K.-I.; Shimizu, N.; Arai, A.; Miura, O.; Watanabe, K.; Nakamura, H.; Nonoyama, S.; Yamamoto, K.; Fujiwara, S. Coexpression of CD40 and CD40 Ligand in Epstein-Barr Virus–Infected T and NK Cells and Their Role in Cell Survival. J. Infect. Dis. 2005, 192, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, M.; Imadome, K.-I.; Komatsu, H.; Wang, L.; Saitoh, Y.; Yamaoka, S.; Fukuda, T.; Kurata, M.; Koyama, T.; Shimizu, N.; et al. CD137 Expression Is Induced by Epstein-Barr Virus Infection through LMP1 in T or NK Cells and Mediates Survival Promoting Signals. PLoS ONE 2014, 9, e112564. [Google Scholar] [CrossRef]
- Chen, H.; Hutt-Fletcher, L.; Cao, L.; Hayward, S.D. A Positive Autoregulatory Loop of LMP1 Expression and STAT Activation in Epithelial Cells Latently Infected with Epstein-Barr Virus. J. Virol. 2003, 77, 4139–4148. [Google Scholar] [CrossRef]
- Onozawa, E.; Shibayama, H.; Takada, H.; Imadome, K.-I.; Aoki, S.; Yoshimori, M.; Shimizu, N.; Fujiwara, S.; Koyama, T.; Miura, O.; et al. STAT3 is constitutively activated in chronic active Epstein-Barr virus infection and can be a therapeutic target. Oncotarget 2018, 9, 31077–31089. [Google Scholar] [CrossRef]
- Omi, N.; Tokuda, Y.; Ikeda, Y.; Ueno, M.; Mori, K.; Sotozono, C.; Kinoshita, S.; Nakano, M.; Tashiro, K. Efficient and reliable establishment of lymphoblastoid cell lines by Epstein-Barr virus transformation from a limited amount of peripheral blood. Sci. Rep. 2017, 7, 43833. [Google Scholar] [CrossRef]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV Persistence in Memory B Cells In Vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef]
- Brink, A.A.; Dukers, D.F.; Brule, A.J.V.D.; Oudejans, J.J.; Middeldorp, J.M.; Meijer, C.J.; Jiwa, M. Presence of Epstein-Barr virus latency type III at the single cell level in post-transplantation lymphoproliferative disorders and AIDS related lymphomas. J. Clin. Pathol. 1997, 50, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Roughan, J.E.; Thorley-Lawson, D.A. The Intersection of Epstein-Barr Virus with the Germinal Center. J. Virol. 2009, 83, 3968–3976. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, G.; Hamilton-Dutoit, S.J.; Rowe, M.; Young, L. Expression of Epstein-Barr virus latent gene products in tumour cells of Hodgkin’s disease. Lancet 1991, 337, 320–322. [Google Scholar] [CrossRef]
- Brooks, L.; Yao, Q.Y.; Rickinson, A.B.; Young, L.S. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: Coexpression of EBNA1, LMP1, and LMP2 transcripts. J. Virol. 1992, 66, 2689–2697. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.; Rowe, D.T.; Gregory, C.D.; Young, L.S.; Farrell, P.J.; Rupani, H.; Rickinson, A.B. Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt’s lymphoma cells. EMBO J. 1987, 6, 2743–2751. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef]
- Sugiura, M.; Imai, S.; Tokunaga, M.; Koizumi, S.; Uchizawa, M.; Okamoto, K.; Osato, T. Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: Unique viral latency in the tumour cells. Br. J. Cancer 1996, 74, 625–631. [Google Scholar] [CrossRef]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.H.J.; Rowe, M. The Epstein-Barr Virus G-Protein-Coupled Receptor Contributes to Immune Evasion by Targeting MHC Class I Molecules for Degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef]
- Zuo, J.; Quinn, L.L.; Tamblyn, J.; Thomas, W.A.; Feederle, R.; Delecluse, H.-J.; Hislop, A.D.; Rowe, M. The Epstein-Barr Virus-Encoded BILF1 Protein Modulates Immune Recognition of Endogenously Processed Antigen by Targeting Major Histocompatibility Complex Class I Molecules Trafficking on both the Exocytic and Endocytic Pathways. J. Virol. 2010, 85, 1604–1614. [Google Scholar] [CrossRef]
