Open Conformation of the Escherichia coli Periplasmic Murein Tripeptide Binding Protein, MppA, at High Resolution

Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression of 6-His Tagged MppA
2.2. Purification of MppA
2.3. Crystallization of MppA and X-Ray Data Collection
2.4. Structure Solution by Molecular Replacement
2.5. Structure Refinement
3. Results
3.1. Expression, Purification, Crystallization, and Structure Solution of MppA
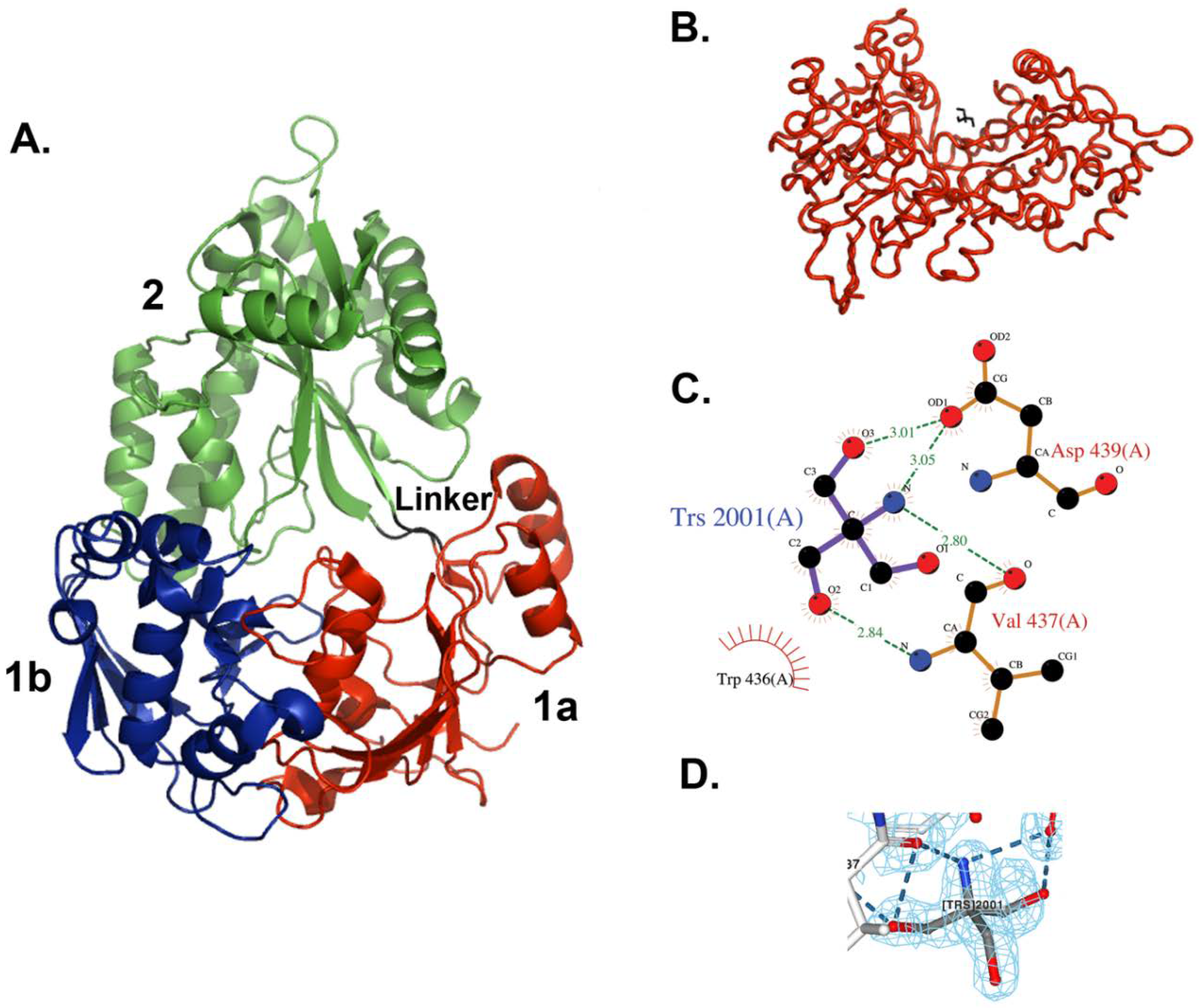
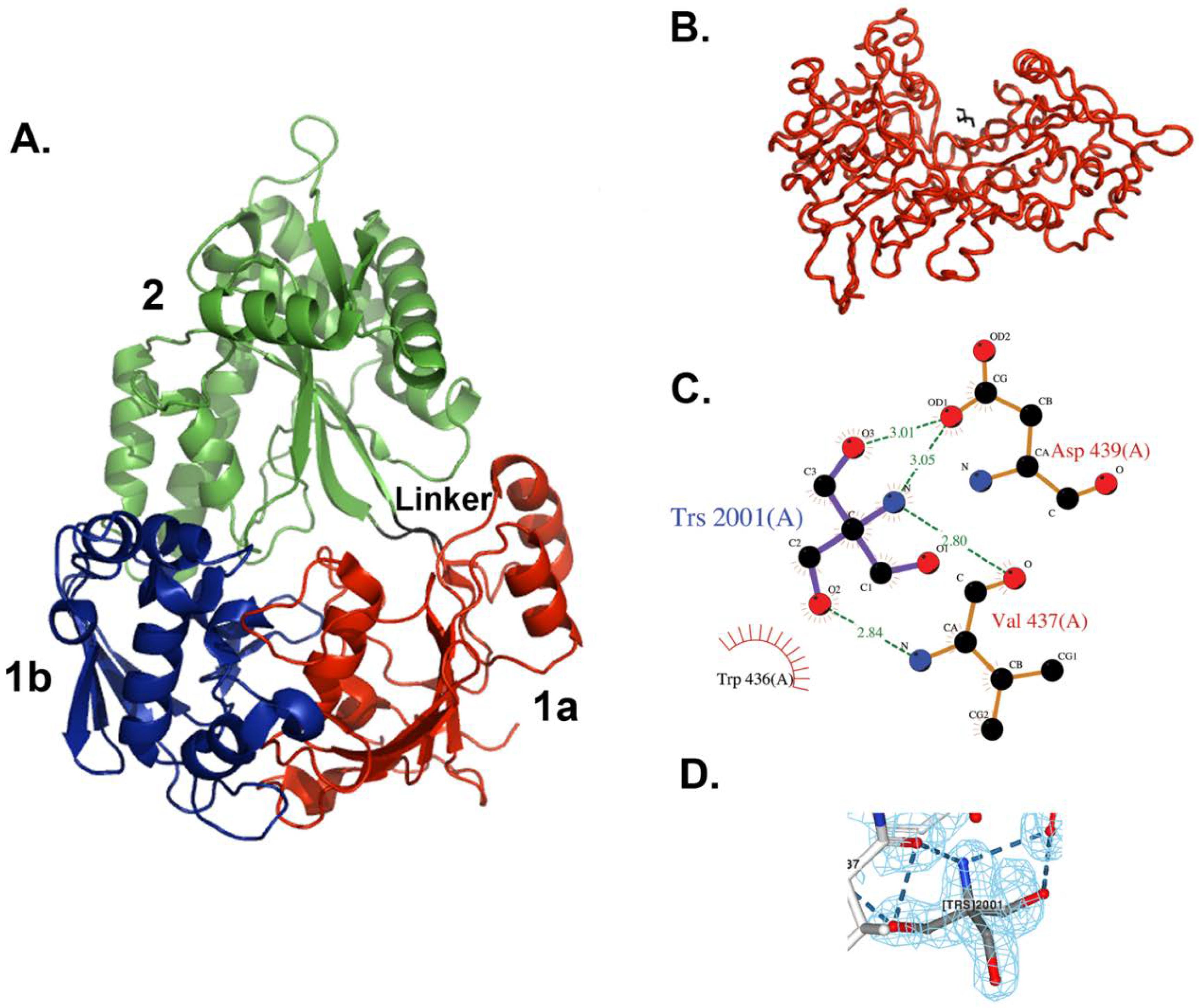
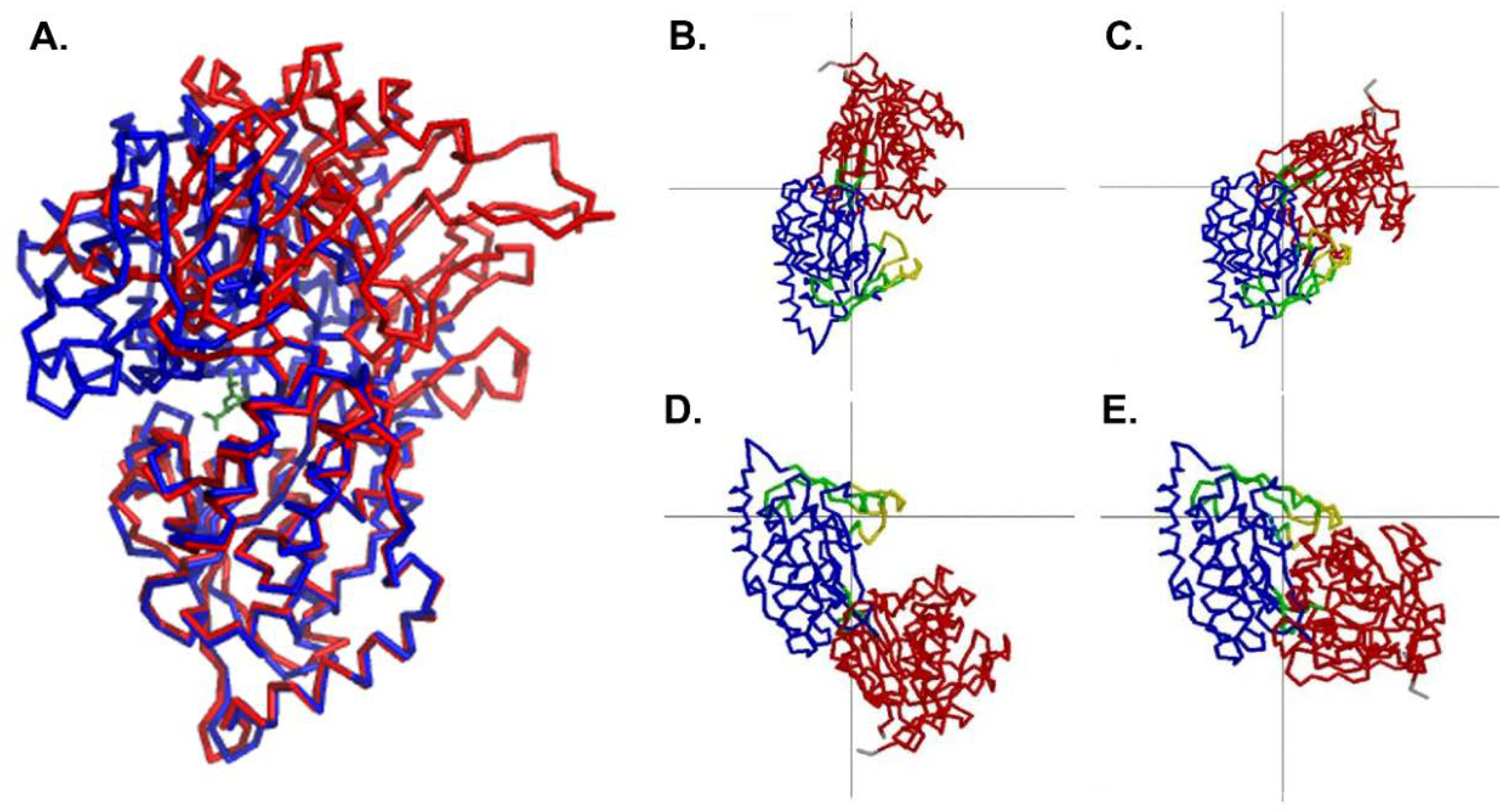
3.2. Overall Structure of MppA
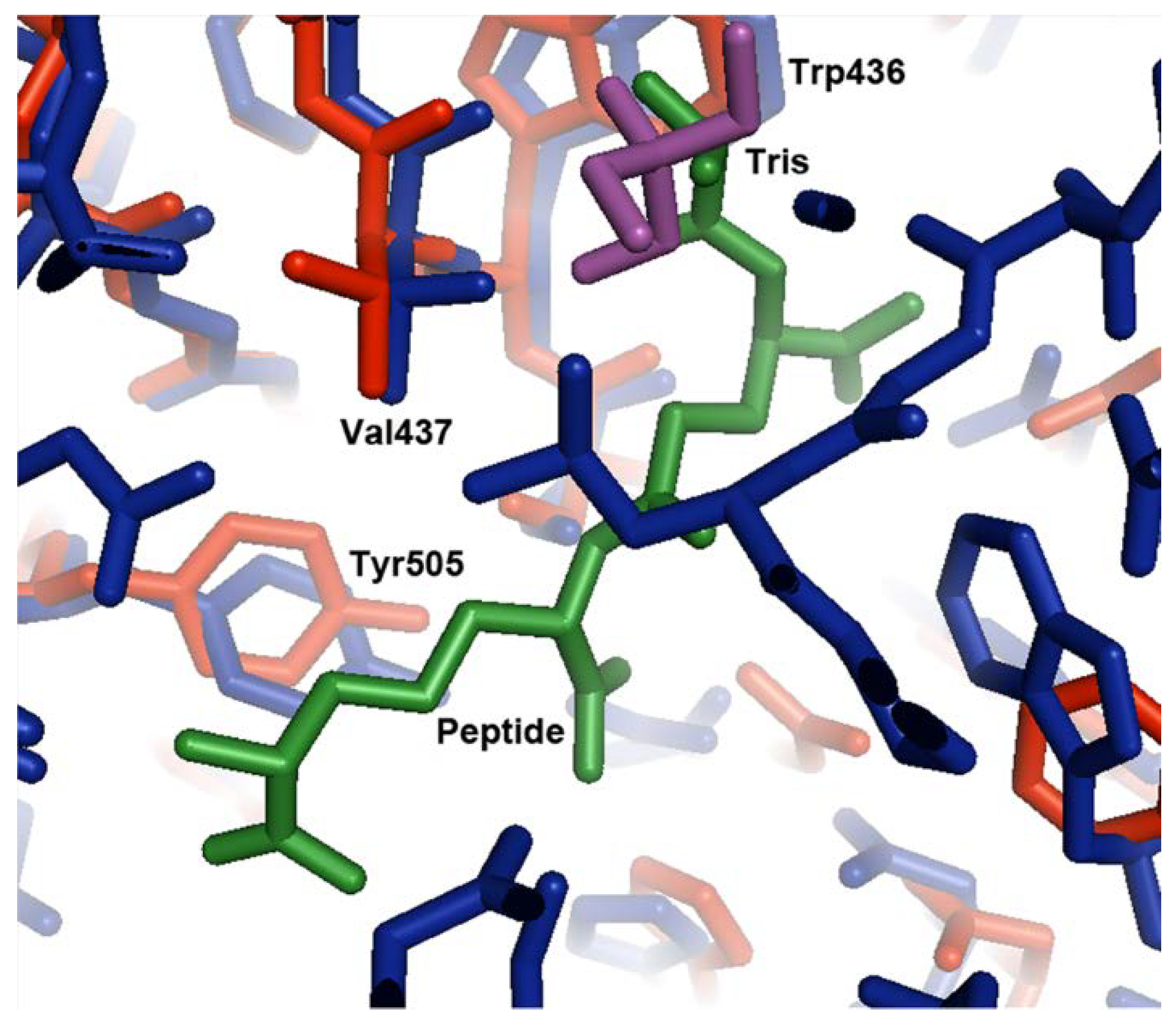
3.3. Tris Binding Site
4. Discussion
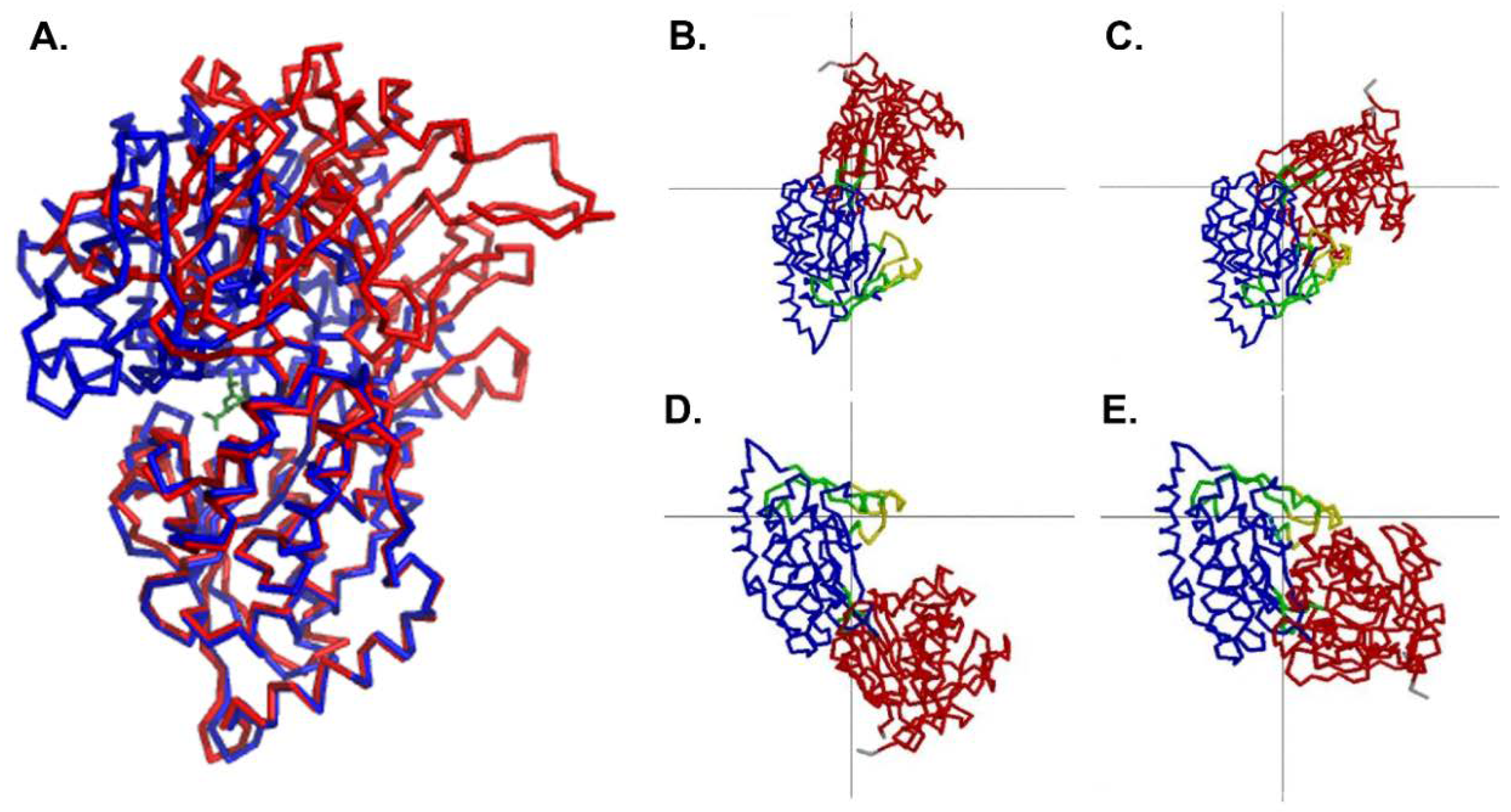
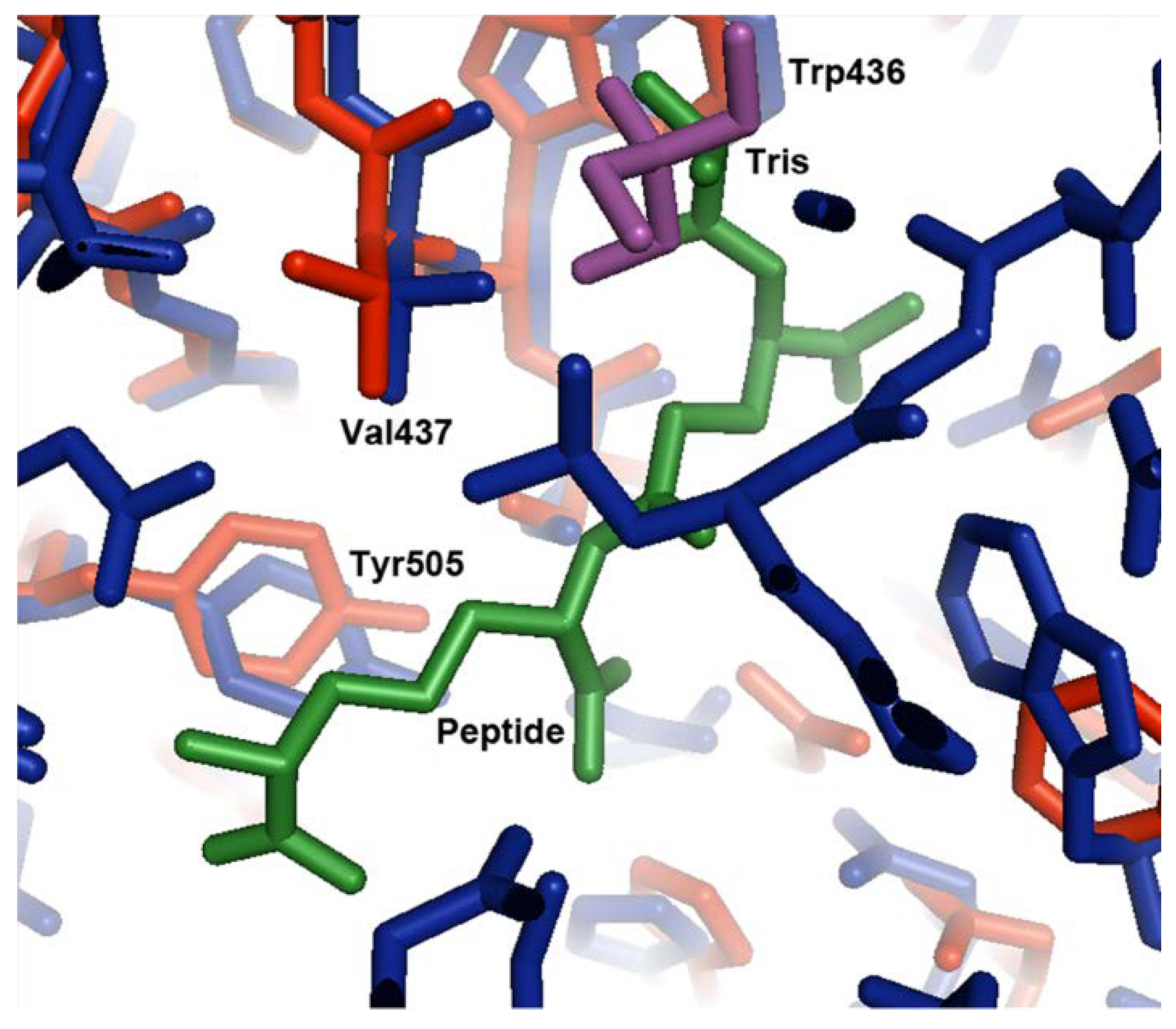
Comparison to the Ligand-Bound Conformation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ames, G.F. Structure and mechanism of bacterial periplasmic transport systems. J. Bioenerg. Biomembr. 1988, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tam, R.; Saier, M.H., Jr. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 1993, 57, 320–346. [Google Scholar] [PubMed]
- Quiocho, F.A.; Wilson, D.K.; Vyas, N.K. Substrate specificity and affinity of a protein modulated by bound water molecules. Nature 1989, 340, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.K.; Vyas, M.N.; Quiocho, F.A. Sugar and signal-transducer binding sites of the Escherichia coli