Age-Dependence and Aging-Dependence: Neuronal Loss and Lifespan in a C. elegans Model of Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Parkinson’s Disease Model
2.2. General Methods and Strains
2.3. Construction of Strains
2.4. Lifespan Assays
2.5. Microscopy
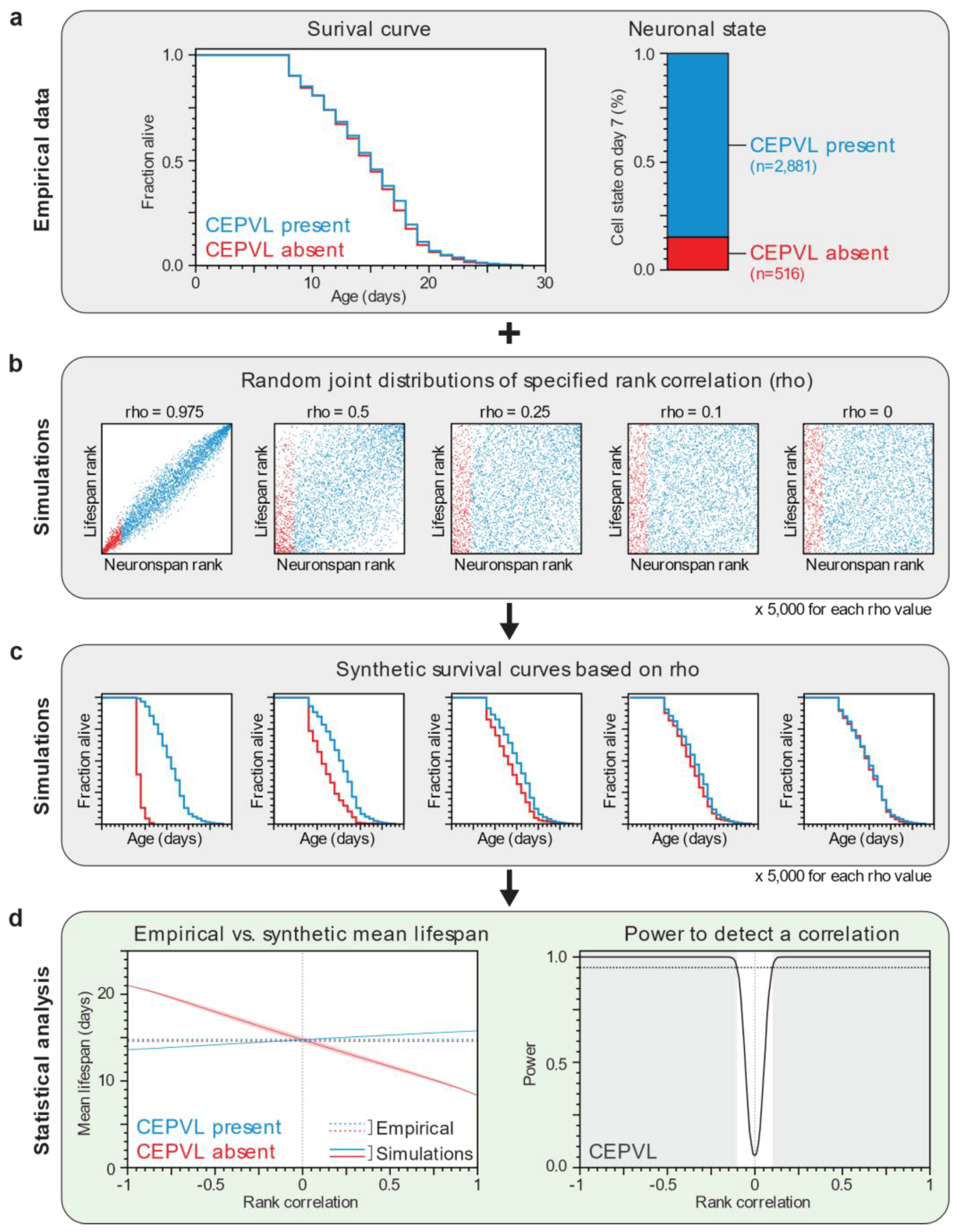
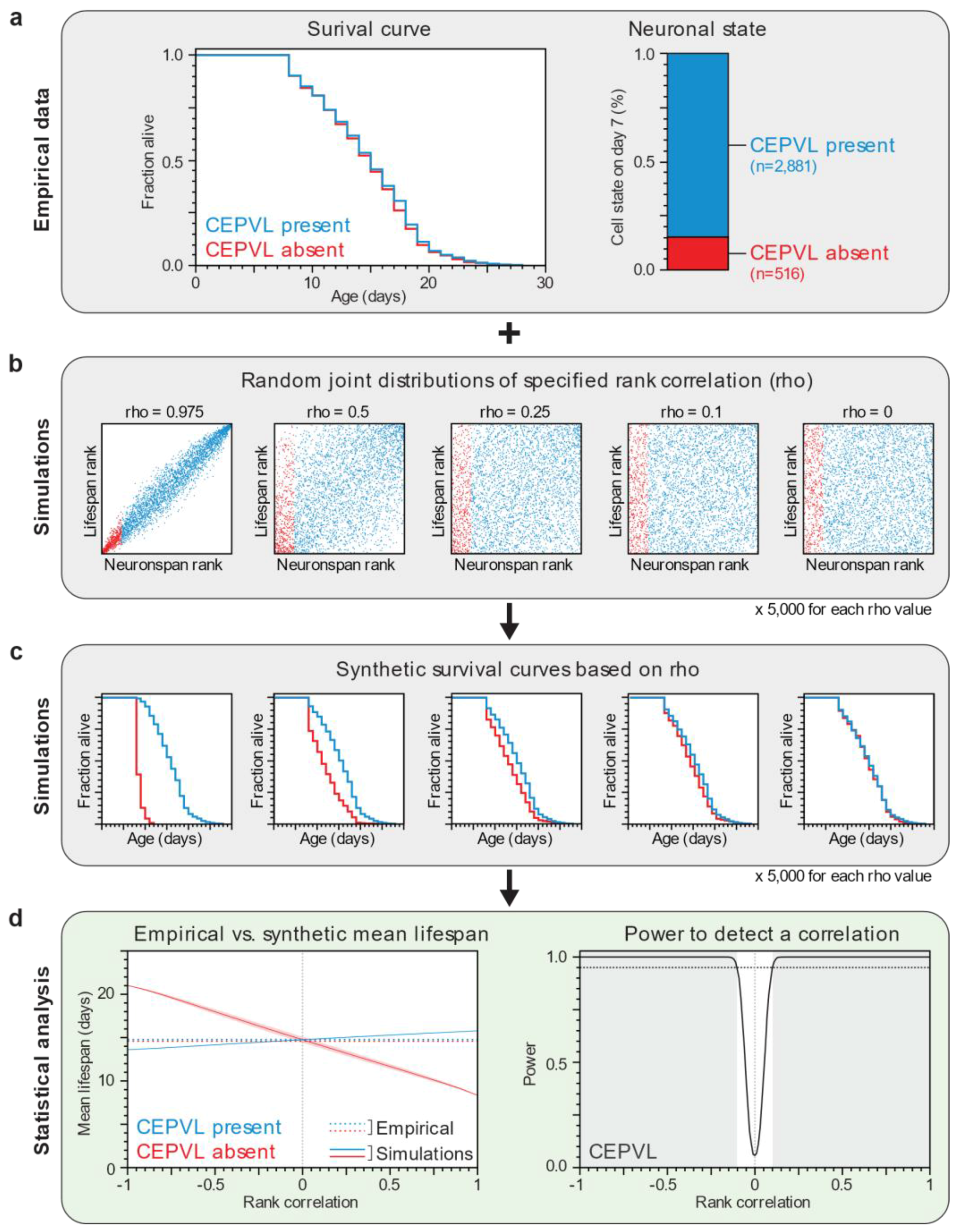
2.6. Computational Power Analysis
2.7. Statistical Analysis
3. Results
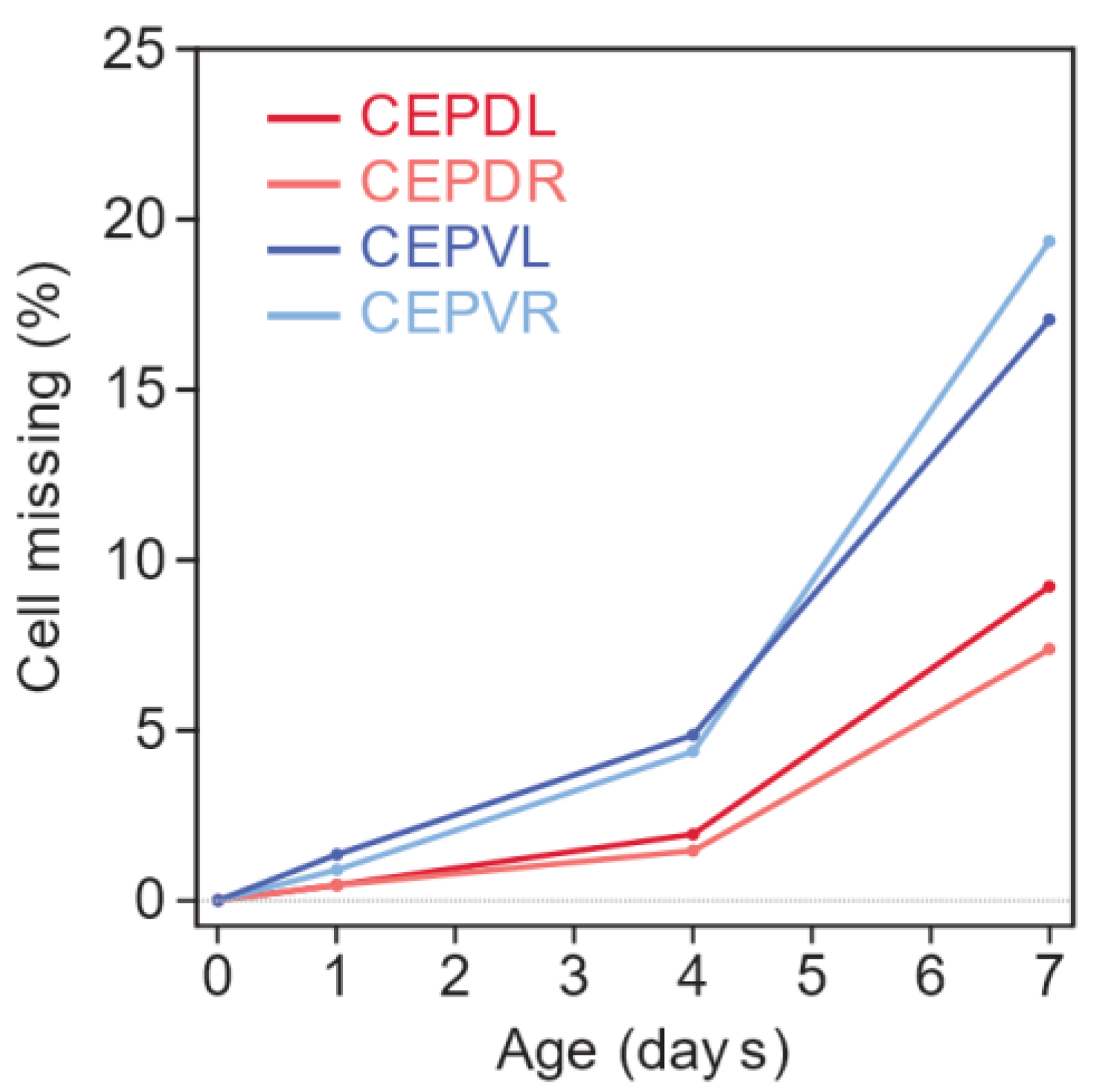
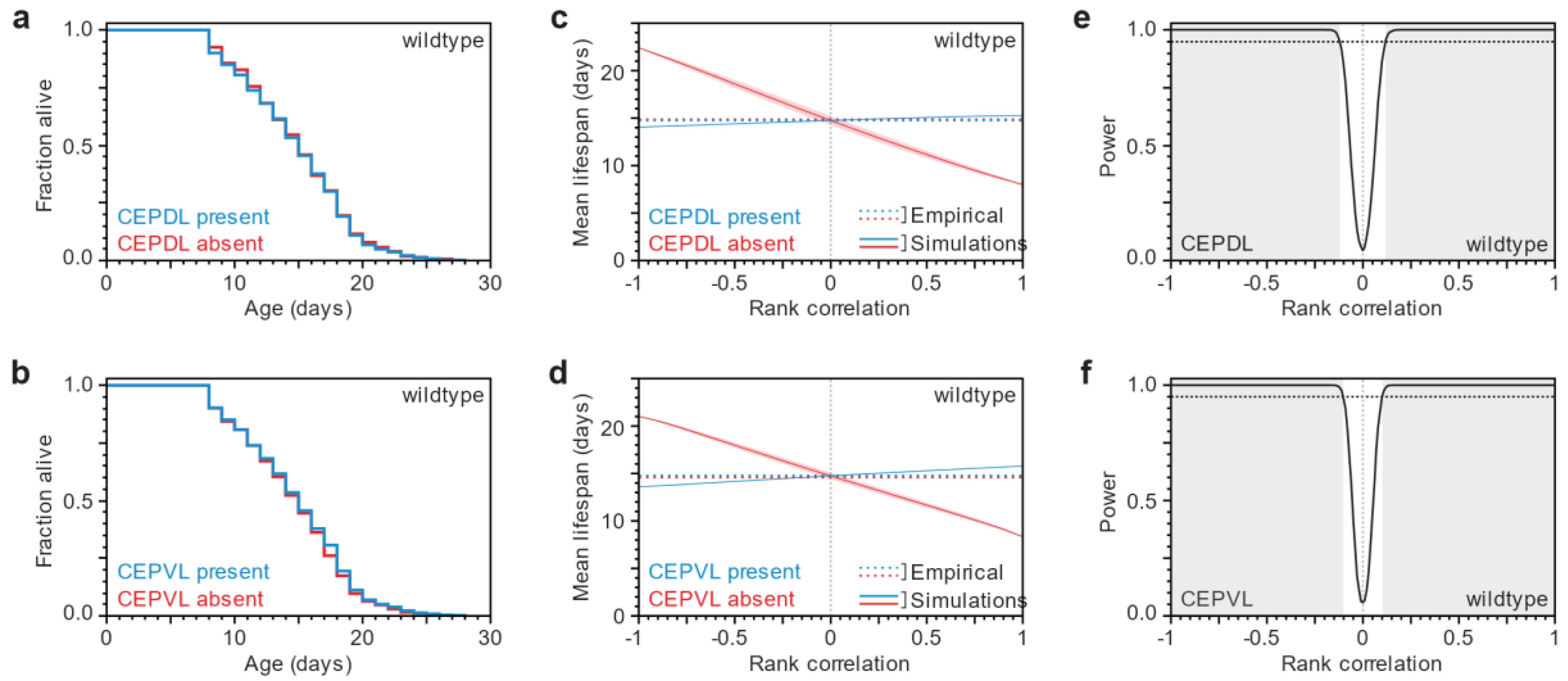
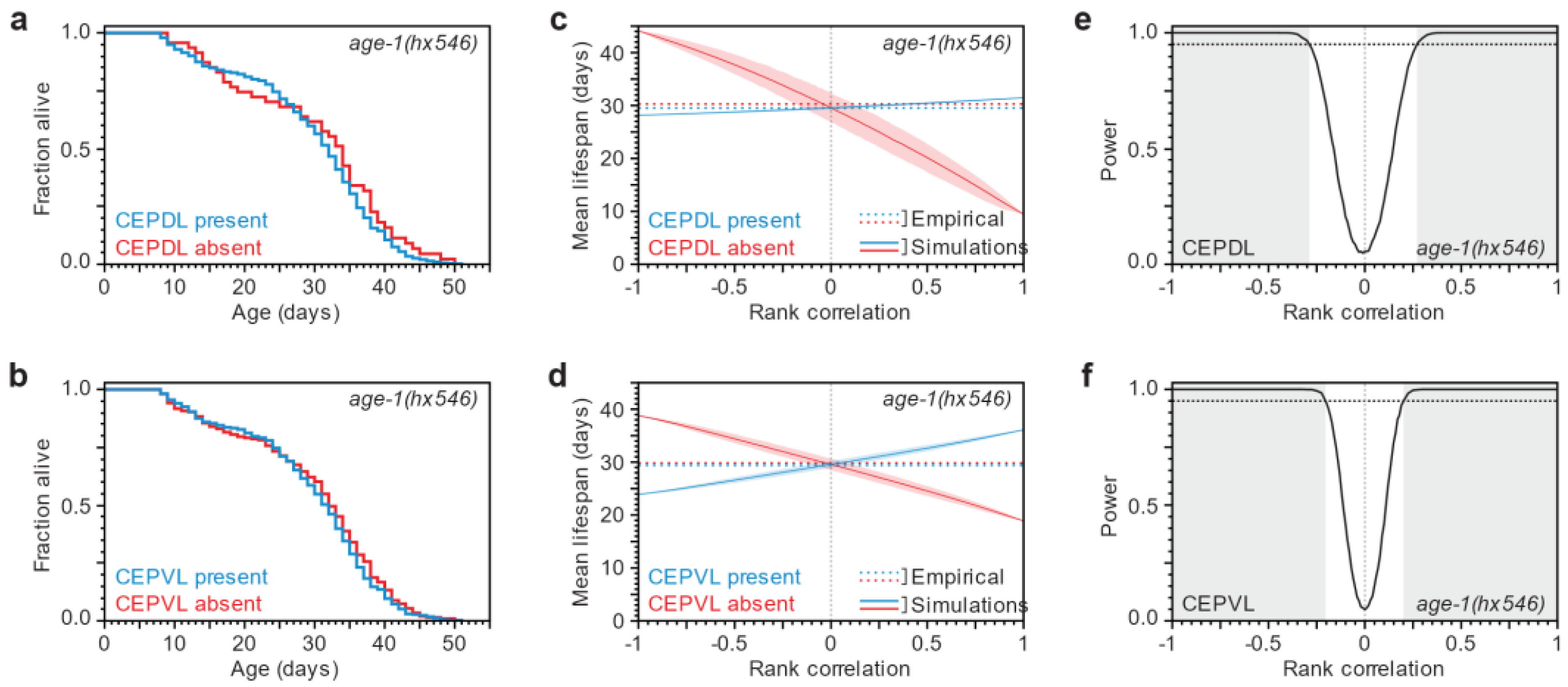
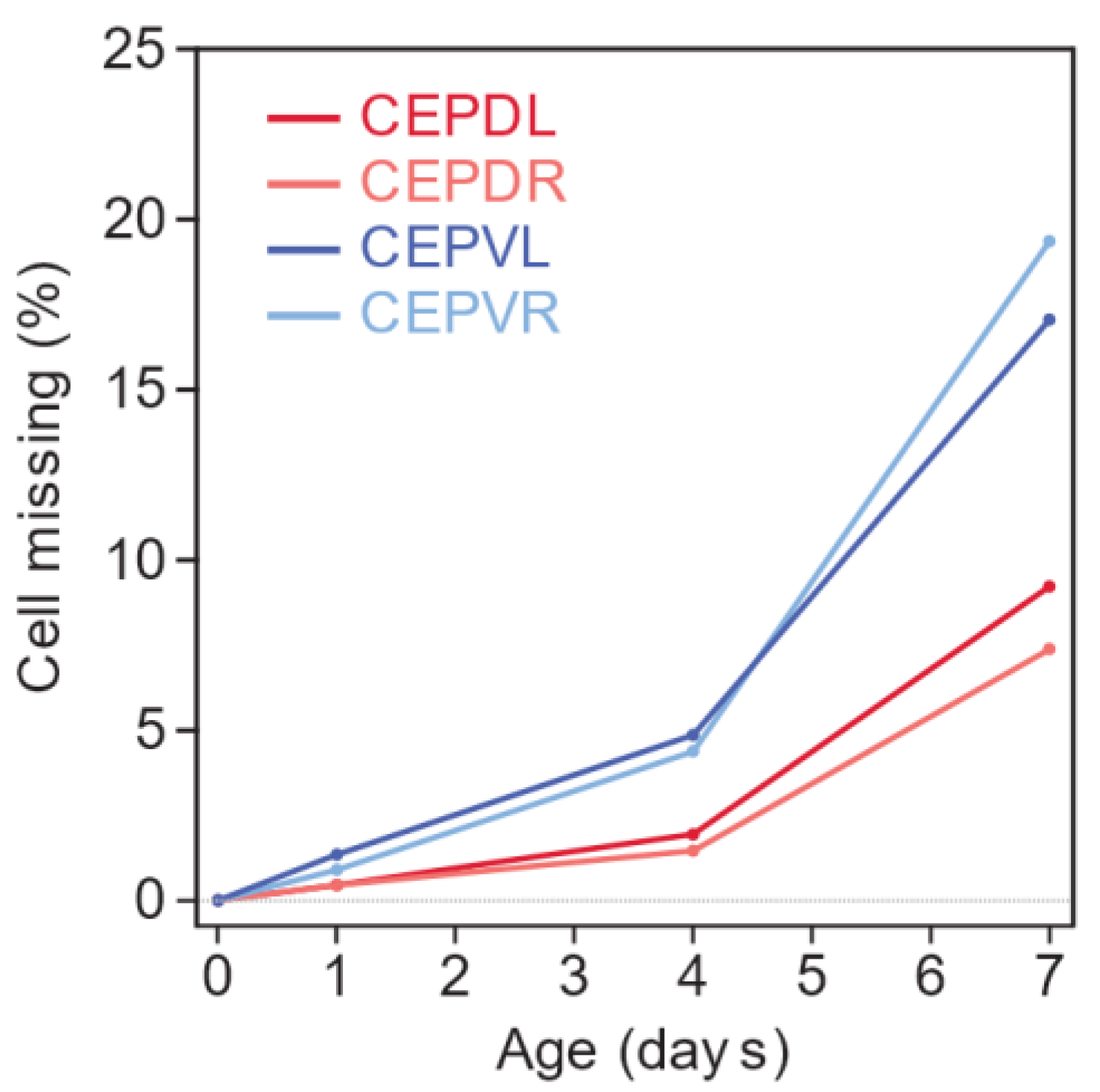
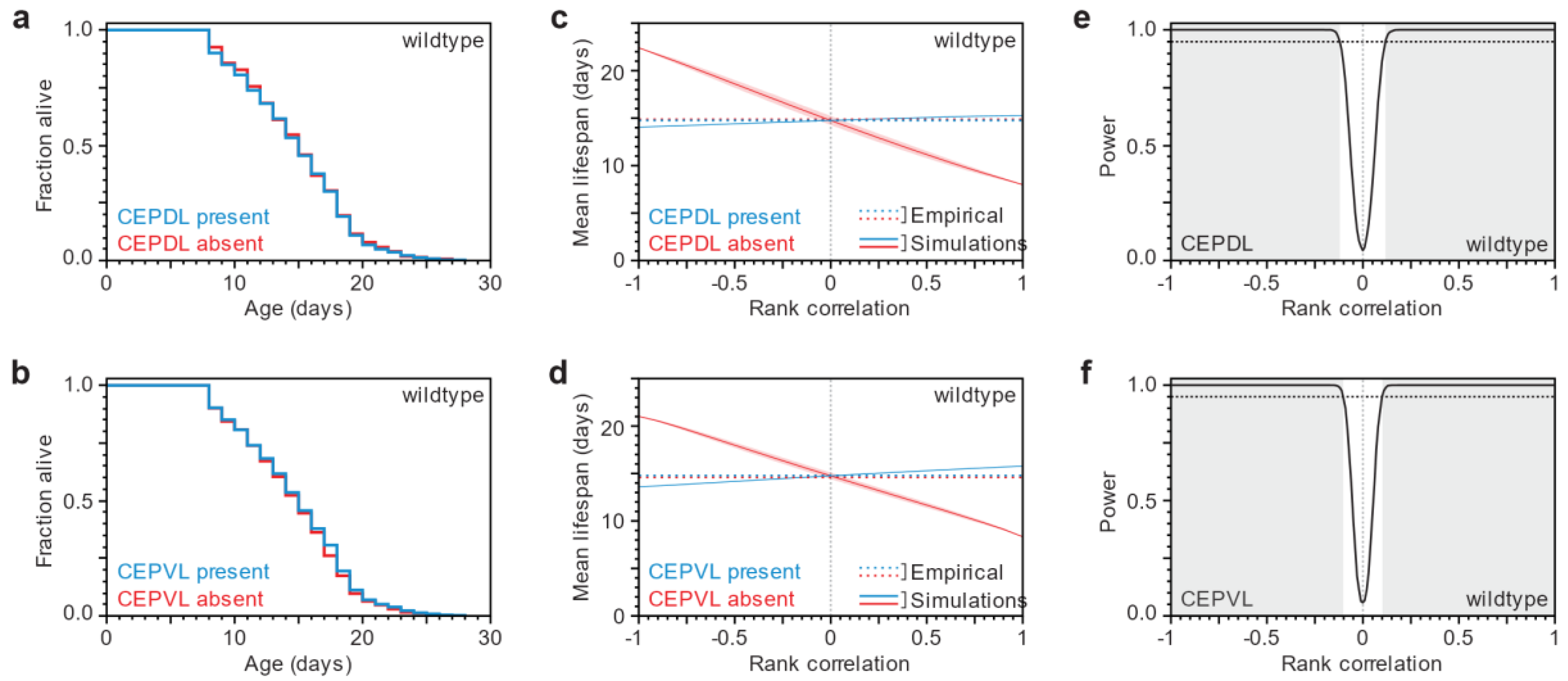
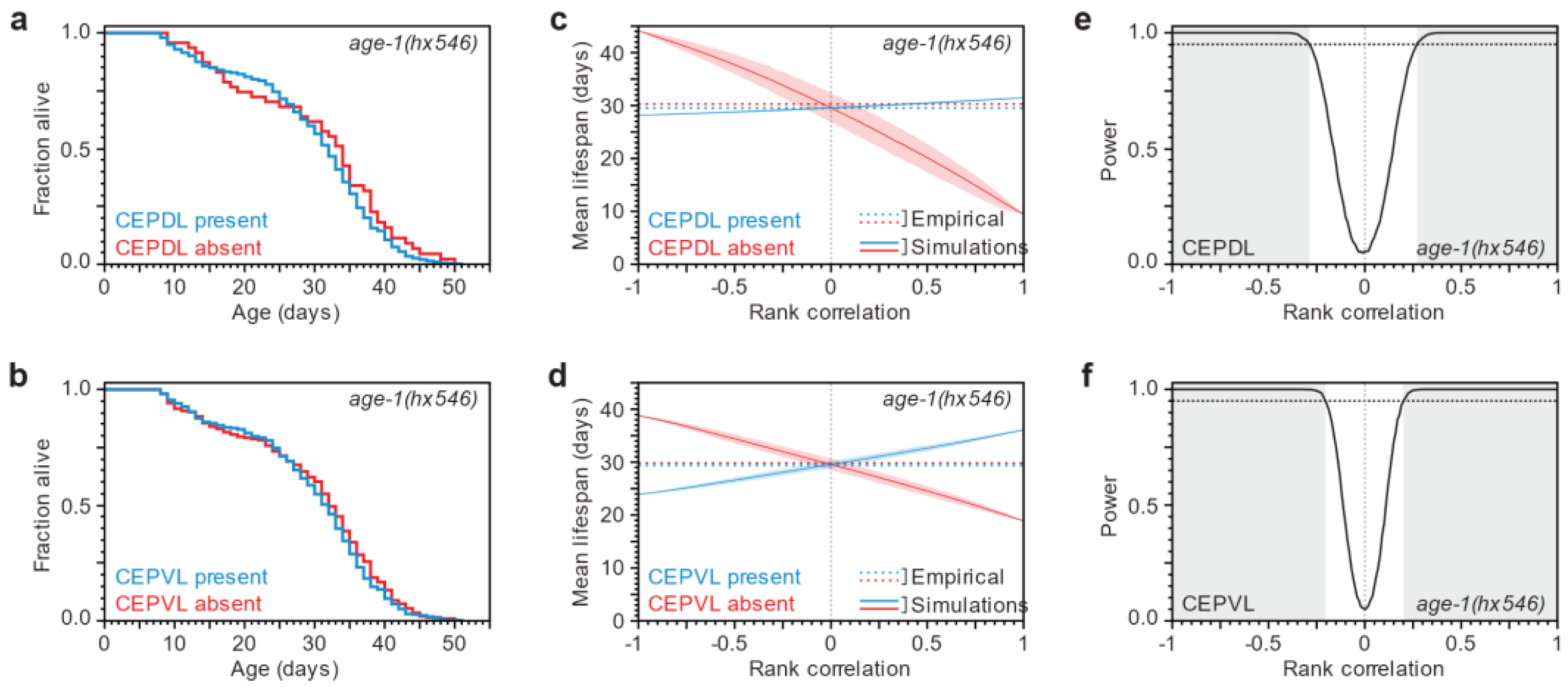
3.1. Lifespan and Neurodegeneration are the Outcomes of Independent Processes
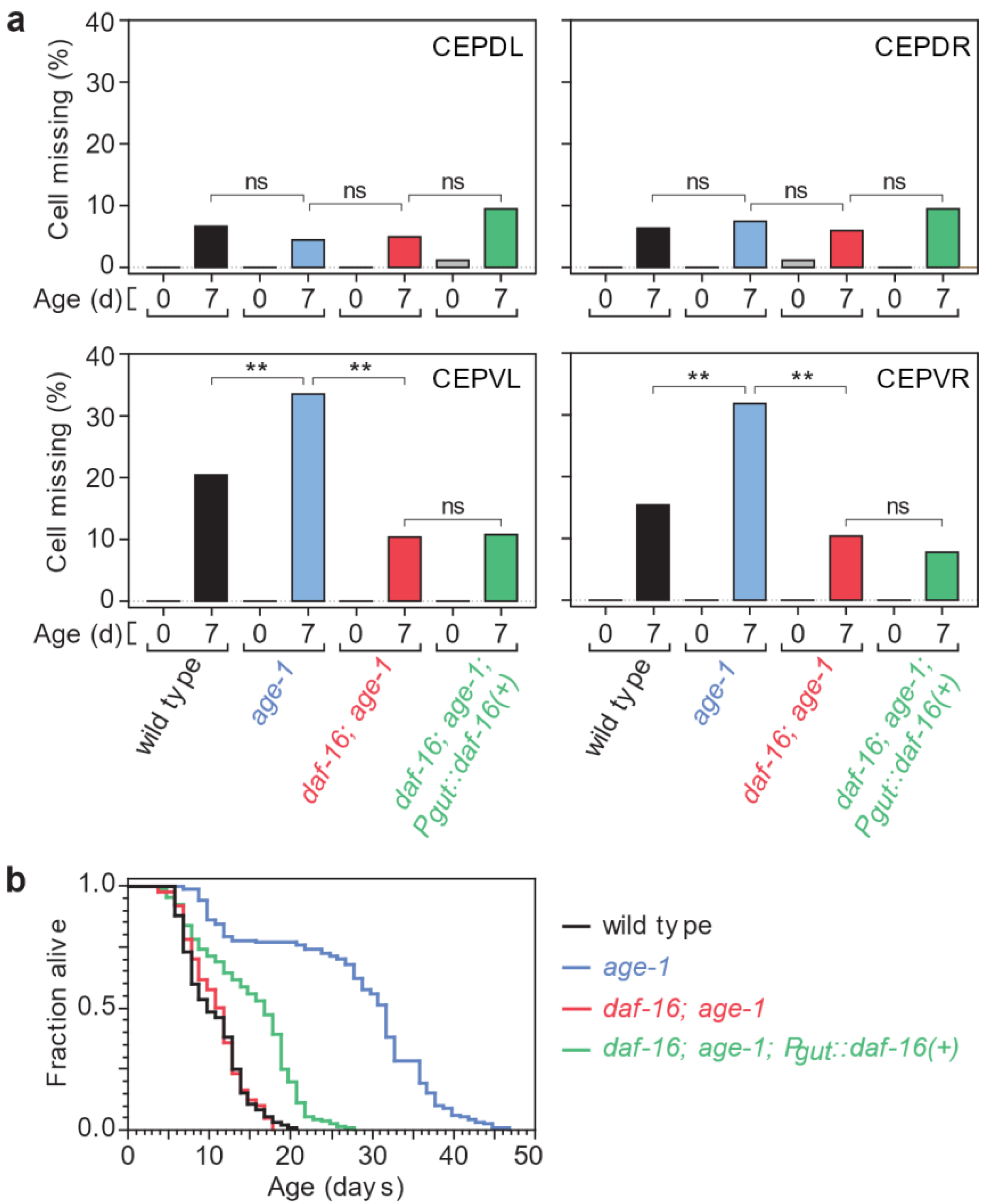
3.2. Tissue-Specific Insulin/IGF1 Signaling Has Neuron-Specific Effects on Age-Dependent Dopaminergic Loss
3.3. After a Lifespan-Extending Intervention, Lifespan and Neurodegeneration Are Still Independent
4. Discussion
4.1. Age-Dependence of Neuronal Loss in a C. elegans Parkinson’s Disease Model
4.2. Aging-Independence of Neuronal Loss in a C. elegans Parkinson’s Disease Model
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heron, M. Deaths: Leading causes for 2010. Natl. Vital Stat. Rep. 2013, 62, 1–96. [Google Scholar] [PubMed]