- Griffin, B.D.; Gram, A.M.; Mulder, A.; Van Leeuwen, D.; Claas, F.H.J.; Wang, F.; Ressing, M.E.; Wiertz, E. EBV BILF1 Evolved To Downregulate Cell Surface Display of a Wide Range of HLA Class I Molecules through Their Cytoplasmic Tail. J. Immunol. 2013, 190, 1672–1684. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Tan, Y.-N.; Wang, Z.-L.; Zeng, L.; Lu, Z.-X.; Li, L.-L.; Luo, W.; Tang, M.; Cao, Y. Phosphorylation and nuclear translocation of STAT3 regulated by the Epstein-Barr virus latent membrane protein 1 in nasopharyngeal carcinoma. Int. J. Mol. Med. 2008, 21, 153–162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kung, C.-P.; Meckes, D.G.; Raab-Traub, N. Epstein-Barr Virus LMP1 Activates EGFR, STAT3, and ERK through Effects on PKC. J. Virol. 2011, 85, 4399–4408. [Google Scholar] [CrossRef] [PubMed]
- Suarez, A.A.R.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef]
- Sharp, T.V.; Wang, H.-W.; Koumi, A.; Hollyman, D.; Endo, Y.; Ye, H.; Du, M.-Q.; Boshoff, C. K15 Protein of Kaposi’s Sarcoma-Associated Herpesvirus Is Latently Expressed and Binds to HAX-1, a Protein with Antiapoptotic Function. J. Virol. 2002, 76, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Ganem, D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2010, 84, 5565–5573. [Google Scholar] [CrossRef]
- Azzi, S.; Smith, S.S.; Dwyer, J.; Leclair, H.M.; Alexia, C.; Hebda, J.K.; Dupin, N.; Bidere, N.; Gavard, J. YGLF motif in the Kaposi sarcoma herpes virus G-protein-coupled receptor adjusts NF-kappaB activation and paracrine actions. Oncogene 2014, 33, 5609–5618. [Google Scholar] [CrossRef]
- Prakash, O.; Tang, Z.-Y.; Peng, X.; Coleman, R.; Gill, J.; Farr, G.; Samaniego, F. Tumorigenesis and Aberrant Signaling in Transgenic Mice Expressing the Human Herpesvirus-8 K1 Gene. J. Natl. Cancer Inst. 2002, 94, 926–935. [Google Scholar] [CrossRef]
- Brinkmann, M.M.; Glenn, M.; Rainbow, L.; Kieser, A.; Henke-Gendo, C.; Chulz, T.F. Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 2003, 77, 9346–9358. [Google Scholar] [CrossRef]
- Vallath, S.; Sage, E.K.; Kolluri, K.K.; Lourenco, S.N.; Teixeira, V.S.; Chimalapati, S.; George, P.J.; Janes, S.M.; Giangreco, A. CADM1 inhibits squamous cell carcinoma progression by reducing STAT3 activity. Sci. Rep. 2016, 6, 24006. [Google Scholar] [CrossRef]
- Burger, M.; Hartmann, T.; Burger, J.A.; Schraufstatter, I.U. KSHV-GPCR and CXCR2 transforming capacity and angiogenic responses are mediated through a JAK2-STAT3-dependent pathway. Oncogene 2005, 24, 2067–2075. [Google Scholar] [CrossRef]
- Wu, J.; Xu, Y.; Mo, D.; Huang, P.; Sun, R.; Huang, L.; Pan, S.; Xu, J. Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 promotes cell proliferation and migration by upregulating DNMT1 via STAT3 activation. PLoS ONE 2014, 9, e93478. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gao, Y.; Nicholas, J. Human Herpesvirus 8 Interleukin-6 Contributes to Primary Effusion Lymphoma Cell Viability via Suppression of Proapoptotic Cathepsin D, a Cointeraction Partner of Vitamin K Epoxide Reductase Complex Subunit 1 Variant 2. J. Virol. 2013, 88, 1025–1038. [Google Scholar] [CrossRef][Green Version]
- Mohanty, S.; Harhaj, E.W. Mechanisms of Oncogenesis by HTLV-1 Tax. Pathogens 2020, 9, 543. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, E.W. Role of post-translational modifications of HTLV-1 Tax in NF-kappaB activation. World J. Boil. Chem. 2010, 1, 13–20. [Google Scholar] [CrossRef]
- Pujari, R.; Hunte, R.; Thomas, R.; van der Weyden, L.; Rauch, D.; Ratner, L.; Nyborg, J.K.; Ramos, J.C.; Takai, Y.; Shembade, N. Human T-cell leukemia virus type 1 (HTLV-1] tax requires CADM1/TSLC1 for inactivation of the NF-kappaB inhibitor A20 and constitutive NF-kappaB signaling. PLoS Pathog. 2015, 11, e1004721. [Google Scholar] [CrossRef] [PubMed]