galactose chemoreceptor protein. Science 1988, 242, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Björkman, A.J.; Binnie, R.A.; Cole, L.B.; Zhang, H.; Hermodson, M.A.; Mowbray, S.L. Identical mutations at corresponding positions in two homologous proteins with nonidentical effects. J. Biol. Chem. 1994, 269, 11196–11200. [Google Scholar] [PubMed]
- Sharff, A.J.; Rodseth, L.E.; Quiocho, F.A. Refined 1.8-A structure reveals the mode of binding of beta-cyclodextrin to the maltodextrin binding protein. Biochemistry 1993, 32, 10553–10559. [Google Scholar] [CrossRef] [PubMed]
- He, J.J.; Quiocho, F.A. Dominant role of local dipoles in stabilizing uncompensated charges on a sulfate sequestered in a periplasmic active transport protein. Protein Sci. 1993, 2, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Choudhary, A.; Ledvina, P.S.; Quiocho, F.A. Fine tuning the specificity of the periplasmic phosphate transport receptor. Site-directed mutagenesis, ligand binding, and crystallographic studies. J. Biol. Chem. 1994, 269, 25091–25094. [Google Scholar] [PubMed]
- Yao, N.; Trakhanov, S.; Quiocho, F.A. Refined 1.89-A structure of the histidine-binding protein complexed with histidine and its relationship with many other active transport/chemosensory proteins. Biochemistry 1994, 33, 4769–4779. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.H.; Kang, C.H.; De Bondt, H.; Kim, S.H.; Nikaido, K.; Joshi, A.K.; Ames, G.F. The bacterial periplasmic histidine-binding protein. structure/function analysis of the ligand-binding site and comparison with related proteins. J. Biol. Chem. 1994, 269, 4135–4143. [Google Scholar] [PubMed]
- Sack, J.S.; Trakhanov, S.D.; Tsigannik, I.H.; Quiocho, F.A. Structure of the L-leucine-binding protein refined at 2.4 A resolution and comparison with the Leu/Ile/Val-binding protein structure. J. Mol. Biol. 1989, 206, 193–207. [Google Scholar] [CrossRef]
- Sack, J.S.; Saper, M.A.; Quiocho, F.A. Periplasmic binding protein structure and function. Refined X-ray structures of the leucine/isoleucine/valine-binding protein and its complex with leucine. J. Mol. Biol. 1989, 206, 171–191. [Google Scholar] [CrossRef]
- Oh, B.H.; Pandit, J.; Kang, C.H.; Nikaido, K.; Gokcen, S.; Ames, G.F.; Kim, S.H. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J. Biol. Chem. 1993, 268, 11348–11355. [Google Scholar] [PubMed]
- Tame, J.R.; Murshudov, G.N.; Dodson, E.J.; Neil, T.K.; Dodson, G.G.; Higgins, C.F.; Wilkinson, A.J. The structural basis of sequence-independent peptide binding by OppA protein. Science 1994, 264, 1578–1581, PMID: 8202710. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, S.H.; Tame, J.R.; Dodson, E.J.; Wilkinson, A.J. Peptide binding in OppA, the crystal structures of the periplasmic oligopeptide binding protein in the unliganded form and in complex with lysyllysine. Biochemistry 1997, 36, 9747–9758. [Google Scholar] [CrossRef] [PubMed]
