
SNP Discovery Using a Pangenome: Has the Single Reference Approach Become Obsolete?


{kind=link}
{kind=link}
Abstract
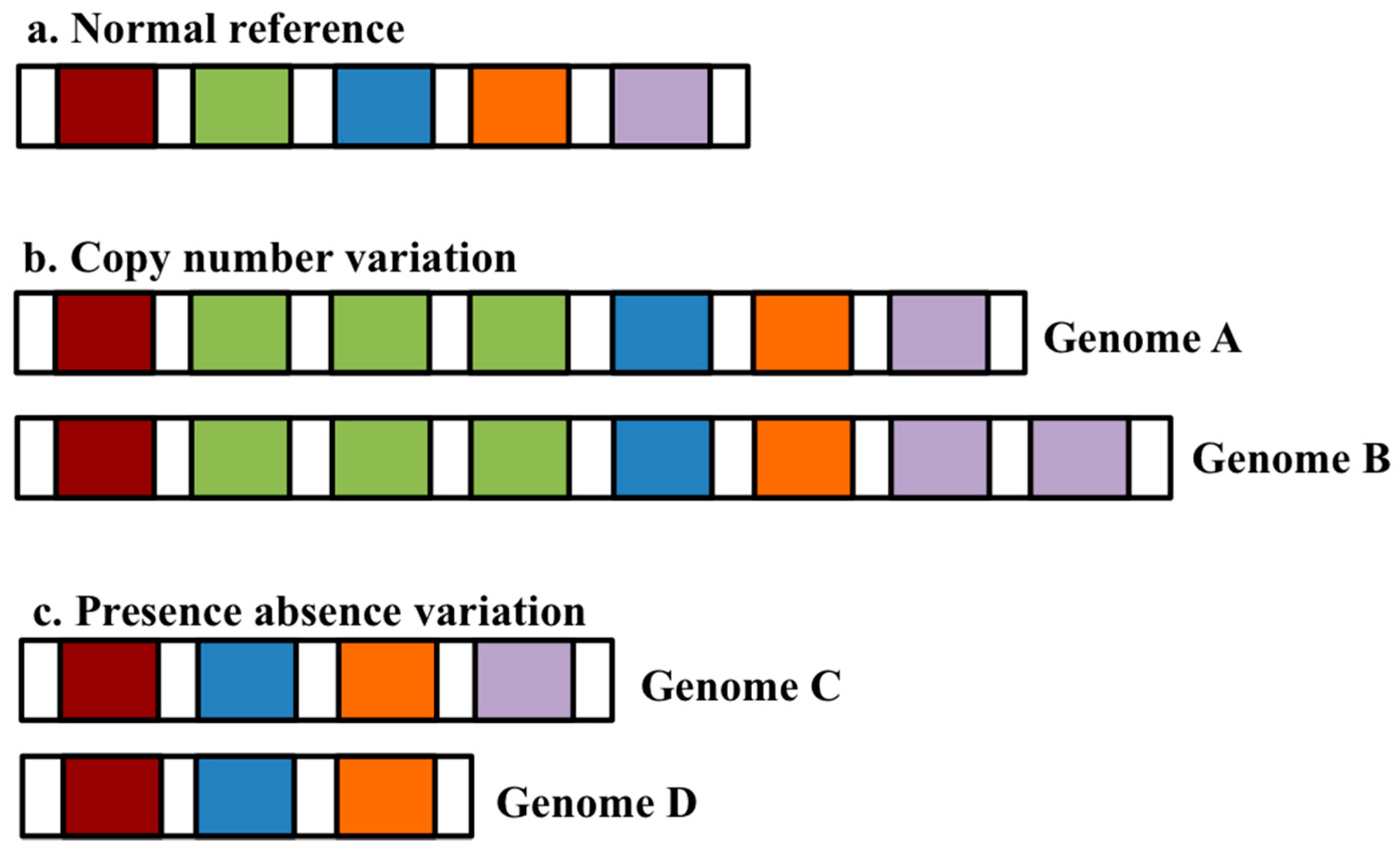
:1. The Pangenome Concept
2. Single Nucleotide Polymorphisms
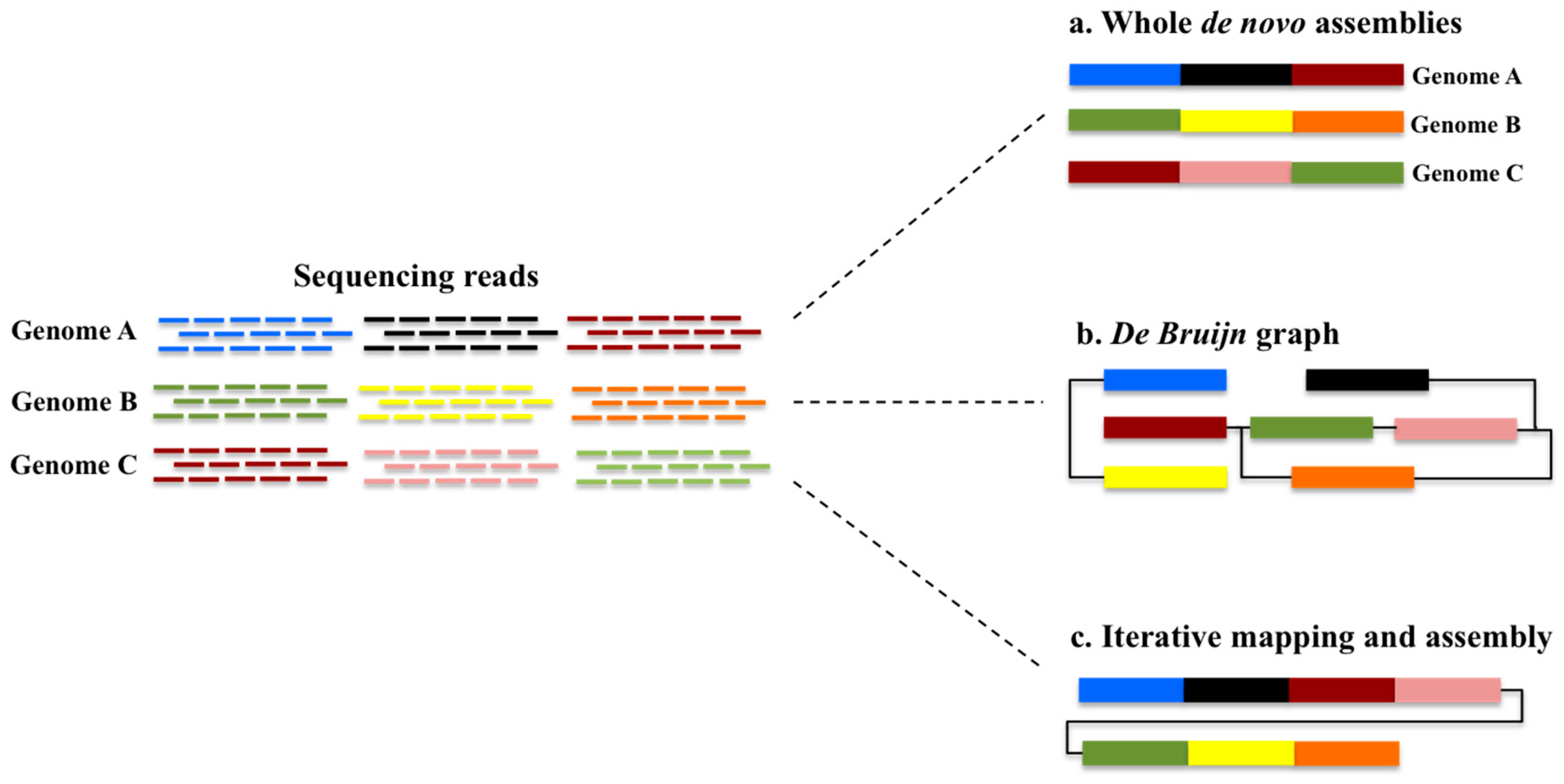
3. How Can the Availability of a Pangenome Increase the Efficiency of SNP Discovery?
4. Applications of Discovered SNPs in Relation to Pangenomes
5. Challenges and Future Directions
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saxena, R.K.; Edwards, D.; Varshney, R.K. Structural variations in plant genomes. Brief. Funct. Genom. 2014, 13, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Structural variation: The genome’s hidden architecture. Nat. Methods 2012, 9, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Baddam, R.; Kumar, N.; Shaik, S.; Lankapalli, A.K.; Ahmed, N. Genome dynamics and evolution of Salmonella typhi strains from the typhoid-endemic zones. Sci. Rep. 2014, 4, 7457. [Google Scholar] [CrossRef] [PubMed]
- Donati, C.; Hiller, N.L.; Tettelin, H.; Muzzi, A.; Croucher, N.J.; Angiuoli, S.V.; Oggioni, M.; Dunning Hotopp, J.C.; Hu, F.Z.; Riley, D.R.; et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 2010, 11, R107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Burnham, C.A.; Hink, T.; Chen, L.; Shaikh, N.; Wollam, A.; Sodergren, E.; Weinstock, G.M.; Tarr, P.I.; Dubberke, E.R. Phenotypic and genotypic analysis of Clostridium difficile isolates: A single-center study. J. Clin. Microbiol. 2014, 52, 4260–4266. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhu, Y.; Yi, Y.; Lu, N.; Zhu, B.; Hu, Y. Comparative genomic analysis of Acinetobacter baumannii clinical isolates reveals extensive genomic variation and diverse antibiotic resistance determinants. BMC Genom. 2014, 15, 1163. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Penagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.A.; Chia, J.-M.; Elshire, R.J.; Sun, Q.; Ersoz, E.S.; Hurwitz, B.L.; Peiffer, J.A.; McMullen, M.D.; Grills, G.S.; Ross-Ibarra, J.; et al. A first-generation haplotype map of maize. Science 2009, 326, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.-M.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.L.; Li, M.W.; He, W.; Qin, N.; Wang, B. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Hernandez Wences, A.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 506. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, G.; Zhao, H.; Wang, G.; Lian, X.; Xie, W. Exploring the rice dispensable genome using a metagenome-like assembly strategy. Genome Biol. 2015, 16, 187. [Google Scholar] [CrossRef] [PubMed]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Zhang, N.; Severing, E.I.; Nijveen, H.; Cheng, F.; Visser, R.G.; Wang, X.; de Ridder, D.; Bonnema, G. Beyond genomic variation—Comparison and functional annotation of three Brassica rapa genomes: A turnip, a rapid cycling and a chinese cabbage. BMC Genom. 2014, 15, 250. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Golicz, A.A.; Batley, J.; Edwards, D. Towards plant pangenomics. Plant Biotechnol. J. 2016, 14, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Nguyen, S.H.; Ganesamoorthy, D.; Elliott, A.; Cooper, M.; Coin, L.J. Scaffolding and completing genome assemblies in real-time with nanopore sequencing. bioRxiv 2016. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. Cegma: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Busco: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Caccamo, M.; Turner, I.; Flicek, P.; McVean, G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 2012, 44, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Marcus, S.; Lee, H.; Schatz, M.C. Splitmem: A graphical algorithm for pan-genome analysis with suffix skips. Bioinformatics 2014, 30, 3476–3483. [Google Scholar] [CrossRef] [PubMed]
- Jehan, T.; Lakhanpaul, S. Single nucleotide polymorphism (SNP)–methods and applications in plant genetics: A review. Indian J. Biotechnol. 2006, 4, 435–459. [Google Scholar]
- Hayward, A.; Mason, A.; Dalton-Morgan, J.; Zander, M.; Edwards, D.; Batley, J. SNP discovery and applications in Brassica napus. Plant Biotechnol. 2012. [Google Scholar] [CrossRef]
- Batley, J.; Barker, G.; O’Sullivan, H.; Edwards, K.J.; Edwards, D. Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data. Plant Physiol. 2003, 132, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. Varscan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Lorenc, M.T.; Hayashi, S.; Stiller, J.; Lee, H.; Manoli, S.; Ruperao, P.; Visendi, P.; Berkman, P.J.; Lai, K.; Batley, J. Discovery of single nucleotide polymorphisms in complex genomes using SGSautoSNP. Biology 2012, 1, 370–382. [Google Scholar] [CrossRef] [PubMed]
- You, N.; Murillo, G.; Su, X.; Zeng, X.; Xu, J.; Ning, K.; Zhang, S.; Zhu, J.; Cui, X. SNP calling using genotype model selection on high-throughput sequencing data. Bioinformatics 2012, 28, 643–650. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013. [Google Scholar] [CrossRef]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Wilkie, A.O.M.; McVean, G.; Lunter, G.; Consortium, W.G.S. Integrating mapping, assembly and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D. Bioinformatics and plant genomics for staple crops improvement. In Breeding Major Food Staples; Kang, M.S., Priyadarshan, P.M., Eds.; Blackwell Publishing: Oxford, UK, 2007; pp. 93–106. [Google Scholar]