- Arpin-André, C.; Mesnard, J.-M. The PDZ Domain-binding Motif of the Human T Cell Leukemia Virus Type 1 Tax Protein Induces Mislocalization of the Tumor Suppressor hScrib in T cells. J. Biol. Chem. 2007, 282, 33132–33141. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Yamamoto, N.; Dewan, Z.; Takahashi, Y.; Yamashita, A.; Yoshida, T.; Nowell, M.A.; Richards, P.; Jones, S.A.; Yamamoto, N. Human T-cell leukemia virus type-I Tax induces expression of interleukin-6 receptor (IL-6R): Shedding of soluble IL-6R and activation of STAT3 signaling. Int. J. Cancer 2006, 119, 823–830. [Google Scholar] [CrossRef]
- Satou, Y.; Yasunaga, J.-I.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef]
- Usui, T.; Yanagihara, K.; Tsukasaki, K.; Murata, K.; Hasegawa, H.; Yamada, Y.; Kamihira, S. Characteristic expression of HTLV-1 basic zipper factor (HBZ) transcripts in HTLV-1 provirus-positive cells. Retrovirology 2008, 5, 34. [Google Scholar] [CrossRef]
- Zhao, T.; Matsuoka, M. HBZ and its roles in HTLV-1 oncogenesis. Front. Microbiol. 2012, 3, 247. [Google Scholar] [CrossRef]
- Satou, Y.; Yasunaga, J.-I.; Zhao, T.; Yoshida, M.; Miyazato, P.; Takai, K.; Shimizu, K.; Ohshima, K.; Green, P.L.; Ohkura, N.; et al. HTLV-1 bZIP Factor Induces T-Cell Lymphoma and Systemic Inflammation In Vivo. PLoS Pathog. 2011, 7, e1001274. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Yasunaga, J.-I.; Mitagami, Y.; Tsukamoto, H.; Nakashima, K.; Ohshima, K.; Matsuoka, M. HTLV-1 induces T cell malignancy and inflammation by viral antisense factor-mediated modulation of the cytokine signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 13740–13749. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. CYLD: A tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, N.S.; Liebl, D.J.; Harhaj, E.W. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 2007, 26, 3910–3922. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, N.S.; Parvatiyar, K.; Copeland, N.G.; Jenkins, N.A.; Matesic, L.E.; Harhaj, E.W. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol. 2008, 9, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Whang, M.I.; Kim, F.S.; Nakamura, M.C.; Sun, X.; Advincula, R.; Turnbaugh, J.A.; Pendse, M.; Tanbun, P.; Achacoso, P.; et al. Non-catalytic ubiquitin binding by A20 prevents psoriatic arthritis–like disease and inflammation. Nat. Immunol. 2020, 21, 422–433. [Google Scholar] [CrossRef]
- Skaug, B.; Chen, J.; Du, F.; He, J.; Ma, A.; Chen, Z.J. Direct, Noncatalytic Mechanism of IKK Inhibition by A20. Mol. Cell 2011, 44, 559–571. [Google Scholar] [CrossRef]
- Shembade, N.; Ma, A.; Harhaj, E.W. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 2010, 327, 1135–1139. [Google Scholar] [CrossRef]
- Wu, X.-F.; Zhang, M.; Sun, S.-C. Mutual regulation between deubiquitinase CYLD and retroviral oncoprotein Tax. Cell Biosci. 2011, 1, 27. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, E.W. A20 inhibition of NFκB and inflammation: Targeting E2:E3 ubiquitin enzyme complexes. Cell Cycle 2010, 9, 2481–2482. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harhaj, E.W.; Shembade, N. Lymphotropic Viruses: Chronic Inflammation and Induction of Cancers. Biology 2020, 9, 390. https://doi.org/10.3390/biology9110390
Harhaj EW, Shembade N. Lymphotropic Viruses: Chronic Inflammation and Induction of Cancers. Biology. 2020; 9(11):390. https://doi.org/10.3390/biology9110390
Chicago/Turabian StyleHarhaj, Edward W., and Noula Shembade. 2020. "Lymphotropic Viruses: Chronic Inflammation and Induction of Cancers" Biology 9, no. 11: 390. https://doi.org/10.3390/biology9110390
APA StyleHarhaj, E. W., & Shembade, N. (2020). Lymphotropic Viruses: Chronic Inflammation and Induction of Cancers. Biology, 9(11), 390. https://doi.org/10.3390/biology9110390