Cholesterol Homeostasis: An In Silico Investigation into How Aging Disrupts Its Key Hepatic Regulatory Mechanisms

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Network Diagram Construction and Model Assembly
2.2. Model Analysis
2.3. Hypothesis Exploration
3. Results
3.1. The ROS and HMG-CoA Reductase Hypothesis
3.2. Exploring the Effects of a Decrease in ACAT2 Activity with Age
3.3. ROS Combined with a Decrease in ACAT2 with Age
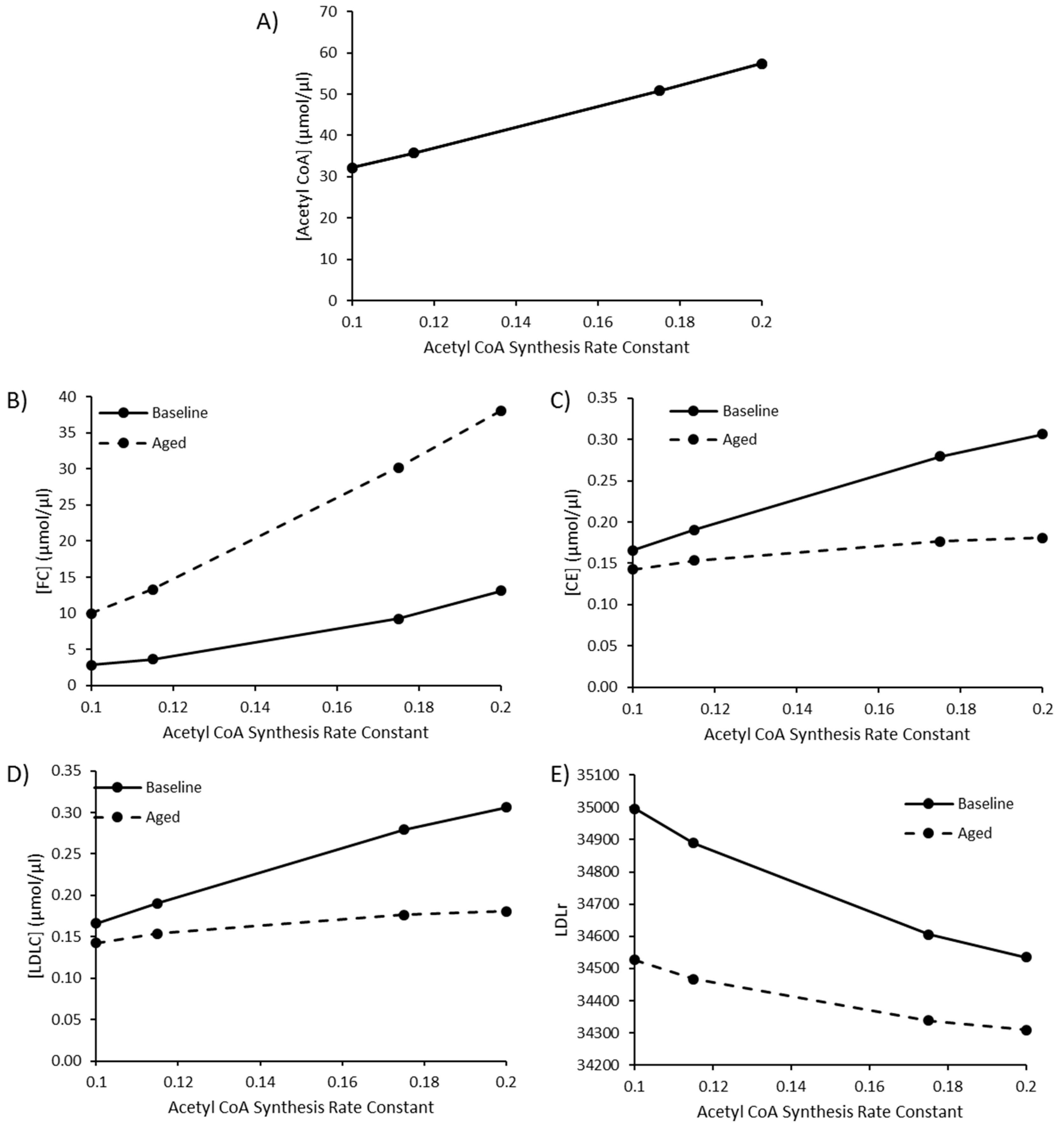
3.4. The Impact of Increasing Acetyl CoA Synthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Homan, R.; Krause, B.R. Established and emerging strategies for inhibition of cholesterol absorption. Curr. Pharm. Des. 1997, 3, 29–44. [Google Scholar]
- van der Wulp, M.Y.; Verkade, H.J.; Groen, A.K. Regulation of cholesterol homeostasis. Mol. Cell. Endocrinol. 2013, 368, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Javitt, N.B. Bile acid synthesis from cholesterol: Regulatory and auxiliary pathways. FASEB J. 1994, 8, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin d metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Castelli, W.P.; Anderson, K.; Wilson, P.W.; Levy, D. Lipids and risk of coronary heart disease. The framingham study. Ann. Epidemiol. 1992, 2, 23–28. [Google Scholar] [CrossRef]
- O’Donnell, C.J.; Elosua, R. Cardiovascular risk factors. Insights from framingham heart study. Rev. Esp. Cardiol. 2008, 61, 299–310. [Google Scholar]
- Mc Auley, M.T.; Mooney, K.M. Lipid metabolism and hormonal interactions: Impact on cardiovascular disease and healthy aging. Expert Rev. Endocrinol. Metab. 2014, 9, 357–367. [Google Scholar] [CrossRef]
- Mooney, K.M.; Mc Auley, M.T. Cardiovascular disease and healthy ageing. J. Integr. Cardiol. 2016, 1, 76–78. [Google Scholar] [CrossRef]
- Fajemiroye, J.O.; Cunha, L.C.d.; Saavedra-Rodríguez, R.; Rodrigues, K.L.; Naves, L.M.; Mourão, A.A.; Silva, E.F.d.; Williams, N.E.E.; Martins, J.L.R.; Sousa, R.B. Aging-induced biological changes and cardiovascular diseases. Biomed. Res. Int. 2018, 2018, 7156435. [Google Scholar] [CrossRef]
- Austin, M.A.; Breslow, J.L.; Hennekens, C.H.; Buring, J.E.; Willett, W.C.; Krauss, R.M. Low-density lipoprotein subclass patterns and risk of myocardial infarction. JAMA 1988, 260, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the european atherosclerosis society consensus panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.E.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.A.; Mc Auley, M.T. Cholesterol metabolism: A review of how ageing disrupts the biological mechanisms responsible for its regulation. Ageing Res. Rev. 2016, 27, 108–124. [Google Scholar] [CrossRef]
- Kreisberg, R.A.; Kasim, S. Cholesterol metabolism and aging. Am. J. Med. 1987, 82, 54–60. [Google Scholar] [CrossRef]
- Berrougui, H.; Khalil, A. Age-associated decrease of high-density lipoprotein-mediated reverse cholesterol transport activity. Rejuvenation Res. 2009, 12, 117–126. [Google Scholar] [CrossRef]
- Holzer, M.; Trieb, M.; Konya, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Aging affects high-density lipoprotein composition and function. Biochim. Biophys. Acta 2013, 1831, 1442–1448. [Google Scholar] [CrossRef]
- Parini, P.; Angelin, B.; Rudling, M. Cholesterol and lipoprotein metabolism in aging: Reversal of hypercholesterolemia by growth hormone treatment in old rats. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 832–839. [Google Scholar] [CrossRef]