- Dunten, P.; Mowbray, S.L. Crystal structure of the dipeptide binding protein from Escherichia coli involved in active transport and chemotaxis. Protein Sci. 1995, 4, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Nickitenko, A.V.; Trakhanov, S.; Quiocho, F.A. 2 A resolution structure of DppA, a periplasmic dipeptide transport/chemosensory receptor. Biochemistry 1995, 34, 16585–16595. [Google Scholar] [CrossRef] [PubMed]
- Heddle, J.; Scott, D.J.; Unzai, S.; Park, S.Y.; Tame, J.R. Crystal structures of the liganded and unliganded nickel-binding protein NikA from Escherichia coli. J. Biol. Chem. 2003, 278, 50322–50329. [Google Scholar] [CrossRef] [PubMed]
- Sharff, A.J.; Rodseth, L.E.; Spurlino, J.C.; Quiocho, F.A. Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry 1992, 31, 10657–10663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Conway, C.; Rosato, M.; Suh, Y.; Manson, M.D. Maltose chemotaxis involves residues in the N-terminal and C-terminal domains on the same face of maltose-binding protein. J. Biol. Chem. 1992, 267, 22813–22820. [Google Scholar] [PubMed]
- Dwyer, M.A.; Hellinga, H.W. Periplasmic binding proteins: A versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 2004, 14, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Vallée-Bélisle, A.; Plaxco, K.W. Structure-switching biosensors: Inspired by Nature. Curr. Opin. Struct. Biol. 2010, 20, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Engineering periplasmic ligand binding proteins as glucose nanosensors. Nano Rev. 2011, 2, 5743. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Raychaudhuri, D.; Li, H.; Normark, S.; Mengin-Lecreulx, D. MppA, a periplasmic binding protein essential for import of the bacterial cell wall peptide l-alanyl-gamma-d-glutamyl-meso-diaminopimelate. J. Bacteriol. 1998, 180, 1215–1223. [Google Scholar] [PubMed]
- Maqbool, A.; Levdikov, V.M.; Blagova, E.V.; Hervé, M.; Horler, R.S.; Wilkinson, A.J.; Thomas, G.H. Compensating stereochemical changes allow murein tripeptide to be accommodated in a conventional peptide-binding protein. J. Biol. Chem. 2011, 286, 31512–31521. [Google Scholar] [CrossRef] [PubMed]
- Létoffé, S.; Delepelaire, P.; Wandersman, C. The housekeeping dipeptide permease is the Escherichia coli heme transporter and functions with two optional peptide binding proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 12891–12896. [Google Scholar] [CrossRef] [PubMed]
- Huard, C.; Miranda, G.; Redko, Y.; Wessner, F.; Foster, S.J.; Chapot-Chartier, M.P. Analysis of the peptidoglycan hydrolase complement of Lactococcus lactis: Identification of a third N-acetylglucosaminidase, AcmC. Appl. Environ. Microbiol. 2004, 70, 3493–3499. [Google Scholar] [CrossRef] [PubMed]
