Abstract
Next generation sequencing and bioinformatic approaches are increasingly used to quantify microorganisms within populations by analysis of ‘meta-barcode’ data. This approach relies on comparison of amplicon sequences of ‘barcode’ regions from a population with public-domain databases of reference sequences. However, for many organisms relevant ‘barcode’ regions may not have been identified and large databases of reference sequences may not be available. A workflow and software pipeline, ‘MetaGaAP,’ was developed to identify and quantify genotypes through four steps: shotgun sequencing and identification of polymorphisms in a metapopulation to identify custom ‘barcode’ regions of less than 30 polymorphisms within the span of a single ‘read’, amplification and sequencing of the ‘barcode’, generation of a custom database of polymorphisms, and quantitation of the relative abundance of genotypes. The pipeline and workflow were validated in a ‘wild type’ Alphabaculovirus isolate, Helicoverpa armigera single nucleopolyhedrovirus (HaSNPV-AC53) and a tissue-culture derived strain (HaSNPV-AC53-T2). The approach was validated by comparison of polymorphisms in amplicons and shotgun data, and by comparison of predicted dominant and co-dominant genotypes with Sanger sequences. The computational power required to generate and search the database effectively limits the number of polymorphisms that can be included in a barcode to 30 or less. The approach can be used in quantitative analysis of the ecology and pathology of non-model organisms.
1. Introduction
Culture-independent molecular techniques to identify and quantify components of microbial communities have been facilitated by the use of next generation sequencing (NGS) [1,2].
Shotgun sequencing and whole or partial genome assembly uses algorithms comparing sequence data to public sequence databases (such as Genbank) [2,3,4,5,6]. ‘Barcode’ analysis uses PCR amplification of well-characterized regions (e.g., the 16S rRNA sub-unit in bacteria, internal transcribed space (ITS) of fungi or cytochrome oxidase) and comparison to sequence databases specific to those regions to determine taxonomic assignment and relative abundance of taxa in the community [2,7,8,9,10,11].
Both approaches are limited by available sequencing technology that relies on partial genome ‘reads’, and by the scope and accuracy of sequences in the reference databases. Shotgun sequencing and partial genome assembly is biased towards identification of dominant genotypes or taxa as a result of the limited read depth across multiple whole genomes [10,12,13]. Amplicon sequencing introduces bias resulting from gene copy number, selection of primers, and classification based on limited span of the genome [2,7,12,14]. Both depend on reference databases which contain sequences from the small proportion of organisms that have been sequenced and variable standards of validation. Furthermore, non-model organisms, for which sequence databases are not available or for which marker regions have not been identified, require custom solutions. This is a particular issue in analysis of viral metapopulations and quasispecies [15].
Baculoviruses (Baculoviridae) are invertebrate-specific double-stranded DNA viruses with a genome of between 80 kb to 180kb [16]. The nucleopolyhedroviruses (Alphabaculoviruses) are known to contain high levels of genotypic and phenotypic diversity within a single isolate [17,18,19,20,21].
Previous studies on within-isolate diversity used techniques such as in vitro and in vivo isolation of sub-populations to identify strains [19,22,23,24,25], but such culture-dependent approaches themselves select a sub-set of strains that are adapted to the selection method, such as growth in tissue culture [19,26,27]. Molecular approaches include restriction fragment length polymorphism (RFLP) in combination with quantitative polymerase chain reaction (qPCR) [28,29,30,31], and denaturing gradient gel electrophoresis (DGGE) [32,33,34]. DGGE cannot be used reliably to quantify relative abundance and both qPCR and DGGE rely on primers that may not detect all variants [14,33,34,35,36,37].
Shotgun sequencing can be used to assemble a consensus sequence for an isolate containing multiple strains, and the same data can then be used to identify polymorphisms across the genome to determine the relative abundance of a single polymorphism [38,39,40]. Shotgun data can also be used to infer an approximate total number of strains within an isolate and the relative abundance of taxonomic clusters of strains within this population [13,19], but cannot determine the relative abundance of individual strains or abundance of strains that may contain multiple polymorphisms distributed across fragmented reads.