- Uchida, K.; Nomura, Y.; Kadowaki, M.; Takase, H.; Takano, K.; Takeuchi, N. Age-related changes in cholesterol and bile acid metabolism in rats. J. Lipid Res. 1978, 19, 544–552. [Google Scholar]
- Liu, H.H.; Li, J.J. Aging and dyslipidemia: A review of potential mechanisms. Ageing Res. Rev. 2015, 19, 43–52. [Google Scholar] [CrossRef]
- Bertolotti, M.; Abate, N.; Bertolotti, S.; Loria, P.; Concari, M.; Messora, R.; Carubbi, F.; Pinetti, A.; Carulli, N. Effect of aging on cholesterol 7 alpha-hydroxylation in humans. J. Lipid Res. 1993, 34, 1001–1007. [Google Scholar]
- Mc Auley, M.; Jones, J.; Wilkinson, D.; Kirkwood, T. Modelling lipid metabolism to improve healthy ageing. BMC Bioinform. 2005, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.D.; Lacher, D.A.; Sorlie, P.D.; Cleeman, J.I.; Gordon, D.J.; Wolz, M.; Grundy, S.M.; Johnson, C.L. Trends in serum lipids and lipoproteins of adults, 1960–2002. JAMA 2005, 294, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hou, X.; Hu, W.; Chen, L.; Chen, S. Serum lipid and lipoprotein levels of middle-aged and elderly chinese men and women in shandong province. Lipids Health Dis. 2019, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Farzadfar, F.; Finucane, M.M.; Danaei, G.; Pelizzari, P.M.; Cowan, M.J.; Paciorek, C.J.; Singh, G.M.; Lin, J.K.; Stevens, G.A.; Riley, L.M.; et al. National, regional, and global trends in serum total cholesterol since 1980: Systematic analysis of health examination surveys and epidemiological studies with 321 country-years and 3.0 million participants. Lancet 2011, 377, 578–586. [Google Scholar] [CrossRef]
- Duncan, M.S.; Vasan, R.S.; Xanthakis, V. Trajectories of blood lipid concentrations over the adult life course and risk of cardiovascular disease and all-cause mortality: Observations from the framingham study over 35 years. J. Am. Heart Assoc. 2019, 8, e011433. [Google Scholar] [CrossRef]
- Felix-Redondo, F.J.; Grau, M.; Fernandez-Berges, D. Cholesterol and cardiovascular disease in the elderly. Facts and gaps. Aging Dis. 2013, 4, 154–169. [Google Scholar]
- Ferrara, A.; Barrett-Connor, E.; Shan, J. Total, ldl, and hdl cholesterol decrease with age in older men and women. The rancho bernardo study 1984–1994. Circulation 1997, 96, 37–43. [Google Scholar] [CrossRef]
- Garry, P.J.; Hunt, W.C.; Koehler, K.M.; VanderJagt, D.J.; Vellas, B.J. Longitudinal study of dietary intakes and plasma lipids in healthy elderly men and women. Am. J. Clin. Nutr. 1992, 55, 682–688. [Google Scholar] [CrossRef]
- Postmus, I.; Deelen, J.; Sedaghat, S.; Trompet, S.; de Craen, A.J.; Heijmans, B.T.; Franco, O.H.; Hofman, A.; Dehghan, A.; Slagboom, P.E.; et al. Ldl cholesterol still a problem in old age? A mendelian randomization study. Int. J. Epidemiol. 2015, 44, 604–612. [Google Scholar] [CrossRef]
- Weverling-Rijnsburger, A.W.; Jonkers, I.J.; van Exel, E.; Gussekloo, J.; Westendorp, R.G. High-density vs. low-density lipoprotein cholesterol as the risk factor for coronary artery disease and stroke in old age. Arch. Intern. Med. 2003, 163, 1549–1554. [Google Scholar] [CrossRef]