- Jancarik, J.; Kim, S.-H. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Cryst. 1991, 24, 409–411. [Google Scholar] [CrossRef]
- Teng, T.-Y. Mounting of Crystals for Macromolecular Crystallography in a Free-Standing Thin Film. J. Appl. Cryst. 1990, 23, 387–391. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzym. 1997, 276, 307–326. [Google Scholar]
- Navaza, J. Implementation of molecular replacement in AMoRe. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Computation Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- Brünger, A.T. Assessment of phase accuracy by cross validation: The free R value. Methods and applications. Acta Crystallogr. D Biol. Crystallogr. 1993, 49, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Lamzin, V.S.; Wilson, K.S. Automated refinement of protein models. Acta Crystallogr. D Biol. Crystallogr. 1993, 49, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [PubMed]
- Jones, T.A.; Zou, J.Y.; Cowan, S.W.; Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 1991, 47, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Smart, O.S.; Horský, V.; Gore, S.; Svobodová Vařeková, R.; Bendová, V.; Kleywegt, G.J.; Velankar, S. Worldwide Protein Data Bank validation information: Usage and trends Acta Cryst. D 2018, 74, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, M.M.; Kovermann, M.; Löw, C.; Balbach, J.; Permentier, H.P.; Fusetti, F.; de Gier, J.W.; Slotboom, D.J.; Berntsson, R.P. Escherichia coli peptide binding