In this paper, we describe the application and validation of stepwise sequencing and a metabarcoding software pipeline to identify and quantify within-isolate strain variants within a baculovirus model.
2. Materials and Methods
2.1. Viruses
The baculovirus isolate HaSNPV-AC53 was obtained from AgBiTech Pty Ltd., passaged once in H. armigera larvae and DNA extracted as previously described [17,39].
The strain variant HaSNPV-AC53-T2 was derived from the AC53 wild type by passage in tissue culture and DNA extracted as previously described [19,41].
2.2. Identification of High Density Polymorphic Regions in Shotgun Data
DNA extraction from the HaSNPV-AC53 wild-type isolate, shotgun sequence generation using the Ion Torrent PGM, and assembly of a consensus sequence (Genbank accession: KJ909666) were completed as previously described [42]. The Genome Analysis Toolkit v3.5 (GATK) (Broad Institute, Cambridge, MA, USA) ‘best practices’ pipeline was used to identify substitutions, insertions and deletions (polymorphisms) in the shotgun data which were filtered to exclude those with a minimum genotype quality of below 60 (0.0001% error) and minimum read depth of 20x coverage [38]. Polymorphisms were annotated using Geneious R9.1.5 (Biomatters, Auckland, New Zealand) and snpEff 4.2 [43,44].
2.3. Amplicon Sequencing and Validation of Sequence Polymorphisms
Primers were designed to amplify custom ‘barcode’ regions of 325 and 365 bp (i.e., less than the span of a single Ion Torrent PGM read) within each of two ORFs with different polymorphism density (Table 1): Baculovirus Repeated ORF-A (BRO-A) and DNA polymerase.

Table 1.
Primers used for amplification of selected regions within the ORFs BRO-A and DNA polymerase.
Both BRO-A and DNA Polymerase ‘barcode’ regions of the AC53 wild-type isolate and the BRO-A region of the HaSNPV-AC53-T2 strain were amplified from DNA using the Platinum Taq High Fidelity Super Mix kit (Life Technologies, Thermo-Fisher, Waltham, MA, USA) and an Eppendorf Pro S thermocycler (Eppendorf, Hamburg, Germany) as per the Platinum Taq standard method (Life Technologies, Thermo-Fisher, Waltham, MA, USA). NGS amplicon preparation and clean-up was completed as per the Life Technologies (Thermo-Fisher, Waltham, MA, USA) Ion Torrent PGM fusion primer manual. Shotgun sequencing was completed using an Ion Torrent PGM with a 318v2 chip and 400 bp chemistry.
Read quality was determined using FastQC 0.11.4 (Babraham Institute, Cambridge, UK) and any reads containing artefacts and/or quality less than Q20 were removed. Reads were trimmed to the expected amplicon size (Table 1) to remove primer regions using Fastx-toolkit 0.0.14 (Hannon Laboratory, Cold Spring Harbor, New York, NY, USA) [45,46]. Polymorphisms within the amplicon reads data were identified as described for the shotgun data and validated by comparison using vcf-compare within the VCFtools package (version 0.1.14) [47].
2.4. Sanger Sequencing
Both ‘barcode’ regions of the AC53 isolate and the BRO-A region of the HaSNPV-AC53-T2 strain were amplified using the forward primer in Table 1, the Mango Taq kit (Bioline, Meridian Bioscience, Cincinnati, OH, USA) and an Eppendorf Pro S thermocycler (Eppendorf, Hamburg, Germany). PCR products were then cleaned using an Isolate II PCR clean-up kit (Bioline, Alexandria, Australia) and labelled using a Big Dye Terminator (BDT) v3.1 kit (Applied Biosystems, Thermo-Fisher, Waltham, MA, USA). Labelled products were then precipitated using EDTA/ethanol as per the BDT v3.1 kit insert. Products were then sequenced using an ABI 3500 Genetic Analyzer (Applied Biosystems, Thermo-Fisher, Waltham, MA, USA).
