Factors that May Protect the Native Hibernator Syrian Hamster Renal Tubular Epithelial Cells from Ferroptosis Due to Warm Anoxia-Reoxygenation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
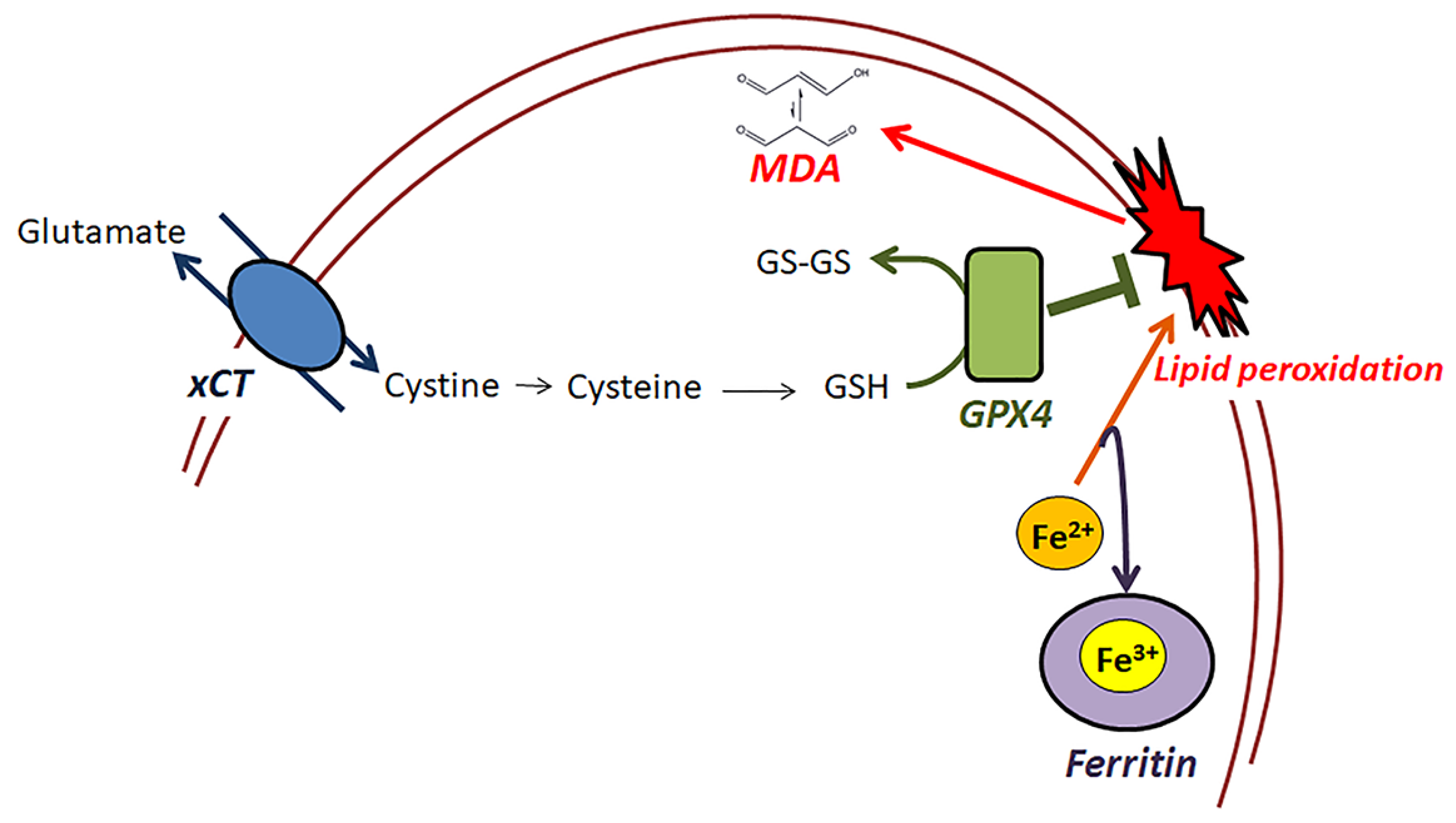
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Assessment of Cell Death and Lipid Peroxidation Due to Anoxia-Reoxygenation
2.3. Assessment of Cell Survival Due to Anoxia Reoxygenation and the Effect of α-Tocopherol
2.4. Assessment of xCT, Ferritin and GPX4 Expression after Anoxia or Reoxygenation
2.5. Statistical Analysis
3. Results
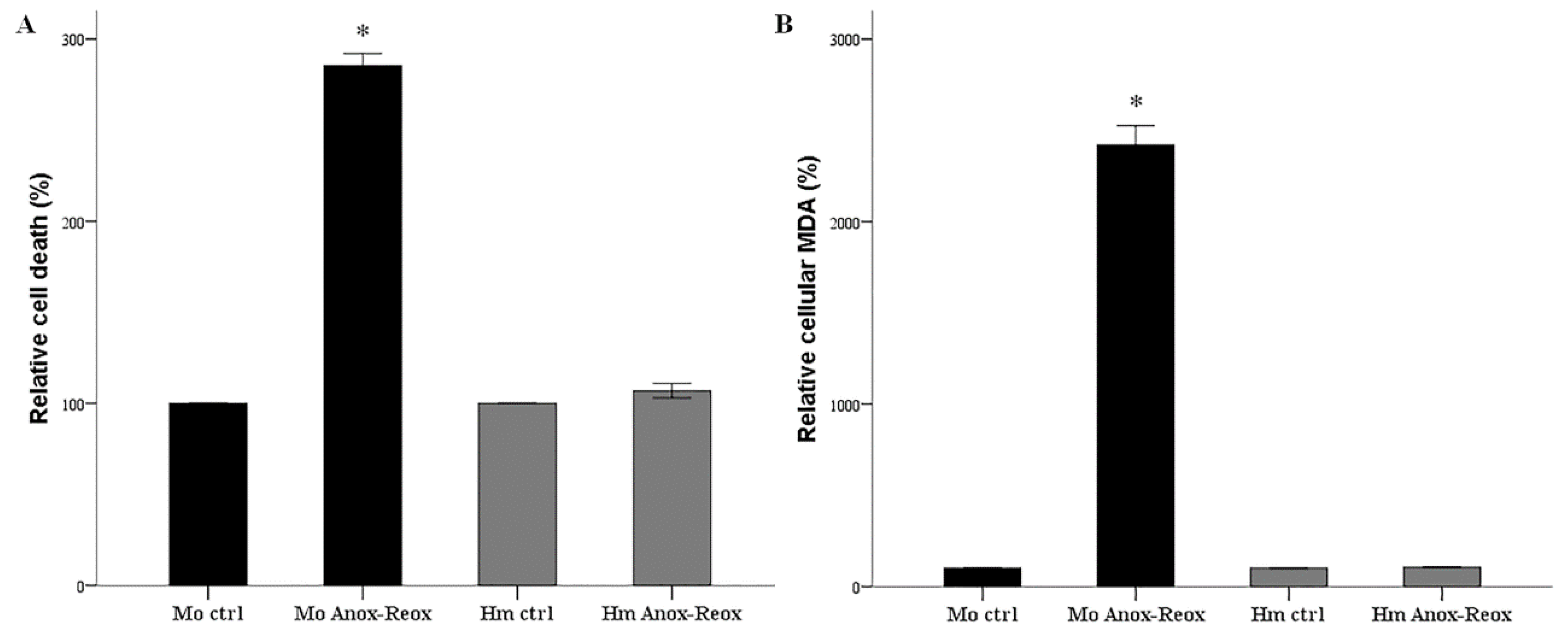
3.1. Anoxia-Reoxygenation Induces Lipid Peroxidation and Cell Death in Mouse RPTECs, but Not in Hamster RPTECs
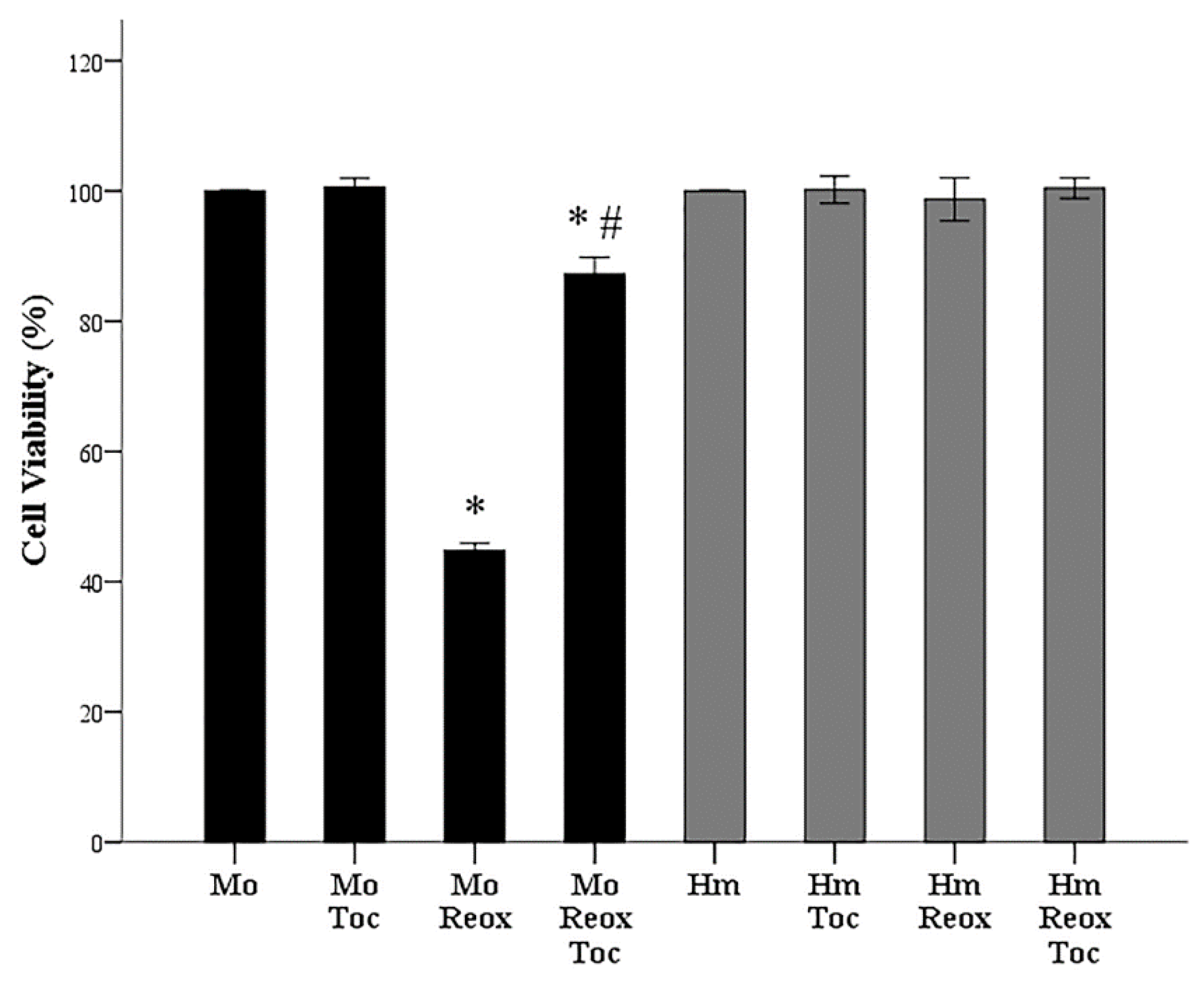
3.2. α-Tocopherol Increases Cell Survival of Mouse RPTECs Subjected to Anoxia-Reoxygenation
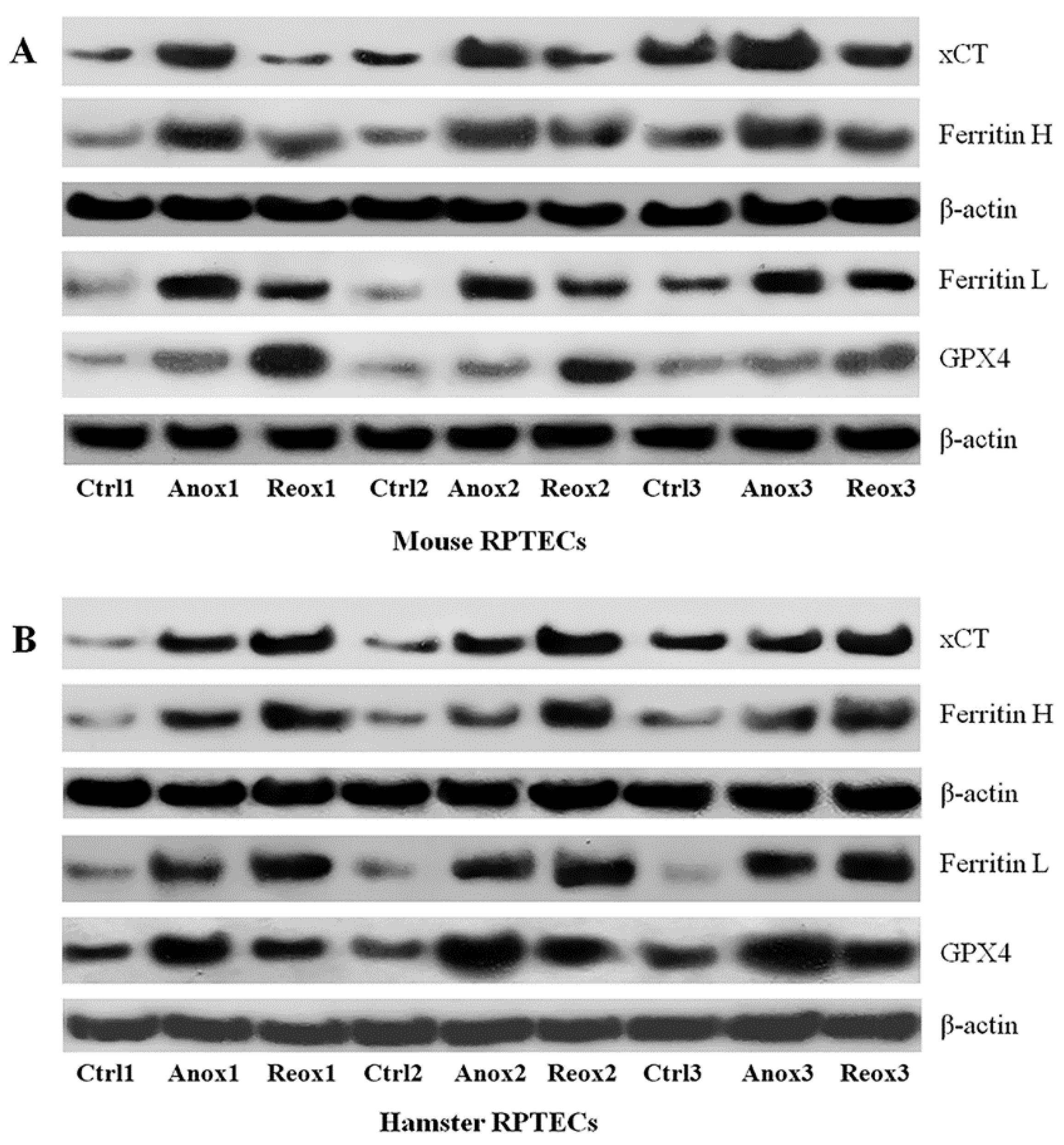
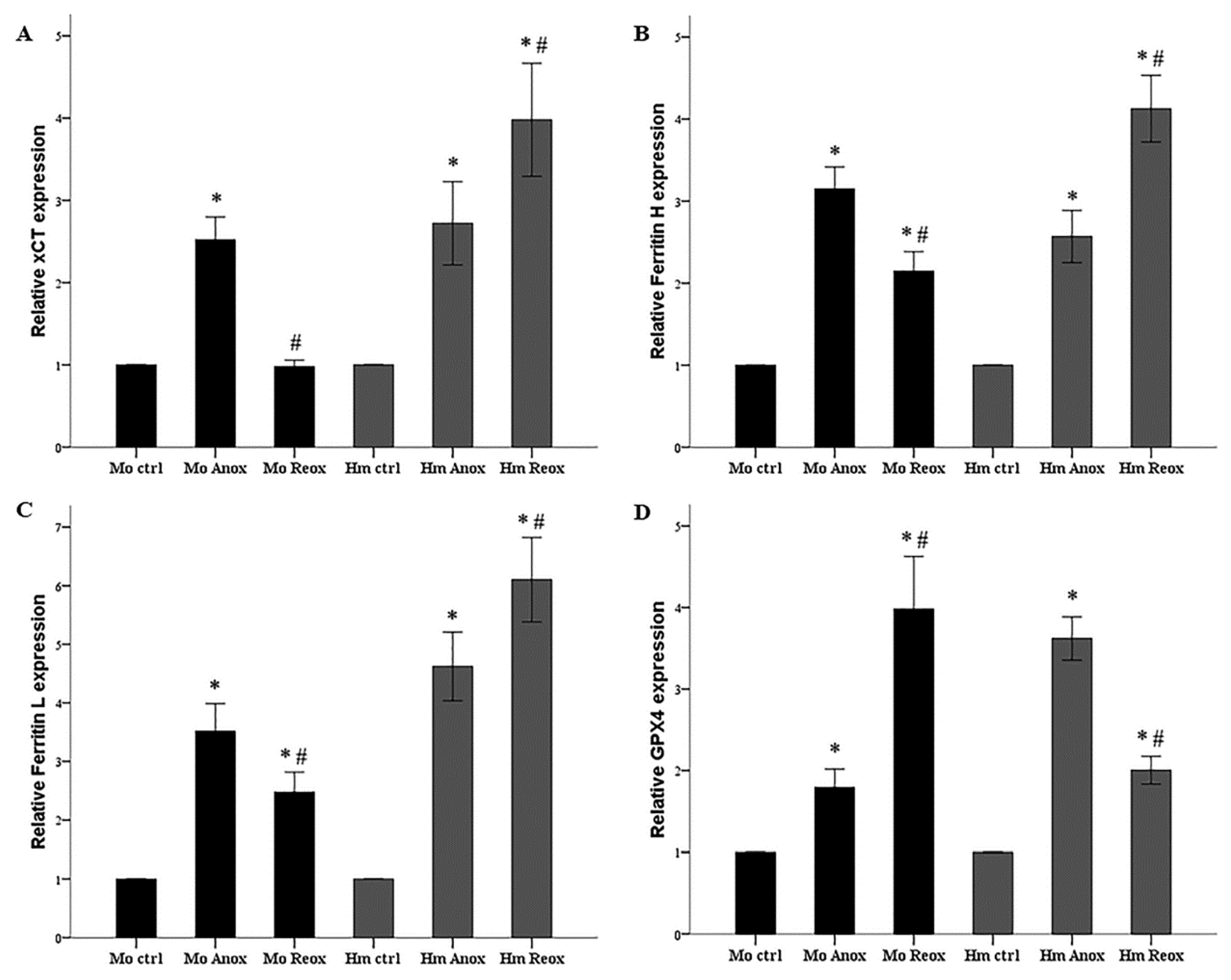
3.3. Different Effect of Anoxia or Reoxygenation on the Expression of xCT, Ferritin, and GPX4 in Mouse and Syrian Hamster RPTECs
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef] [PubMed]
- Bakthavachalam, P.; Shanmugam, P.S.T. Mitochondrial dysfunction - Silent killer in cerebral ischemia. J. Neurol. Sci. 2017, 375, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Chanthaphavong, R.S.; Pape, H.C. Current