protein OppA has a preference for positively charged peptides. J. Mol. Biol. 2011, 414, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. Web-based molecular graphics for large complexes. In Proceedings of the ACM 21st International Conference on Web3D Technology (Web3D ‘16), Anaheim, CA, USA, 22–24 July 2016; pp. 185–186. [Google Scholar] [CrossRef]
- Rose, A.S.; Hildebrand, P.W. NGL Viewer: A web application for molecular visualization. Nucleic Acids Res. 2015, 43, W576–W579. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.; Berendsen, H.J. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 1998, 30, 144–154. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| A. Data statistics | ||
| Resolution | 50–1.5 Å | |
| Space group | P21 | |
| Unit cell dimensions | a = 74.97, b = 76.81, c = 89.57, β = 91.58 | |
| Number of unique reflections | 161,107 | |
| Completeness | 99.6% | |
| I/σ(I) | 6.82 | |
| B. Refinement | ||
| Resolution range (refinement) | 35–1.5 Å | |
| Crystallographic R-factor a (Rwork) | 0.18 | |
| Free R-factor b (Rfree) | 0.205 | |
| Number per asymmetric unit | ||
| MppA molecules | 2 | |
| Tris molecules | 1 | |
| Protein atoms | 8150 | |
| Solvent atoms | 1647 | |
| Deviation from standard geometry (Gfactors c) | ||
| Dihedral angles | ||
| Phi-psi distribution | −0.10 | |
| Chi1-chi2 distribution | 0.12 | |
| Chi1 only | 0.19 | |
| Chi3 and Chi4 | 0.59 | |
| Omega | 0.54 | |