- Driver, J.A.; Logroscino, G.; Gaziano, J.M.; Kurth, T. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology 2009, 72, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Stern, M.B.; Sethi, K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology 2009, 72, S1–S136. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W. The natural history of Parkinson’s disease. J. Neurol. 2006, 253, VII2–VII6. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Gelwix, C.C.; Caldwell, K.A.; Caldwell, G.A. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 3801–3812. [Google Scholar] [CrossRef] [PubMed]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAI screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes park9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Sawin, E.R.; Ranganathan, R.; Horvitz, H.R. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 2000, 26, 619–631. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R.A. C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.B.; Johnson, T.E. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 1988, 118, 75–86. [Google Scholar] [PubMed]
- Clancy, D.J.; Gems, D.; Harshman, L.G.; Oldham, S.; Stocker, H.; Hafen, E.; Leevers, S.J.; Partridge, L. Extension of life-span by loss of chico, a drosophila insulin receptor substrate protein. Science 2001, 292, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Dorman, J.B.; Rodan, A.; Kenyon, C. Daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 1997, 278, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The fork head transcription factor daf-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Giannakou, M.E.; Goss, M.; Junger, M.A.; Hafen, E.; Leevers, S.J.; Partridge, L. Long-lived drosophila with overexpressed dFOXO in adult fat body. Science 2004, 305. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, D.S.; Gersham, B.; Tu, M.P.; Palmer, M.; Tatar, M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 2004, 429, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [PubMed]
- Flachsbart, F.; Caliebe, A.; Kleindorp, R.; Blanche, H.; von Eller-Eberstein, H.; Nikolaus, S.; Schreiber, S.; Nebel, A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. USA 2009, 106, 2700–2705. [Google Scholar] [CrossRef] [PubMed]
- Apfeld, J.; Kenyon, C. Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell 1998, 95, 199–210. [Google Scholar] [CrossRef]
- Libina, N.; Berman, J.R.; Kenyon, C. Tissue-specific activities of C. elegans daf-16 in the regulation of lifespan. Cell 2003, 115, 489–502. [Google Scholar] [CrossRef]
- Iser, W.B.; Gami, M.S.; Wolkow, C.A. Insulin signaling in Caenorhabditis elegans regulates both endocrine-like and cell-autonomous outputs. Dev. Biol. 2007, 303, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Alic, N.; Tullet, J.M.; Niccoli, T.; Broughton, S.; Hoddinott, M.P.; Slack, C.; Gems, D.; Partridge, L. Cell-nonautonomous effects of dFOXO/DAF-16 in aging. Cell Rep. 2014, 6, 608–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappeler, L.; De