- Laing, C.; Pegg, C.; Yawney, D.; Ziebell, K.; Steele, M.; Johnson, R.; Thomas, J.E.; Taboada, E.N.; Zhang, Y.; Gannon, V.P. Rapid determination of Escherichia coli o157: H7 lineage types and molecular subtypes by using comparative genomic fingerprinting. Appl. Environ. Microbiol. 2008, 74, 6606–6615. [Google Scholar] [CrossRef] [PubMed]
- Laing, C.; Buchanan, C.; Taboada, E.N.; Zhang, Y.; Kropinski, A.; Villegas, A.; Thomas, J.E.; Gannon, V.P. Pan-genome sequence analysis using Panseq: An online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinform. 2010. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, J.A. Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci. 2002, 162, 329–333. [Google Scholar] [CrossRef]
- Batley, J.; Edwards, D. SNP applications in plants. In Association Mapping in Plants; Springer: Haarlem, Netherlands, 2007; pp. 95–102. [Google Scholar]
- Hyten, D.L.; Song, Q.; Zhu, Y.; Choi, I.Y.; Nelson, R.L.; Costa, J.M.; Specht, J.E.; Shoemaker, R.C.; Cregan, P.B. Impacts of genetic bottlenecks on soybean genome diversity. Proc. Natl. Acad. Sci. USA 2006, 103, 16666–16671. [Google Scholar] [CrossRef] [PubMed]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The molecular genetics of crop domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Narita, N.N.; Hayashi, K.; Asada, J.; Hamada, T.; Sonobe, S.; Nakajima, K.; Hashimoto, T. Plant-specific microtubule-associated protein SPIRAL2 is required for anisotropic growth in Arabidopsis. Plant Physiol. 2004, 136, 3933–3944. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Roy, J.; Prasad, M. Single nucleotide polymorphisms: A new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants. Curr. Sci. 2001, 80, 524–535. [Google Scholar]
- Ao, S.I.; Yip, K.; Ng, M.; Cheung, D.; Fong, P.Y.; Melhado, I.; Sham, P.C. Clustag: Hierarchical clustering and graph methods for selecting tag SNPs. Bioinformatics 2005, 21, 1735–1736. [Google Scholar] [CrossRef] [PubMed]
- Lewontin, R. On measures of gametic disequilibrium. Genetics 1988, 120, 849–852. [Google Scholar] [PubMed]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [PubMed]