2.5. Genotyping and Abundance Pipeline
Amplicon reads were mapped to the relevant consensus sequence for the 2 ORFs in the HaSNPV-AC53 genome and the BRO-A sequence for the HaSNPV-AC53-T2 strain using the BWA mem 0.7.12 algorithm with default settings to produce unsorted SAM files [48]. These unsorted SAM files were converted to sorted BAM files using SAMtools 1.3 [49]. The BAM headers were then corrected and the reference sequences and BAM files were indexed and a sequence dictionary was produced using samtools 1.3 and picard-tools 2.5.0 (Broad Institute, Cambridge, Massachusetts, USA) [50].
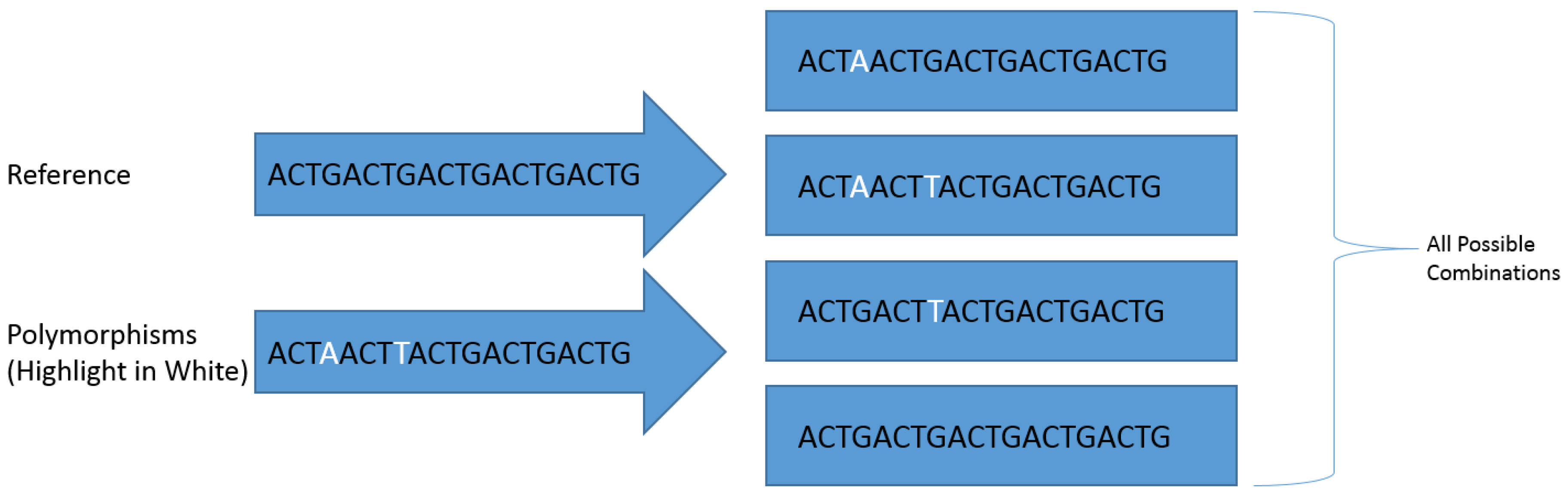
Polymorphisms were identified within the genome using the GATK HaplotypeCaller to produce a ‘genomic variant call format’ (gVCF) file with the following parameters: maximum read depth per site of 300,000 reads, 100 maximum alternate alleles per site, genotyping mode set to ‘discovery’,down-sampling set to ‘none’ and emit reference confidence set to ‘GVCF’. These files were then sorted to genotype and converted to a standard ‘variant call format’ (VCF) file using the GATK GenotypeGVCFs tool and hard-filtered using the GATK VariantFiltration tool to include only polymorphisms with a genotype quality (GQ) score greater than 60 (minimum 0.0001% error on a Phred scale). The final VCF file containing all the filtered polymorphisms within each ORF and the consensus sequence of each ORF were imported respectively into the Biostars 175929 tool as part of the JVarkit package [51] to produce a compressed fasta file database for each amplicon containing generated references sequences with every possible combination of identified polymorphisms (Figure 1). All generated reference sequences were renamed using the BBmap Renamer tool [52] to include the ORF from which they were derived and a numerical identification number.
