The H2S–Nrf2–Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
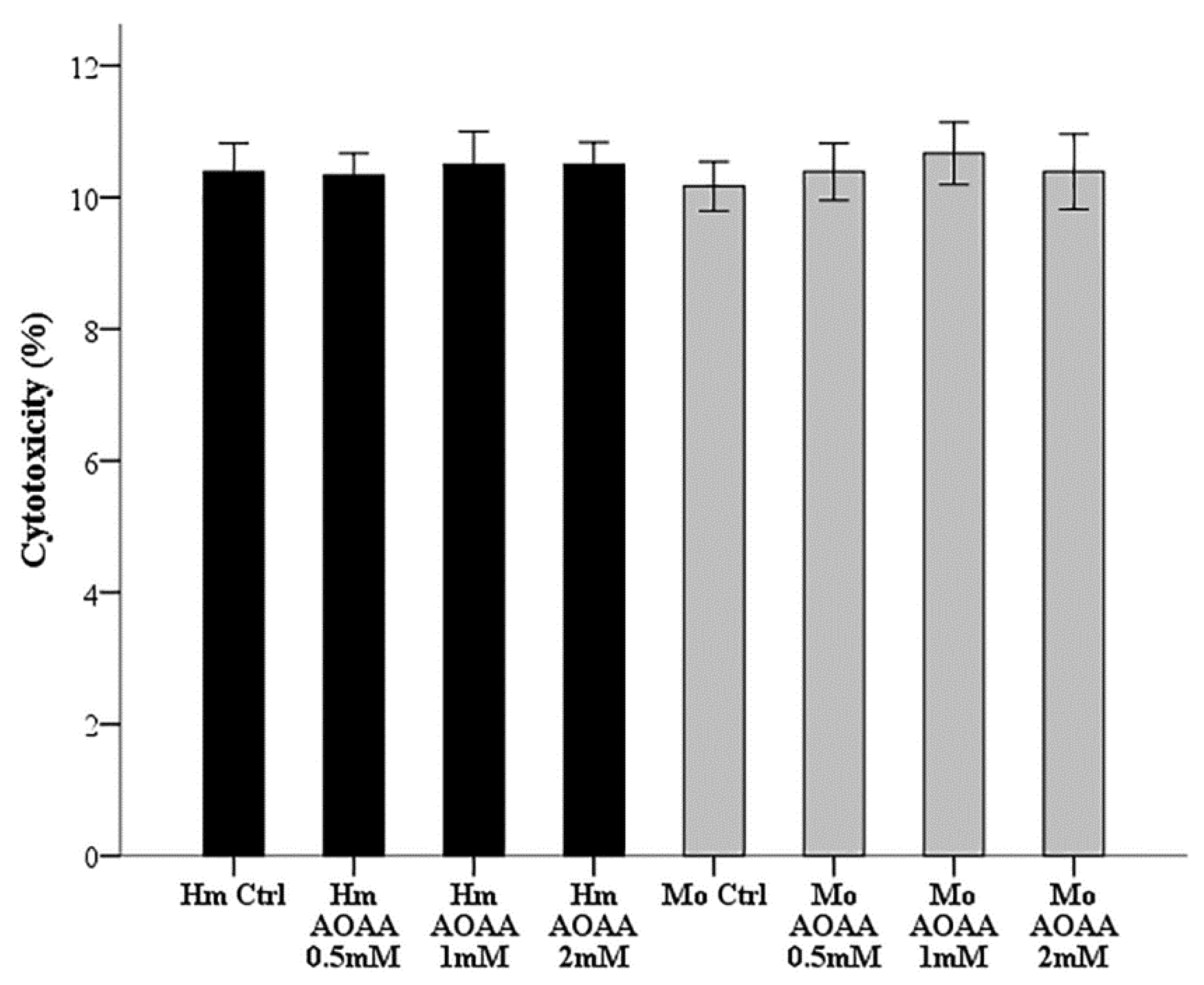
2.2. Assessment of AOAA Toxicity in RPTECs
2.3. Assessment of Proteins of Interest
2.4. Measurement of H2S Production
2.5. Assessment of ROS Production
2.6. Evaluation of Cell Death
2.7. Statistical Analysis
3. Results
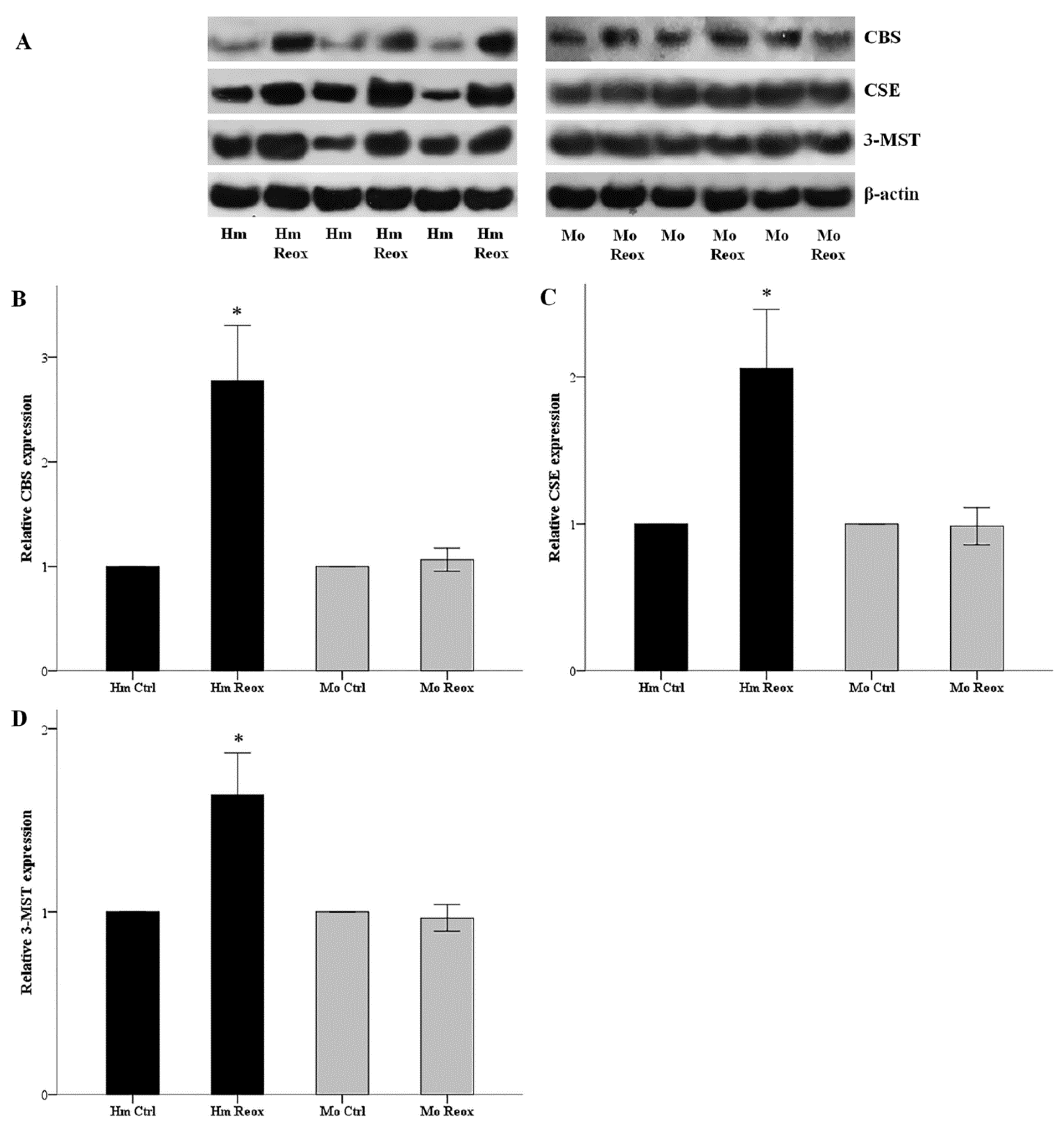
3.1. Reoxygenation Increases the Level of H2S-Producing Enzymes in the Hamster, but Not in Mouse RPRECs
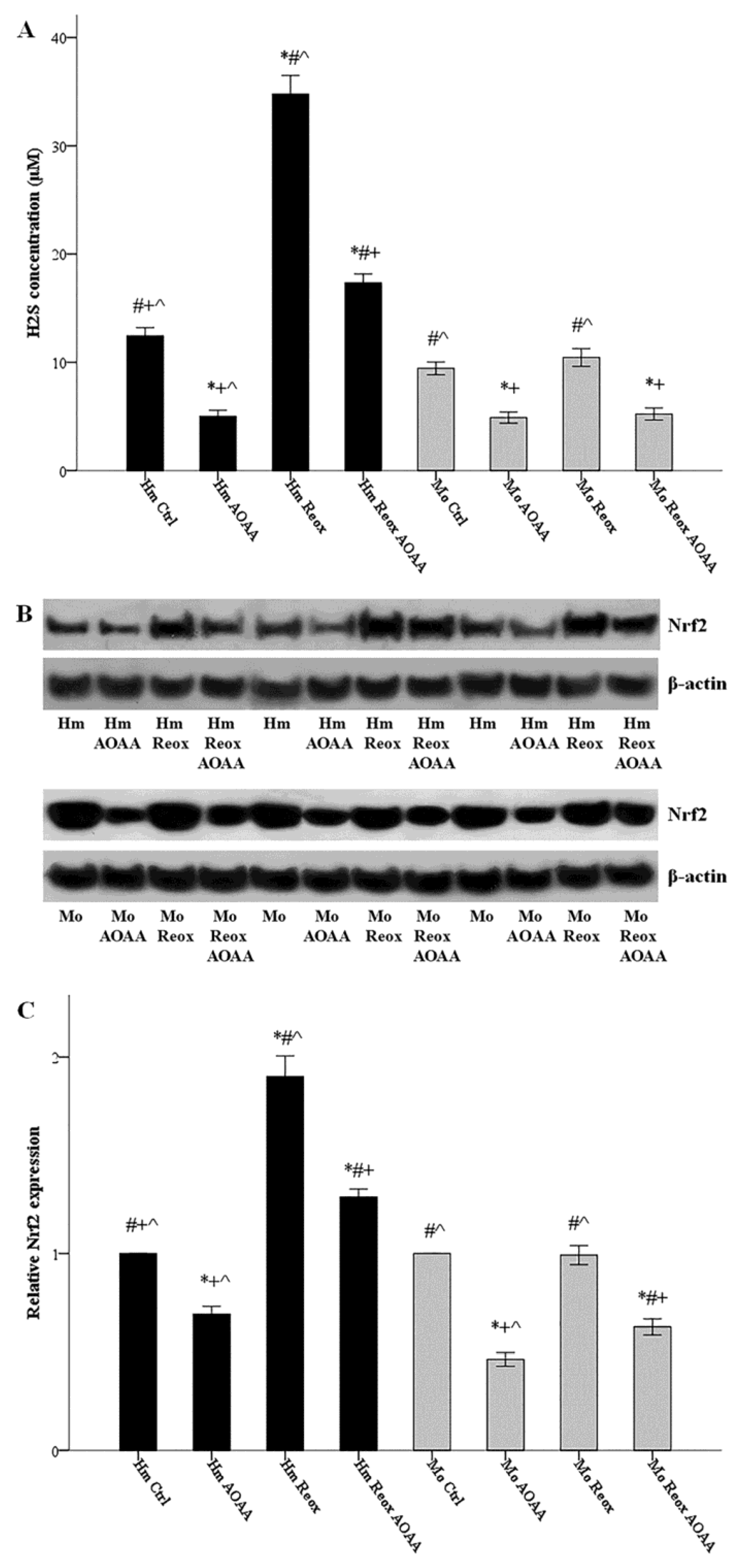
3.2. Reoxygenation Increases H2S Production in a H2S-Producing Enzymes-Dependent Way in the Hamster, but Not in Mouse RPTECs
3.3. Reoxygenation Increases the Nrf2 Level in a H2S-Producing Enzymes-Dependent Way in the Hamster, but Not in Mouse RPTECs
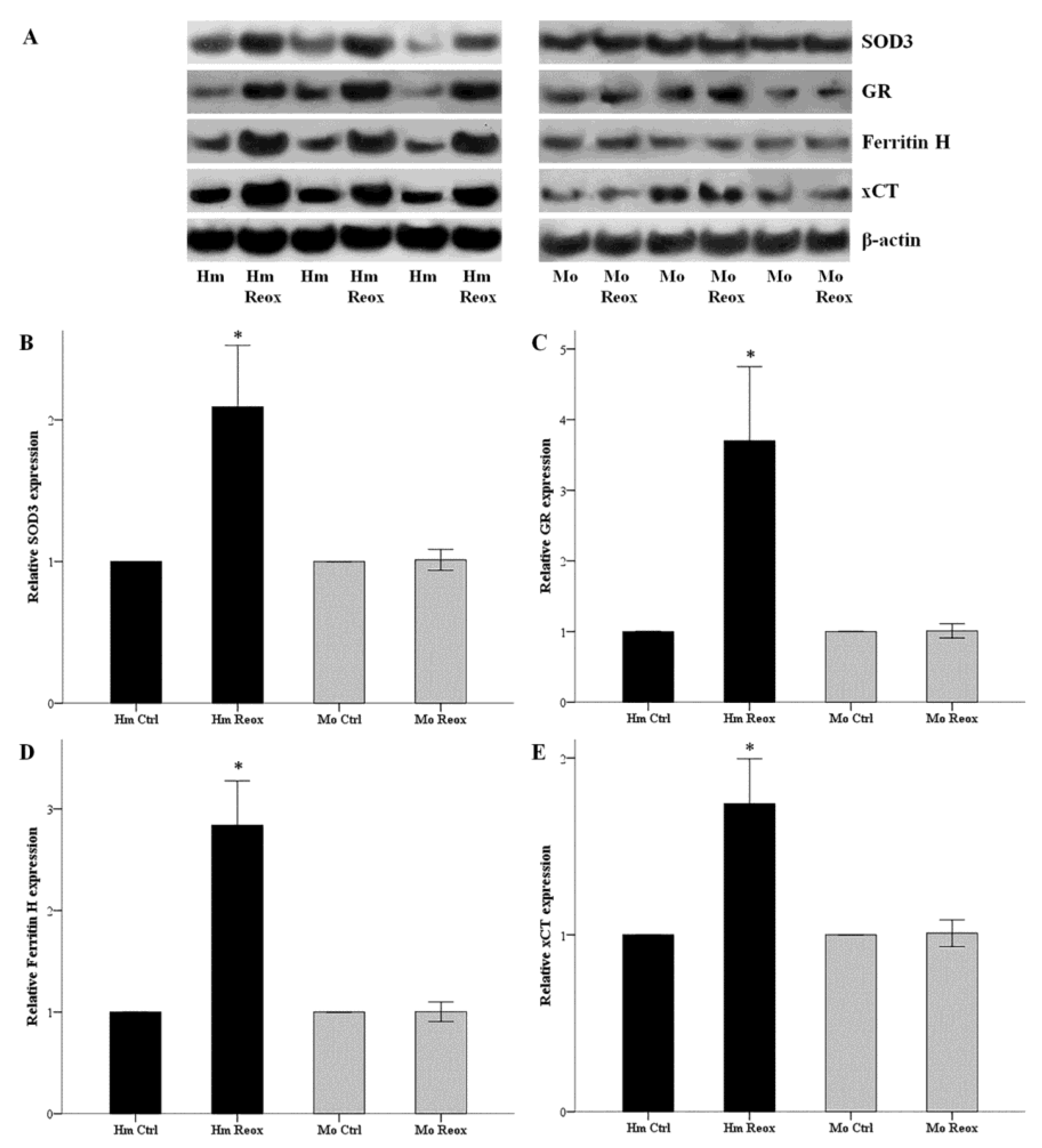
3.4. Reoxygenation Increases the Expression of SOD3, GR, Ferritin H, and xCT in the Hamster, but Not in Mouse RPTECs
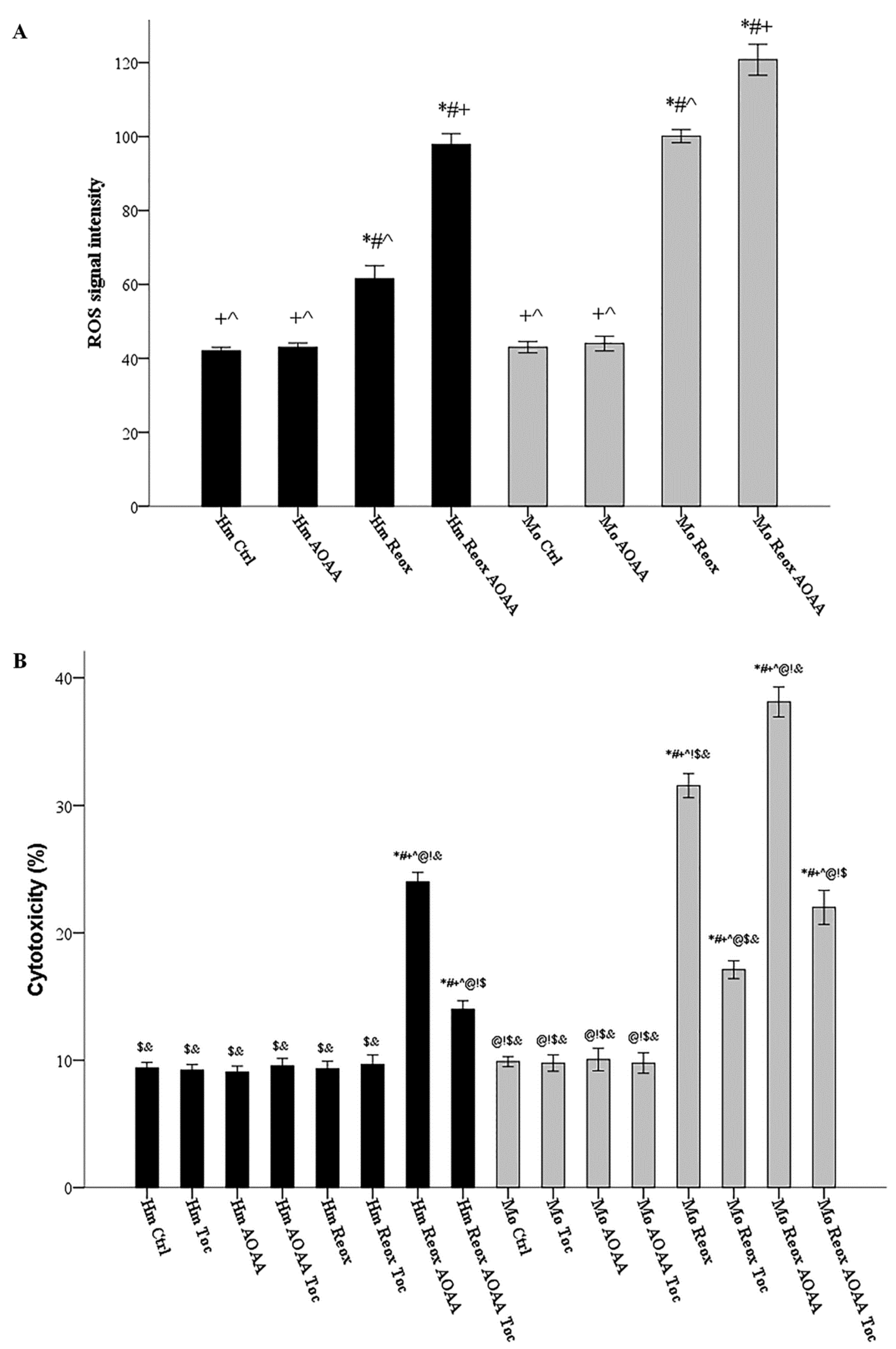
3.5. Under Reoxygenation, the H2S-Producing Enzymes Play a Significant Role in Controlling ROS Production in Both Hamster and Mouse Cells
3.6. H2S-Producing Enzymes Rescue Hamster RPTECs from Reoxygenation-Induced, Lipid Peroxidation-Mediated Cell Death and Also Decrease Reoxygenation Cytotoxicity in Mouse RPTECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Availability of Data and Materials
Conflicts of Interest
References