theories on the pathophysiology of multiple organ failure after trauma. Injury 2010, 41, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Gilbo, N.; Monbaliu, D. Temperature and oxygenation during organ preservation: Friends or foes? Curr. Opin. Organ. Transplant. 2017, 22, 290–299. [Google Scholar] [CrossRef]
- Carey, H.V.; Andrews, M.T.; Martin, S.L. Mammalian hibernation: Cellular and molecular responses to depressed metabolism and low temperature. Physiol. Rev. 2003, 83, 1153–1181. [Google Scholar] [CrossRef] [PubMed]
- Storey, K.B.; Storey, J.M. Metabolic rate depression: The biochemistry of mammalian hibernation. Adv. Clin. Chem. 2010, 52, 77–108. [Google Scholar] [PubMed]
- Dugbartey, G.J.; Bouma, H.R.; Strijkstra, A.M.; Boerema, A.S.; Henning, R.H. Induction of a Torpor-Like State by 5’-AMP Does Not Depend on H2S Production. PLoS ONE 2015, 10, e0136113. [Google Scholar] [CrossRef]
- Zancanaro, C.; Malatesta, M.; Mannello, F.; Vogel, P.; Fakan, S. The kidney during hibernation and arousal from hibernation. A natural model of organ preservation during cold ischaemia and reperfusion. Nephrol. Dial. Transplant. 1999, 14, 1982–1990. [Google Scholar] [CrossRef]
- Levin, E.; Plotnik, B.; Amichai, E.; Braulke, L.J.; Landau, S.; Yom-Tov, Y.; Kronfeld-Schor, N. Subtropical mouse-tailed bats use geothermally heated caves for winter hibernation. Proc. Biol. Sci. 2015, 282, 20142781. [Google Scholar] [CrossRef] [PubMed]