- Ravnskov, U. High cholesterol may protect against infections and atherosclerosis. QJM 2003, 96, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Han, R. Plasma lipoproteins are important components of the immune system. Microbiol. Immunol. 2010, 54, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Ravnskov, U.; de Lorgeril, M.; Diamond, D.M.; Hama, R.; Hamazaki, T.; Hammarskjold, B.; Hynes, N.; Kendrick, M.; Langsjoen, P.H.; Mascitelli, L.; et al. Ldl-c does not cause cardiovascular disease: A comprehensive review of the current literature. Expert Rev. Clin. Pharmacol. 2018, 11, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Ravnskov, U.; Diamond, D.M.; Hama, R.; Hamazaki, T.; Hammarskjöld, B.; Hynes, N.; Kendrick, M.; Langsjoen, P.H.; Malhotra, A.; Mascitelli, L. Lack of an association or an inverse association between low-density-lipoprotein cholesterol and mortality in the elderly: A systematic review. BMJ Open 2016, 6, e010401. [Google Scholar] [CrossRef] [PubMed]
- Mc Auley, M.T.; Mooney, K.M. Ldl-c levels in older people: Cholesterol homeostasis and the free radical theory of ageing converge. Med. Hypotheses 2017, 104, 15–19. [Google Scholar] [CrossRef]
- Mc Auley, M.T. The interplay between cholesterol metabolism and intrinsic ageing. Subcell. Biochem. 2018, 90, 99–118. [Google Scholar]
- Tiwari, S.; Siddiqi, S.A. Intracellular trafficking and secretion of vldl. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef]
- Goldberg, I.J. Lipoprotein lipase and lipolysis: Central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res. 1996, 37, 693–707. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. The ldl receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on cholesterol homeostasis: The central role of scap. Annu. Rev. Biochem. 2018, 87, 783–807. [Google Scholar] [CrossRef]
- Sato, R. Sterol metabolism and srebp activation. Arch. Biochem. Biophys. 2010, 501, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. Srebp transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Lee, H.-S.; Kim, K.-S.; Kim, Y.-K.; Yoon, D.; Park, S.W. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J. Lipid Res. 2008, 49, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Lagace, T.A. Pcsk9 and ldlr degradation: Regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Shah, N.; Muhieddine, D.; Zhen, J.; Yudkevich, J.; Kasselman, L.J.; DeLeon, J. Pcsk9 in cholesterol metabolism: From bench to bedside. Clin. Sci. 2018, 132, 1135–1153. [Google Scholar] [CrossRef]
- Chaudhary, R.; Garg, J.; Shah, N.; Sumner, A. Pcsk9 inhibitors: A new era of lipid lowering therapy. World J. Cardiol. 2017, 9, 76–91. [Google Scholar] [CrossRef]
- Parini, P.; Davis, M.; Lada, A.T.; Erickson, S.K.; Wright, T.L.; Gustafsson, U.; Sahlin, S.; Einarsson, C.; Eriksson, M.; Angelin, B.; et al. Acat2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation 2004, 110, 2017–2023. [Google Scholar] [CrossRef]
- Semsei, I.; Rao, G.; Richardson, A. Changes in the expression of superoxide dismutase and catalase as a function of age and dietary restriction. Biochem. Biophys. Res. Commun. 1989, 164, 620–625. [Google Scholar] [CrossRef]
- Ji, L.L. Antioxidant enzyme response to exercise and aging. Med. Sci. Sports Exerc. 1993, 25, 225–231. [Google Scholar] [CrossRef]
- Pallottini, V.; Martini, C.; Bassi, A.M.; Romano, P.; Nanni, G.; Trentalance, A. Rat hmgcoa reductase activation in thioacetamide-induced liver injury is related to an increased reactive oxygen species content. J. Hepatol. 2006, 44, 368–374. [Google Scholar] [CrossRef]