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Bakthavachalam, P.; Shanmugam, P.S.T. Mitochondrial dysfunction—Silent killer in cerebral ischemia. J. Neurol. Sci. 2017, 375, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Chanthaphavong, R.S.; Pape, H.C. Current theories on the pathophysiology of multiple organ failure after trauma. Injury 2010, 41, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Carey, H.V.; Andrews, M.T.; Martin, S.L. Mammalian hibernation: Cellular and molecular responses to depressed metabolism and low temperature. Physiol. Rev. 2003, 83, 1153–1181. [Google Scholar] [CrossRef] [PubMed]
- Storey, K.B.; Storey, J.M. Metabolic rate depression: The biochemistry of mammalian hibernation. Adv. Clin. Chem. 2010, 52, 77–108. [Google Scholar] [PubMed]
- Levin, E.; Plotnik, B.; Amichai, E.; Braulke, L.J.; Landau, S.; Yom-Tov, Y.; Kronfeld-Schor, N. Subtropical mouse-tailed bats use geothermally heated caves for winter hibernation. Proc. Biol. Sci. 2015, 282, 20142781. [Google Scholar] [CrossRef]
- Dausmann, K.H.; Glos, J.; Ganzhorn, J.U.; Heldmaier, G. Physiology: Hibernation in a tropical primate. Nature 2004, 429, 825–826. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.B.; Dausmann, K.H.; Ranaivoarisoa, J.F.; Yoder, A.D. Underground hibernation in a primate. Sci. Rep. 2013, 3, 1768. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Dugbartey, G.J.; Bouma, H.R.; Strijkstra, A.M.; Boerema, A.S.; Henning, R.H. Induction of a Torpor-Like State by 5'-AMP Does Not Depend on H2S Production. PLoS ONE 2015, 10, e0136113. [Google Scholar] [CrossRef]