- Dausmann, K.H.; Glos, J.; Ganzhorn, J.U.; Heldmaier, G. Physiology: Hibernation in a tropical primate. Nature 2004, 429, 825–826. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.B.; Dausmann, K.H.; Ranaivoarisoa, J.F.; Yoder, A.D. Underground hibernation in a primate. Sci. Rep. 2013, 3, 1768. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.R.; Prado, R.; Raval, A.P.; Drew, K.L.; Perez-Pinzon, M.A. The arctic ground squirrel brain is resistant to injury from cardiac arrest during euthermia. Stroke 2006, 37, 1261–1265. [Google Scholar] [CrossRef]
- Quinones, Q.J.; Zhang, Z.; Ma, Q.; Smith, M.P.; Soderblom, E.; Moseley, M.A.; Bain, J.; Newgard, C.B.; Muehlbauer, M.J.; Hirschey, M.; et al. Proteomic Profiling Reveals Adaptive Responses to Surgical Myocardial Ischemia-Reperfusion in Hibernating Arctic Ground Squirrels Compared to Rats. Anesthesiology 2016, 124, 1296–1310. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Cell Death Patterns Due to Warm Ischemia or Reperfusion in Renal Tubular Epithelial Cells Originating from Human, Mouse, or the Native Hibernator Hamster. Biology 2018, 7, 48. [Google Scholar] [CrossRef]