- Pallottini, V.; Martini, C.; Cavallini, G.; Bergamini, E.; Mustard, K.J.; Hardie, D.G.; Trentalance, A. Age-related hmg-coa reductase deregulation depends on ros-induced p38 activation. Mech. Ageing Dev. 2007, 128, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Pallottini, V.; Martini, C.; Pascolini, A.; Cavallini, G.; Gori, Z.; Bergamini, E.; Incerpi, S.; Trentalance, A. 3-hydroxy-3-methylglutaryl coenzyme a reductase deregulation and age-related hypercholesterolemia: A new role for ros. Mech. Ageing Dev. 2005, 126, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Trapani, L.; Pallottini, V. Age-related hypercholesterolemia and hmg-coa reductase dysregulation: Sex does matter (a gender perspective). Curr. Gerontol. Geriatr. Res. 2010, 2010, 420139. [Google Scholar] [CrossRef] [PubMed]
- Trapani, L.; Violo, F.; Pallottini, V. Hypercholesterolemia and 3-hydroxy-3-methylglutaryl coenzyme a reductase regulation in aged female rats. Exp. Gerontol. 2010, 45, 119–128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.S. Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef]
- Mulas, M.F.; Demuro, G.; Mulas, C.; Putzolu, M.; Cavallini, G.; Donati, A.; Bergamini, E.; Dessi, S. Dietary restriction counteracts age-related changes in cholesterol metabolism in the rat. Mech. Ageing Dev. 2005, 126, 648–654. [Google Scholar] [CrossRef]
- Ståhlberg, D.; Angelin, B.; Einarsson, K. Age-related changes in the metabolism of cholesterol in rat liver microsomes. Lipids 1991, 26, 349–352. [Google Scholar] [CrossRef]
- Shiomi, M.; Ito, T.; Fujioka, T.; Tsujita, Y. Age-associated decrease in plasma cholesterol and changes in cholesterol metabolism in homozygous watanabe heritable hyperlipidemic rabbits. Metabolism 2000, 49, 552–556. [Google Scholar] [CrossRef]
- Ioannou, G.N. The role of cholesterol in the pathogenesis of nash. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Hagstrom, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stal, P.; Askling, J.; Hultcrantz, R.; Kechagias, S. Cardiovascular risk factors in non-alcoholic fatty liver disease. Liver Int. 2019, 39, 197–204. [Google Scholar] [CrossRef]
- Morgan, A.E.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.A.; Mc Auley, M.T. Investigating cholesterol metabolism and ageing using a systems biology approach. Proc. Nutr. Soc. 2017, 76, 378–391. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Mooney, K.M. Computationally modeling lipid metabolism and aging: A mini-review. Comput. Struct. Biotechnol. J. 2015, 13, 38–46. [Google Scholar] [CrossRef]
- Mc Auley, M.; Mooney, K. Chapter 7—Using computational models to study aging. In Conn’s Handbook of Models for Human Aging, 2nd ed.; Ram, J.L., Conn, P.M., Eds.; Academic Press: London, UK, 2018; pp. 79–91. [Google Scholar]
- Neumann, S.J.; Berceli, S.A.; Sevick, E.M.; Lincoff, A.M.; Warty, V.S.; Brant, A.M.; Herman, I.M.; Borovetz, H.S. Experimental determination and mathematical model of the transient incorporation of cholesterol in the arterial wall. Bull. Math Biol. 1990, 52, 711–732. [Google Scholar] [CrossRef]
- Lu, J.; Hubner, K.; Nanjee, M.N.; Brinton, E.A.; Mazer, N.A. An in-silico model of lipoprotein metabolism and kinetics for the evaluation of targets and biomarkers in the reverse cholesterol transport pathway. PLoS Comput. Biol. 2014, 10, e1003509. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Kenny, R.A.; Kirkwood, T.B.; Wilkinson, D.J.; Jones, J.J.; Miller, V.M. A mathematical model of aging-related and cortisol induced hippocampal dysfunction. BMC Neurosci. 2009, 10, 26. [Google Scholar]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Guimera, A.M.; Hodgson, D.; McDonald, N.; Mooney, K.M.; Morgan, A.E.; Proctor, C.J. Modelling the molecular mechanisms of aging. Biosci. Rep. 2017, 37, BSR20160177. [Google Scholar] [CrossRef]