- Zancanaro, C.; Malatesta, M.; Mannello, F.; Vogel, P.; Fakan, S. The kidney during hibernation and arousal from hibernation. A natural model of organ preservation during cold ischaemia and reperfusion. Nephrol. Dial. Transplant. 1999, 14, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Bogren, L.K.; Olson, J.M.; Carpluk, J.; Moore, J.M.; Drew, K.L. Resistance to systemic inflammation and multi organ damage after global ischemia/reperfusion in the arctic ground squirrel. PLoS ONE 2014, 9, e94225. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Cell Death Patterns Due to Warm Ischemia or Reperfusion in Renal Tubular Epithelial Cells Originating from Human, Mouse, or the Native Hibernator Hamster. Biology (Basel) 2018, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen Sulfide Protects Against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The Gasotransmitter Hydrogen Sulfide Induces Nrf2-Target Genes by Inactivating the Keap1 Ubiquitin Ligase Substrate Adaptor Through Formation of a Disulfide Bond Between Cys-226 and Cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef]
- Miyamoto, R.; Otsuguro, K.-I.; Yamaguchi, S.; Ito, S. Contribution of cysteine aminotransferase and mercaptopyruvate sulfurtransferase to hydrogen sulfide production in peripheral neurons. J. Neurochem. 2014, 130, 29–40. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Talaei, F.; Bouma, H.R.; Van der Graaf, A.C.; Strijkstra, A.M.; Schmidt, M.; Henning, R.H. Serotonin and dopamine protect from hypothermia/rewarming damage through the CBS/H2S pathway. PLoS ONE 2011, 6, e22568. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-Z.; Yang, S.; Wu, G. Free radicals, antioxidants, and nutrition. Nutrition 2002, 18, 872–879. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, T.; Iuchi, Y.; Okada, F.; Kuwata, K.; Yamanobe, T.; Bannai, S.; Tomita, Y.; Sato, H.; Fujii, J. Aggravation of ischemia–reperfusion-triggered acute renal failure in xCT-deficient mice. Arch. Biochem. Biophys. 2009, 490, 63–69. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Hatcher, H.C.; Tesfay, L.; Torti, S.V.; Torti, F.M. Cytoprotective Effect of Ferritin H in Renal Ischemia Reperfusion Injury. PLoS ONE 2015, 10, e0138505. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Factors that May Protect the Native Hibernator Syrian Hamster Renal Tubular Epithelial Cells from Ferroptosis Due to Warm Anoxia-Reoxygenation. Biology 2019, 8, 22. [Google Scholar] [CrossRef]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Linkermann, A.; Brasen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012, 81, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I.; Lawson, B.R. Toll-like receptors and their role in renal pathologies. Inflamm. Allergy Drug Targets 2012, 11, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Talaei, F.; Bouma, H.R.; Hylkema, M.N.; Strijkstra, A.M.; Boerema, A.S.; Schmidt, M.; Henning, R.H. The role of endogenous H2S formation in reversible remodeling of lung tissue during hibernation in the Syrian hamster. J. Exp. Biol. 2012, 215, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Pier, M., Jr.; Ni, Z.; McMullen, D.C.; Storey, K.B. Expression of Nrf2 and its downstream gene targets in hibernating 13-lined ground squirrels, Spermophilus tridecemlineatus. Mol. Cell. Biochem. 2008, 312, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Frigault, J.J.; Gaudet, J.D.; Morin, P., Jr. Investigating Nrf2-associated non-coding RNAs in the hibernating ground squirrel, Ictidomys tridecemlineatus. J. Therm. Biol. 2018, 75, 38–44. [Google Scholar] [CrossRef]
- Hermes-Lima, M.; Yin, Q.; Ge, H.; Liao, C.-C.; Liu, D.; Zhang, S.; Pan, Y.-H. Antioxidant Defenses in the Brains of Bats during Hibernation. PLoS ONE 2016, 11, e0152135. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, J.; Xu, S.; Peng, X.; Yan, X.; Li, X.; Wang, H.; Chang, H.; Gao, Y. Controllable oxidative stress and tissue specificity in major tissues during the torpor–arousal cycle in hibernating Daurian ground squirrels. Open Biol. 2018, 8, 180068. [Google Scholar] [CrossRef] [PubMed]