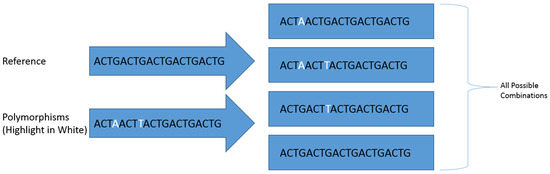
Figure 1.
A visual representation of how the Biostars 175929 tool produces sequences containing all polymorphism combinations.
The generated reference sequences were then indexed and used as the references to map amplicon reads using the BWA mem 0.7.12 algorithm with default settings to produce a SAM file [48]. The SAM files were then imported into Tablet 1.15.09.01 for visual and statistical comparison of the mapped amplicon sequence reads and the generated reference sequences [53,54]. The mapping statistics were produced using samtools 1.3 and sequences that contained less than 20x coverage (equivalent to a 1% error on a phred scale) or sequences with imperfect mapping (containing gaps) were excluded using kentUtils (version 302) and custom R scripts built with Microsoft R Open 3.3.1 [55,56,57]. Relative abundances of each identified genotype were calculated using Microsoft R Open 3.3.1 (Microsoft, Redmond, WA, USA) [55,57].
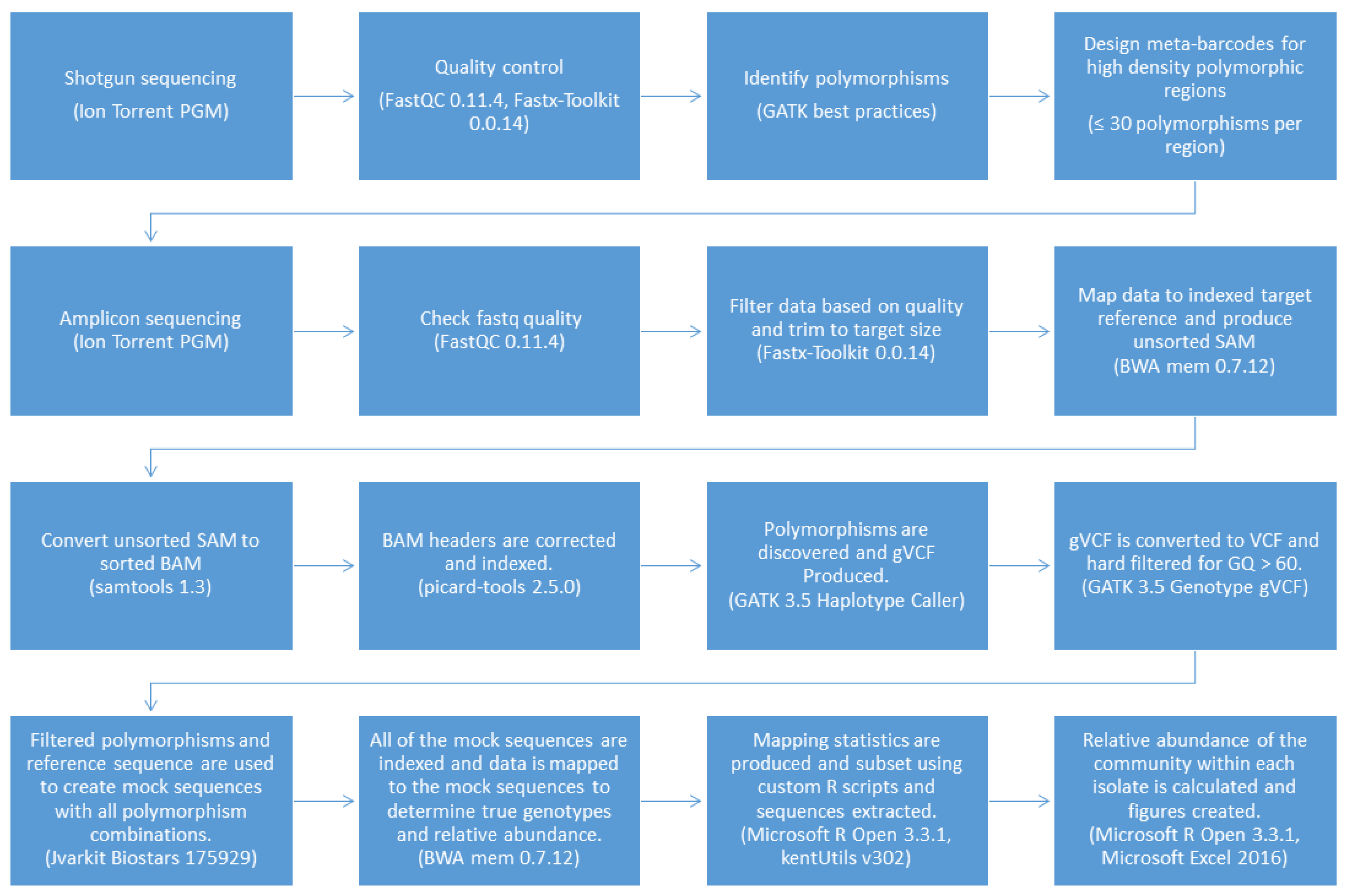
The pipeline was coded using Bash and Microsoft R Open 3.3.1 with a text-based interface to improve versatility and ease of use and named the Meta-barcoding Genotyping and Abundance Pipeline (MetaGaAP) [58]. A schematic of the pipeline is presented in Figure 2.
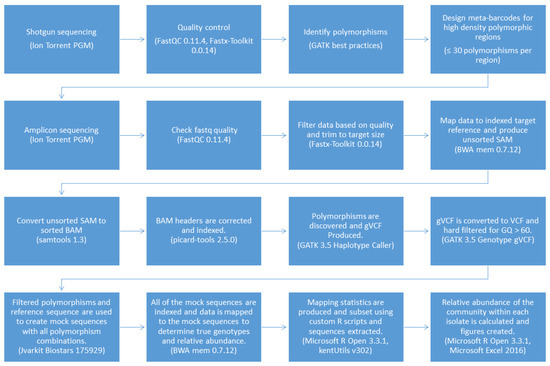
Figure 2.
The MetaGaAP workflow to identify genotypes and the relative abundance of the community composition within a single isolate.