- Mooney, K.M.; Morgan, A.E.; Mc Auley, M.T. Aging and computational systems biology. Wiley interdisciplinary reviews. Syst. Biol. Med. 2016, 8, 123–139. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Proctor, C.J.; Corfe, B.M.; Cuskelly, G.J.; Mooney, K.M. Nutrition research and the impact of computational systems biology. J. Comput. Sci. Syst. Biol. 2013, 6, 271–285. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Mooney, K.M.; Angell, P.J.; Wilkinson, S.J. Mathematical modelling of metabolic regulation in aging. Metabolites 2015, 5, 232–251. [Google Scholar] [CrossRef]
- Kilner, J.; Corfe, B.M.; McAuley, M.T.; Wilkinson, S.J. A deterministic oscillatory model of microtubule growth and shrinkage for differential actions of short chain fatty acids. Mol. Biosyst. 2016, 12, 93–101. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Choi, H.; Mooney, K.; Paul, E.; Miller, V.M. Systems biology and synthetic biology: A new epoch for toxicology research. Adv. Toxicol. 2015, 2015, 575403. [Google Scholar] [CrossRef]
- Saqi, M.; Pellet, J.; Roznovat, I.; Mazein, A.; Ballereau, S.; De Meulder, B.; Auffray, C. Systems medicine: The future of medical genomics, healthcare, and wellness. Methods Mol. Biol. (Clifton N.J.) 2016, 1386, 43–60. [Google Scholar]
- Ostaszewski, M.; Gebel, S.; Kuperstein, I.; Mazein, A.; Zinovyev, A.; Dogrusoz, U.; Hasenauer, J.; Fleming, R.M.T.; Le Novère, N.; Gawron, P.; et al. Community-driven roadmap for integrated disease maps. Brief. Bioinform. 2019, 20, 659–670. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Mooney, K.M. Computational systems biology for aging research. Interdiscip. Top. Gerontol. 2015, 40, 35–48. [Google Scholar]
- Parton, A.; McGilligan, V.; O’Kane, M.; Baldrick, F.R.; Watterson, S. Computational modelling of atherosclerosis. Brief. Bioinform. 2016, 17, 562–575. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Wilkinson, D.J.; Jones, J.J.; Kirkwood, T.B. A whole-body mathematical model of cholesterol metabolism and its age-associated dysregulation. BMC Syst. Biol. 2012, 6, 130. [Google Scholar] [CrossRef]
- Pool, F.; Currie, R.; Sweby, P.K.; Salazar, J.D.; Tindall, M.J. A mathematical model of the mevalonate cholesterol biosynthesis pathway. J. Theor. Biol. 2018, 443, 157–176. [Google Scholar] [CrossRef]
- Bhattacharya, B.S.; Sweby, P.K.; Minihane, A.M.; Jackson, K.G.; Tindall, M.J. A mathematical model of the sterol regulatory element binding protein 2 cholesterol biosynthesis pathway. J. Theor. Biol. 2014, 349, 150–162. [Google Scholar] [CrossRef]
- Watterson, S.; Guerriero, M.L.; Blanc, M.; Mazein, A.; Loewe, L.; Robertson, K.A.; Gibbs, H.; Shui, G.; Wenk, M.R.; Hillston, J.; et al. A model of flux regulation in the cholesterol biosynthesis pathway: Immune mediated graduated flux reduction versus statin-like led stepped flux reduction. Biochimie 2013, 95, 613–621. [Google Scholar] [CrossRef]
- Morgan, A.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.; Mc Auley, M.T. Mathematically modelling the dynamics of cholesterol metabolism and ageing. Biosystems 2016, 145, 19–32. [Google Scholar] [CrossRef]