| Main-chain covalent forces | ||
| Main-chain bond lengths | 0.68 | |
| Main-chain bond angles | 0.45 | |
| Average B value (Å2) | ||
| All atoms | 16.1 | |
| of protein | 13.7 | |
| of Tris ligand | 26.1 | |
| of waters | 28.0 | |
| Ramachandran plot statistics from ProCheck | ||
| Number of residues Percent | ||
| Most favored regions | 817 | 90.5 |
| Additional allowed regions | 84 | 9.3 |
| Generously allowed regions | 1 | 0.1 |
| Disallowed regions | 1 | 0.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatt, F.; Patel, V.; Jeffery, C.J. Open Conformation of the Escherichia coli Periplasmic Murein Tripeptide Binding Protein, MppA, at High Resolution. Biology 2018, 7, 30. https://doi.org/10.3390/biology7020030
Bhatt F, Patel V, Jeffery CJ. Open Conformation of the Escherichia coli Periplasmic Murein Tripeptide Binding Protein, MppA, at High Resolution. Biology. 2018; 7(2):30. https://doi.org/10.3390/biology7020030
Chicago/Turabian StyleBhatt, Forum, Vishal Patel, and Constance J. Jeffery. 2018. "Open Conformation of the Escherichia coli Periplasmic Murein Tripeptide Binding Protein, MppA, at High Resolution" Biology 7, no. 2: 30. https://doi.org/10.3390/biology7020030
APA StyleBhatt, F., Patel, V., & Jeffery, C. J. (2018). Open Conformation of the Escherichia coli Periplasmic Murein Tripeptide Binding Protein, MppA, at High Resolution. Biology, 7(2), 30. https://doi.org/10.3390/biology7020030