- Bogren, L.K.; Olson, J.M.; Carpluk, J.; Moore, J.M.; Drew, K.L. Resistance to systemic inflammation and multi organ damage after global ischemia/reperfusion in the arctic ground squirrel. PLoS ONE 2014, 9, e94225. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Brasen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012, 81, 751–761. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I.; Lawson, B.R. Toll-like receptors and their role in renal pathologies. Inflamm. Allergy Drug Targets 2012, 11, 464–477. [Google Scholar] [CrossRef]
- Kavsan, V.M.; Iershov, A.V.; Balynska, O.V. Immortalized cells and one oncogene in malignant transformation: Old insights on new explanation. BMC Cell Biol. 2011, 12, 23. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Filippidis, G.; Liakopoulos, V.; Stefanidis, I. Renal tubular epithelial cells of the native hibernator Syrian hamster recover more rapidly from endoplasmic reticulum stress compared to those of human or mouse following warm anoxia-reoxygenation, possibly due to increased proteasomal function. World Acad. Sci. J. 2018. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Golfinopoulos, S.; Liakopoulos, V.; Stefanidis, I. Energy handling in renal tubular epithelial cells of the hamster, a native hibernator, under warm anoxia or reoxygenation. Biomed. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Hatcher, H.C.; Tesfay, L.; Torti, S.V.; Torti, F.M. Cytoprotective Effect of Ferritin H in Renal Ischemia Reperfusion Injury. PLoS ONE 2015, 10, e0138505. [Google Scholar] [CrossRef]