- Tindall, M.J.; Wattis, J.A.; O’Malley, B.J.; Pickersgill, L.; Jackson, K.G. A continuum receptor model of hepatic lipoprotein metabolism. J. Theor. Biol. 2009, 257, 371–384. [Google Scholar] [CrossRef]
- August, E.; Parker, K.H.; Barahona, M. A dynamical model of lipoprotein metabolism. Bull. Math Biol. 2007, 69, 1233–1254. [Google Scholar] [CrossRef]
- Pool, F.; Sweby, P.; Tindall, M.J.P. An integrated mathematical model of cellular cholesterol biosynthesis and lipoprotein metabolism. Processes 2018, 6, 134. [Google Scholar] [CrossRef]
- Toroghi, M.K.; Cluett, W.R.; Mahadevan, R.J.C.; Engineering, C. A multi-scale model for low-density lipoprotein cholesterol (ldl-c) regulation in the human body: Application to quantitative systems pharmacology. Comput. Chem. Eng. 2019, 130, 106507. [Google Scholar] [CrossRef]
- Kervizic, G.; Corcos, L. Dynamical modeling of the cholesterol regulatory pathway with boolean networks. BMC Syst. Biol. 2008, 2, 99. [Google Scholar] [CrossRef]
- Benson, H.E.; Watterson, S.; Sharman, J.L.; Mpamhanga, C.P.; Parton, A.; Southan, C.; Harmar, A.J.; Ghazal, P. Is systems pharmacology ready to impact upon therapy development? A study on the cholesterol biosynthesis pathway. Br. J. Pharmacol. 2017, 174, 4362–4382. [Google Scholar] [CrossRef]
- Mazein, A.; Watterson, S.; Hsieh, W.Y.; Griffiths, W.J.; Ghazal, P. A comprehensive machine-readable view of the mammalian cholesterol biosynthesis pathway. Biochem. Pharmacol. 2013, 86, 56–66. [Google Scholar] [CrossRef]
- Bourgin, M.; Labarthe, S.; Kriaa, A.; Lhomme, M.; Gérard, P.; Lesnik, P.; Laroche, B.; Maguin, E.; Rhimi, M. Exploring the bacterial impact on cholesterol cycle: A numerical study. Front. Microbiol. 2020, 11, 1121. [Google Scholar] [CrossRef]
- Gomez-Cabrero, D.; Compte, A.; Tegner, J. Workflow for generating competing hypothesis from models with parameter uncertainty. Interface Focus. 2011, 1, 438–449. [Google Scholar] [CrossRef]
- Parton, A.; McGilligan, V.; Chemaly, M.; O’Kane, M.; Watterson, S. New models of atherosclerosis and multi-drug therapeutic interventions. Bioinformatics 2019, 35, 2449–2457. [Google Scholar] [CrossRef]
- Bekkar, A.; Estreicher, A.; Niknejad, A.; Casals-Casas, C.; Bridge, A.; Xenarios, I.; Dorier, J.; Crespo, I. Expert curation for building network-based dynamical models: A case study on atherosclerotic plaque formation. Database 2018, 2018, bay031. [Google Scholar] [CrossRef]
- Le Novere, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.I.; Wimalaratne, S.M. The systems biology graphical notation. Nat. Biotechnol. 2009, 27, 735–741. [Google Scholar] [CrossRef]
- Junker, B.H.; Klukas, C.; Schreiber, F. Vanted: A system for advanced data analysis and visualization in the context of biological networks. BMC Bioinform. 2006, 7, 109. [Google Scholar]
- Czauderna, T.; Klukas, C.; Schreiber, F. Editing, validating and translating of sbgn maps. Bioinformatics 2010, 26, 2340–2341. [Google Scholar] [CrossRef][Green Version]
- Wildermuth, M.C. Metabolic control analysis: Biological applications and insights. Genome Biol. 2000, 1, reviews1031.1–reviews1031.5. [Google Scholar] [CrossRef]
- Mc Auley, M.T. Model analysis in greater depth. In Computer Modelling for Nutritionists; Springer International Publishing: Cham, Switzerland, 2019; pp. 63–78. [Google Scholar]
- Fell, D.; Cornish-Bowden, A. Understanding the Control of Metabolism; Portland Press: London, UK, 1997; Volume 2. [Google Scholar]