- Dugbartey, G.J.; Bouma, H.R.; Saha, M.N.; Lobb, I.; Henning, R.H.; Sener, A. A Hibernation-Like State for Transplantable Organs: Is Hydrogen Sulfide Therapy the Future of Organ Preservation? Antioxid. Redox Signal. 2018, 28, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kim, J.I.; Park, J.-W.; Park, K.M. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrol. Dial. Transplant. 2015, 30, 1497–1506. [Google Scholar] [CrossRef]
- Sekijima, M.; Sahara, H.; Miki, K.; Villani, V.; Ariyoshi, Y.; Iwanaga, T.; Tomita, Y.; Yamada, K. Hydrogen sulfide prevents renal ischemia-reperfusion injury in CLAWN miniature swine. J. Surg. Res. 2017, 219, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.-Y.; Siow, Y.L.; O, K. Ischemia-reperfusion reduces cystathionine-β-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. Ren. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1215–H1224. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Xue, L.; Cheng, J.; Bai, Y. Preconditioning of H 2 S inhalation protects against cerebral ischemia/reperfusion injury by induction of HSP70 through PI3K/Akt/Nrf2 pathway. Br. Res. Bull. 2016, 121, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Ganster, F.; Burban, M.; de la Bourdonnaye, M.; Fizanne, L.; Douay, O.; Loufrani, L.; Mercat, A.; Cales, P.; Radermacher, P.; Henrion, D.; et al. Effects of hydrogen sulfide on hemodynamics, inflammatory response and oxidative stress during resuscitated hemorrhagic shock in rats. Crit. Care 2010, 14, R165. [Google Scholar] [CrossRef]