- Faherty, S.L.; Villanueva-Canas, J.L.; Blanco, M.B.; Alba, M.M.; Yoder, A.D. Transcriptomics in the wild: Hibernation physiology in free-ranging dwarf lemurs. Mol. Ecol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Larade, K. Accumulation and translation of ferritin heavy chain transcripts following anoxia exposure in a marine invertebrate. J. Exp. Biol. 2004, 207, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Eckl, P. Oxidative Stress and the Homeodynamics of Iron Metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Kayano, T.; Hanaya, T.; Arai, S.; Ikeda, M.; Kurimoto, M. Up-regulation of an extracellular superoxide dismutase-like activity in hibernating hamsters subjected to oxidative stress in mid- to late arousal from torpor. Compar. Biochem. Physiol. Part C Toxicol. Pharmacol. 2006, 144, 47–56. [Google Scholar] [CrossRef]
- Ohta, H.; Okamoto, I.; Hanaya, T.; Arai, S.; Ohta, T.; Fukuda, S. Enhanced antioxidant defense due to extracellular catalase activity in Syrian hamster during arousal from hibernation. Compar. Biochem. Physiol. Part C Toxicol. Pharmacol. 2006, 143, 484–491. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta BBA-Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, T.; Iuchi, Y.; Okada, F.; Kuwata, K.; Yamanobe, T.; Bannai, S.; Tomita, Y.; Sato, H.; Fujii, J. Aggravation of ischemia–reperfusion-triggered acute renal failure in xCT-deficient mice. Arch. Biochem. Biophys. 2009, 490, 63–69. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Basal Levels of eIF2α Phosphorylation Determine Cellular Antioxidant Status by Regulating ATF4 and xCT Expression. J. Biol. Chem. 2009, 284, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Habib, E.; Linher-Melville, K.; Lin, H.-X.; Singh, G. Expression of xCT and activity of system x c—Are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Storey, K.B. Heme oxygenase expression and Nrf2 signaling during hibernation in ground squirrels. Can. J. Physiol. Pharmacol. 2010, 88, 379–387. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, J.; Xu, S.; Peng, X.; Yan, X.; Li, X.; Wang, H.; Chang, H.; Gao, Y. Controllable oxidative stress and tissue specificity in major tissues during the torpor-arousal cycle in hibernating Daurian ground squirrels. Open Biol. 2018, 8, 180068. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Lin, Y.-J.; Chen, W.-L.; Huang, Y.-C.; Chang, C.-W.; Cheng, F.-C.; Liu, R.-S.; Shyu, W.-C. HIF-1α triggers long-lasting glutamate excitotoxicity via system xc- in cerebral ischaemia-reperfusion. J. Pathol. 