Magalhaes Filho, C.; Dupont, J.; Leneuve, P.; Cervera, P.; Perin, L.; Loudes, C.; Blaise, A.; Klein, R.; Epelbaum, J.; et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008, 6, e254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Nass, R.; Hall, D.H.; Miller, D.M., 3rd; Blakely, R.D. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 3264–3269. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Yuan, L. Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res. Rev. 2014, 15C, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302. [Google Scholar] [CrossRef] [PubMed]
- Apfeld, J.; O’Connor, G.; McDonagh, T.; DiStefano, P.S.; Curtis, R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004, 18, 3004–3009. [Google Scholar] [CrossRef] [PubMed]
- Herndon, L.A.; Schmeissner, P.J.; Dudaronek, J.M.; Brown, P.A.; Listner, K.M.; Sakano, Y.; Paupard, M.C.; Hall, D.H.; Driscoll, M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 2002, 419, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.L.; Yan, X.; Hamamichi, S.; Ajjuri, R.R.; Mazzulli, J.R.; Zhang, M.W.; Daigle, J.G.; Zhang, S.; Borom, A.R.; Roberts, L.R.; et al. The glycolytic enzyme, GPI, is a functionally conserved modifier of dopaminergic neurodegeneration in Parkinson’s models. Cell Metab. 2014, 20, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.F.