- Shi, L.; Tu, B.P. Acetyl-coa and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Cline, G.W.; Petersen, K.F.; Shulman, G.I. A non-invasive method to assess hepatic acetyl-coa in vivo. Cell Metab. 2017, 25, 749–756. [Google Scholar] [CrossRef]
- Johnston, T.P.; Palmer, W.K. The effect of pravastatin on hepatic 3-hydroxy-3-methylglutaryl coa reductase obtained from poloxamer 407-induced hyperlipidemic rats. Pharmacotherapy 1997, 17, 342–347. [Google Scholar]
- Pedersen, T.R. The success story of ldl cholesterol lowering. Circ. Res. 2016, 118, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.B.; Yin, Z.X.; Chei, C.L.; Qian, H.Z.; Kraus, V.B.; Zhang, J.; Brasher, M.S.; Shi, X.M.; Matchar, D.B.; Zeng, Y. Low-density lipoprotein cholesterol was inversely associated with 3-year all-cause mortality among chinese oldest old: Data from the chinese longitudinal healthy longevity survey. Atherosclerosis 2015, 239, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Weverling-Rijnsburger, A.W.; Blauw, G.J.; Lagaay, A.M.; Knook, D.L.; Meinders, A.E.; Westendorp, R.G. Total cholesterol and risk of mortality in the oldest old. Lancet 1997, 350, 1119–1123. [Google Scholar] [CrossRef]
- Al-Mallah, M.H.; Hatahet, H.; Cavalcante, J.L.; Khanal, S. Low admission ldl-cholesterol is associated with increased 3-year all-cause mortality in patients with non st segment elevation myocardial infarction. Cardiol. J. 2009, 16, 227–233. [Google Scholar]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Tirosh, O. Hypoxic signaling and cholesterol lipotoxicity in fatty liver disease progression. Oxid. Med. Cell. Longev. 2018, 2018, 2548154. [Google Scholar] [CrossRef]
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-coenzyme a: Cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1–E9. [Google Scholar] [CrossRef]
- Bell, T.A., 3rd; Brown, J.M.; Graham, M.J.; Lemonidis, K.M.; Crooke, R.M.; Rudel, L.L. Liver-specific inhibition of acyl-coenzyme a: Cholesterol acyltransferase 2 with antisense oligonucleotides limits atherosclerosis development in apolipoprotein b100-only low-density lipoprotein receptor-/- mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1814–1820. [Google Scholar] [CrossRef]
- Temel, R.E.; Lee, R.G.; Kelley, K.L.; Davis, M.A.; Shah, R.; Sawyer, J.K.; Wilson, M.D.; Rudel, L.L. Intestinal cholesterol absorption is substantially reduced in mice deficient in both abca1 and acat2. J. Lipid Res. 2005, 46, 2423–2431. [Google Scholar] [CrossRef]
- Lebeau, P.F.; Byun, J.H.; Platko, K.; MacDonald, M.E.; Poon, S.V.; Faiyaz, M.; Seidah, N.G.; Austin, R.C. Diet-induced hepatic steatosis abrogates cell-surface ldlr by inducing de novo pcsk9 expression in mice. J. Biol. Chem. 2019, 294, 9037–9047. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Name | Abbreviation |
|---|---|---|
| R1 | Acetyl coenzyme A (CoA) synthesis | CoAS → ACoA |
| R2 | Interconversion of Acetyl CoA and Acetoacetyl CoA | ACoA = AACoA |
| R3 | 3-hydroxy-3-methylglutaryl (HMG)-CoA formation | ACoA + AACoA → HMGCoA |
| R4 | Mevalonate (MV) formation | HMGCoA → MV |
| R5 | Mevalonate5P (MV5P) formation | MV → MV5P |
| R6 | Mevalonate5PP (MV5PP) formation | MV5P = MV5PP |
| R7 | Isopentenyl-PP (IPP) formation | MV5PP → IPP |
| R8 | Dimethylallyl-PP (DMAPP) interconversion | IPP = DMAPP |
| R9 | GeranylPP (GPP) formation | DMAPP + IPP → GPP |
| R10 | FarnesylPP (FPP) formation | GPP + IPP → FPP |