2017, 241, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Arnold, W.; Ruf, T.; Frey-Roos, F.; Bruns, U. Diet-Independent Remodeling of Cellular Membranes Precedes Seasonally Changing Body Temperature in a Hibernator. PLoS ONE 2011, 6, e18641. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2016, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Factors that May Protect the Native Hibernator Syrian Hamster Renal Tubular Epithelial Cells from Ferroptosis Due to Warm Anoxia-Reoxygenation. Biology 2019, 8, 22. https://doi.org/10.3390/biology8020022
Eleftheriadis T, Pissas G, Liakopoulos V, Stefanidis I. Factors that May Protect the Native Hibernator Syrian Hamster Renal Tubular Epithelial Cells from Ferroptosis Due to Warm Anoxia-Reoxygenation. Biology. 2019; 8(2):22. https://doi.org/10.3390/biology8020022
Chicago/Turabian StyleEleftheriadis, Theodoros, Georgios Pissas, Vassilios Liakopoulos, and Ioannis Stefanidis. 2019. "Factors that May Protect the Native Hibernator Syrian Hamster Renal Tubular Epithelial Cells from Ferroptosis Due to Warm Anoxia-Reoxygenation" Biology 8, no. 2: 22. https://doi.org/10.3390/biology8020022
APA StyleEleftheriadis, T., Pissas, G., Liakopoulos, V., & Stefanidis, I. (2019). Factors that May Protect the Native Hibernator Syrian Hamster Renal Tubular Epithelial Cells from Ferroptosis Due to Warm Anoxia-Reoxygenation. Biology, 8(2), 22. https://doi.org/10.3390/biology8020022