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Senchuk, M.M.; Van Raamsdonk, J.M. Delaying aging is neuroprotective in Parkinson’s disease: A genetic analysis in C. elegans models. NPJ Parkinson’s Dis. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Alavez, S.; Lithgow, G.J. Pharmacological maintenance of protein homeostasis could postpone age-related disease. Aging Cell 2012, 11, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.N.; Miller, R.A.; Perry, D.; Carnes, B.A.; Williams, T.F.; Cassel, C.; Brody, J.; Bernard, M.A.; Partridge, L.; Kirkwood, T.; et al. New model of health promotion and disease prevention for the 21st century. BMJ 2008, 337. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Validation of anti-aging drugs by treating age-related diseases. Aging 2009, 1, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Radical medicine: Treating ageing to cure disease. Nat. Rev. Mol. Cell Biol. 2005, 6, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Sutton, A.J.; Sundermeyer, M.L.; Albert, P.S.; King, K.V.; Edgley, M.L.; Larsen, P.L.; Riddle, D.L. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 1998, 150, 129–155. [Google Scholar] [PubMed]
- Patel, D.S.; Garza-Garcia, A.; Nanji, M.; McElwee, J.J.; Ackerman, D.; Driscoll, P.C.; Gems, D. Clustering of genetically defined allele classes in the Caenorhabditis elegans daf-2 Insulin/IGF-1 receptor. Genetics 2008, 178, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.Z.; Tissenbaum, H.A.; Ruvkun, G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 