| R11 | Squalene formation | FPP → SQ |
| R12 | Squalene epoxide formation | SQ → SQE |
| R13 | Lanosterol formation | SQE → LAN |
| R14 | Free cholesterol (FC) formation | LAN → FC |
| R15 | Conversion of FC to cholesteryl esters (CE) | FC → CE |
| R16 | Conversion of CE to FC | CE → FC |
| R17 | Cholesterol esters flux to low-density lipoprotein cholesterol (LDL-C) | CE → LDLC |
| R18 | LDL-C sink | LDLC → LDLCs |
| R19 | LDL receptor (LDLr) synthesis | sLDLR → LDLR |
| R20 | LDLr degradation | LDLR → dLDLR |
| R21 | Reuptake of LDL-C | LDLC → FC |
| R22 | SREBP synthesis | sSRBP2 → SRBP2 |
| R23 | SREBP degradation | SRBP2 → dSRBP2 |
| R24 | Antioxidant production | sAOX → AOX |
| R25 | Reactive oxygen species (ROS) production | sROS → ROS |
| R26 | ROS degradation | AOX+ROS → ROSsink |
| R27 | HMGCoA reductase synthesis | sHMGCoAR → HMGCoAR |
| R28 | HMGCoA reductase degradation | HMGCoAR → dHMGCoAR |
| R29 | Acetyl-CoA acetyltransferase 2 (ACAT2) synthesis | sACAT2 → ACAT2 |
| R30 | ACAT2 Degradation | ACAT2 → dACAT2 |
| Vmax R15 Conversion of FC to CE (µMoles/min) | Vmax R4 Mevalonate Formation (µMoles/min) | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| FC | |||||
| 0.01 | 22.1333 | 22.2205 | 22.2485 | 22.2623 | 22.2705 |
| 0.02 | 9.93711 | 9.96287 | 9.97106 | 9.97509 | 9.97749 |
| 0.03 | 4.75843 | 4.76165 | 4.76266 | 4.76315 | 4.76345 |
| 0.04 | 2.83914 | 2.8396 | 2.83975 | 2.83981 | 2.83985 |
| 0.05 | 1.99568 | 1.99583 | 1.99587 | 1.99589 | 1.9959 |
| CE | |||||
| 0.01 | 0.084617 | 0.084669 | 0.084685 | 0.084693 | 0.084698 |
| 0.02 | 0.14247 | 0.142576 | 0.14261 | 0.142627 | 0.142637 |
| 0.03 | 0.162901 | 0.162953 | 0.162969 | 0.162976 | 0.162981 |
| 0.04 | 0.166003 | 0.166019 | 0.166024 | 0.166026 | 0.166028 |
| 0.05 | 0.166386 | 0.166394 | 0.166397 | 0.166398 | 0.166399 |
| LDL-C | |||||
| 0.01 | 0.08454 | 0.084591 | 0.084608 | 0.084616 | 0.084621 |
| 0.02 | 0.14232 | 0.142428 | 0.142462 | 0.142479 | 0.142489 |
| 0.03 | 0.162786 | 0.162839 | 0.162855 | 0.162863 | 0.162867 |
| 0.04 | 0.165925 | 0.165941 | 0.165947 | 0.165949 | 0.16595 |
| 0.05 | 0.166317 | 0.166326 | 0.166328 | 0.166329 | 0.16633 |
| LDLr | |||||
| 0.01 | 34,335.9 | 34,334 | 34,333.4 | 34,333.1 | 34,332.9 |
| 0.02 | 34,530.3 | 34,527 | 34,526 | 34,525.5 | 34,525.2 |
| 0.03 | 34,770.5 | 34,766.7 | 34,765.4 | 34,764.8 | 34,764.4 |
| 0.04 | 34,998.3 | 34,994.4 | 34,993.2 | 34,992.5 | 34,992.2 |
| 0.05 | 35,192 | 35,188.2 | 35,187 | 35,186.4 | 35,186 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, A.E.; Mc Auley, M.T. Cholesterol Homeostasis: An In Silico Investigation into How Aging Disrupts Its Key Hepatic Regulatory Mechanisms. Biology 2020, 9, 314. https://doi.org/10.3390/biology9100314
Morgan AE, Mc Auley MT. Cholesterol Homeostasis: An In Silico Investigation into How Aging Disrupts Its Key Hepatic Regulatory Mechanisms. Biology. 2020; 9(10):314. https://doi.org/10.3390/biology9100314
Chicago/Turabian StyleMorgan, Amy Elizabeth, and Mark Tomás Mc Auley. 2020. "Cholesterol Homeostasis: An In Silico Investigation into How Aging Disrupts Its Key Hepatic Regulatory Mechanisms" Biology 9, no. 10: 314. https://doi.org/10.3390/biology9100314
APA StyleMorgan, A. E., & Mc Auley, M. T. (2020). Cholesterol Homeostasis: An In Silico Investigation into How Aging Disrupts Its Key Hepatic Regulatory Mechanisms. Biology, 9(10), 314. https://doi.org/10.3390/biology9100314