- Satterly, S.A.; Salgar, S.; Hoffer, Z.; Hempel, J.; DeHart, M.J.; Wingerd, M.; Raywin, H.; Stallings, J.D.; Martin, M. Hydrogen sulfide improves resuscitation via non-hibernatory mechanisms in a porcine shock model. J. Surg. Res. 2015, 199, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Reddy, N.M.; Higbee, E.M.; Potteti, H.R.; Noel, S.; Racusen, L.; Kensler, T.W.; Sporn, M.B.; Reddy, S.P.; Rabb, H. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia–reperfusion injury in mice. Kidney Int. 2014, 85, 134–141. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eleftheriadis, T.; Pissas, G.; Nikolaou, E.; Liakopoulos, V.; Stefanidis, I. The H2S–Nrf2–Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death. Biology 2019, 8, 74. https://doi.org/10.3390/biology8040074
Eleftheriadis T, Pissas G, Nikolaou E, Liakopoulos V, Stefanidis I. The H2S–Nrf2–Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death. Biology. 2019; 8(4):74. https://doi.org/10.3390/biology8040074
Chicago/Turabian StyleEleftheriadis, Theodoros, Georgios Pissas, Evdokia Nikolaou, Vassilios Liakopoulos, and Ioannis Stefanidis. 2019. "The H2S–Nrf2–Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death" Biology 8, no. 4: 74. https://doi.org/10.3390/biology8040074
APA StyleEleftheriadis, T., Pissas, G., Nikolaou, E., Liakopoulos, V., & Stefanidis, I. (2019). The H2S–Nrf2–Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death. Biology, 8(4), 74. https://doi.org/10.3390/biology8040074