1996, 382, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Slack, C.; Giannakou, M.E.; Foley, A.; Goss, M.; Partridge, L. dFOXO-independent effects of reduced insulin-like signaling in Drosophila. Aging Cell 2011, 10, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Tatar, M. Insulin receptor substrate chico acts with the transcription factor FOXO to extend Drosophila lifespan. Aging Cell 2011, 10, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Judy, M.; Lee, S.J.; Kenyon, C. Direct and indirect gene regulation by a life-extending FOXO protein in C. elegans: Roles for gata factors and lipid gene regulators. Cell Metab. 2013, 17, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Demontis, F.; Perrimon, N. FOXO/4E-BP signaling in drosophila muscles regulates organism-wide proteostasis during aging. Cell 2010, 143, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.T.; Lee, S.J.; Kenyon, C. Tissue entrainment by feedback regulation of insulin gene expression in the endoderm of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 19046–19050. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.B.; Walradt, T.; Gardner, K.E.; Hubbert, A.; Reinke, V.; Hammarlund, M. Insulin/IGF1 signaling inhibits age-dependent axon regeneration. Neuron 2014, 81, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Adachi, H.; Fujiwara, Y.; Ishii, N. Protein carbonyl accumulation in Aging Dauer Formation—Defective (daf) mutants of Caenorhabditis elegans. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1999, 54, B47–B51. [Google Scholar] [CrossRef]