- Cadzow, M.; Boocock, J.; Nguyen, H.T.; Wilcox, P.; Merriman, T.R.; Black, M.A. A bioinformatics workflow for detecting signatures of selection in genomic data. Front. Genet. 2014, 5, 293. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Akey, J.M. Genomic insights into positive selection. Trends Genet. 2006, 22, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Reif, J.C.; Jackson, S.A.; Ma, Y.S.; Chang, R.Z.; Qiu, L.J. Detecting SNPs underlying domestication-related traits in soybean. BMC Plant Biol. 2014, 14, 251. [Google Scholar] [CrossRef] [PubMed]
- Srichumpa, P.; Brunner, S.; Keller, B.; Yahiaoui, N. Allelic series of four powdery mildew resistance genes at the pm3 locus in hexaploid bread wheat. Plant Physiol. 2005, 139, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Clark, T.; Zheng, H.; Vang, S.; Li, R.; Wong, G.K.S.; Wang, J.; Zheng, X. Gene conversion in the rice genome. BMC Genom. 2008, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Roulin, A.; Piegu, B.; Fortune, P.M.; Sabot, F.; D’Hont, A.; Manicacci, D.; Panaud, O. Whole genome surveys of rice, maize and sorghum reveal multiple horizontal transfers of the LTR-retrotransposon Route66 in Poaceae. BMC Evolut. Biol. 2009, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Li, W.H.; Wu, C.I.; Luo, C.C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evolut. 1985, 2, 150–174. [Google Scholar]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evolut. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Kohany, O.; Jurka, M.V. Repetitive sequences in complex genomes: Structure and evolution. Annu. Rev. Genom. Hum. Genet. 2007, 8, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.D.; Vasilinetc, I.; Bankevich, A.; Gurevich, A.; Krivosheeva, T.; Nurk, S.; Pham, S.; Korobeynikov, A.; Lapidus, A.; Pevzner, P.A. Exspander: A universal repeat resolver for DNA fragment assembly. Bioinformatics 2014, 30, i293–i301. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Berlin, K.; Koren, S.; Chin, C.S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurgobin, B.; Edwards, D. SNP Discovery Using a Pangenome: Has the Single Reference Approach Become Obsolete? Biology 2017, 6, 21. https://doi.org/10.3390/biology6010021
Hurgobin B, Edwards D. SNP Discovery Using a Pangenome: Has the Single Reference Approach Become Obsolete? Biology. 2017; 6(1):21. https://doi.org/10.3390/biology6010021
Chicago/Turabian StyleHurgobin, Bhavna, and David Edwards. 2017. "SNP Discovery Using a Pangenome: Has the Single Reference Approach Become Obsolete?" Biology 6, no. 1: 21. https://doi.org/10.3390/biology6010021
APA StyleHurgobin, B., & Edwards, D. (2017). SNP Discovery Using a Pangenome: Has the Single Reference Approach Become Obsolete? Biology, 6(1), 21. https://doi.org/10.3390/biology6010021