- Gerstbrein, B.; Stamatas, G.; Kollias, N.; Driscoll, M. In Vivo spectrofluorimetry reveals endogenous biomarkers that report healthspan and dietary restriction in Caenorhabditis elegans. Aging Cell 2005, 4, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Garigan, D.; Hsu, A.L.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Genetic analysis of tissue aging in Caenorhabditis elegans: A role for heat-shock factor and bacterial proliferation. Genetics 2002, 161, 1101–1112. [Google Scholar] [PubMed]
- Duhon, S.A.; Johnson, T.E. Movement as an index of vitality: Comparing wild type and the age-1 mutant of Caenorhabditis elegans. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1995, 50, B254–B261. [Google Scholar] [CrossRef]
- Huang, C.; Xiong, C.; Kornfeld, K. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2004, 101, 8084–8089. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, A.L.; Ashraf, J.M.; Corces-Zimmerman, M.R.; Landis, J.N.; Murphy, C.T. Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol. 2010, 8, e1000372. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J.H.; Kim, S.; DiLoreto, R.; Shi, C.; Lee, S.J.V.; Murphy, C.T.; Nam, H.G. C. elegans maximum velocity correlates with healthspan and is maintained in worms with an insulin receptor mutation. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.L.; Peng, C.Y.; Chen, C.H.; McIntire, S. Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 9274–9279. [Google Scholar] [CrossRef] [PubMed]
- Tank, E.M.; Rodgers, K.E.; Kenyon, C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 9279–9288. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.L.; Melentijevic, I.; Shah, L.; Bhatia, A.; Lu, K.; Talwar, A.; Naji, H.; Ibanez-Ventoso, C.; Ghose, P.; Jevince, A.; et al. Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 8778–8790. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apfeld, J.; Fontana, W. Age-Dependence and Aging-Dependence: Neuronal Loss and Lifespan in a C. elegans Model of Parkinson’s Disease. Biology 2018, 7, 1. https://doi.org/10.3390/biology7010001
Apfeld J, Fontana W. Age-Dependence and Aging-Dependence: Neuronal Loss and Lifespan in a C. elegans Model of Parkinson’s Disease. Biology. 2018; 7(1):1. https://doi.org/10.3390/biology7010001
Chicago/Turabian StyleApfeld, Javier, and Walter Fontana. 2018. "Age-Dependence and Aging-Dependence: Neuronal Loss and Lifespan in a C. elegans Model of Parkinson’s Disease" Biology 7, no. 1: 1. https://doi.org/10.3390/biology7010001
APA StyleApfeld, J., & Fontana, W. (2018). Age-Dependence and Aging-Dependence: Neuronal Loss and Lifespan in a C. elegans Model of Parkinson’s Disease. Biology, 7(1), 1. https://doi.org/10.3390/biology7010001