2.6. Comparison of amplicon and Sanger sequences
Genotype sequences identified using MetaGaAP and chromatograms from Sanger sequencing were visualized using Geneious R9.1.5 and aligned using MAFFT v7.222 (Kyoto University, Kyoto, Japan) with the FFT-NS-2 algorithm and default settings [59]. The dominant genotype and abundant minor genotypes predicted from the mapped NGS amplicon sequences were compared visually at each predicted SNP locus with the Sanger chromatographs.
3. Results
3.1. Identification of Polymorphisms in Shotgun Sequence Data
A total of 438 polymorphisms were identified within the Ion Torrent PGM shotgun dataset of the AC53 isolate, equivalent to 1 nucleotide change every 297 bases. Within the 139 ORFs in the AC53 consensus genome sequence, 37 ORFs contained no polymorphisms and 102 ORFs contained polymorphisms. Polymorphisms were identified within exons, intergenic regions and all five homologous repeat (Hr) regions (Table S1): 53 were insertions, 339 were deletions and 46 were substitutions. Most ORFs contained 9 or fewer polymorphisms. The ORF with the highest number of polymorphisms was BRO-A (30) and had a mix of substitutions, insertions and deletions and was selected for amplicon sequencing. DNA polymerase contained 5 polymorphisms across the entire 3 kb ORF: a 325 bp region within the ORF that contained no polymorphisms was selected as the negative control.
3.2. Validation by Comparison of Amplicon Sequence Variants to Shotgun Sequence Data
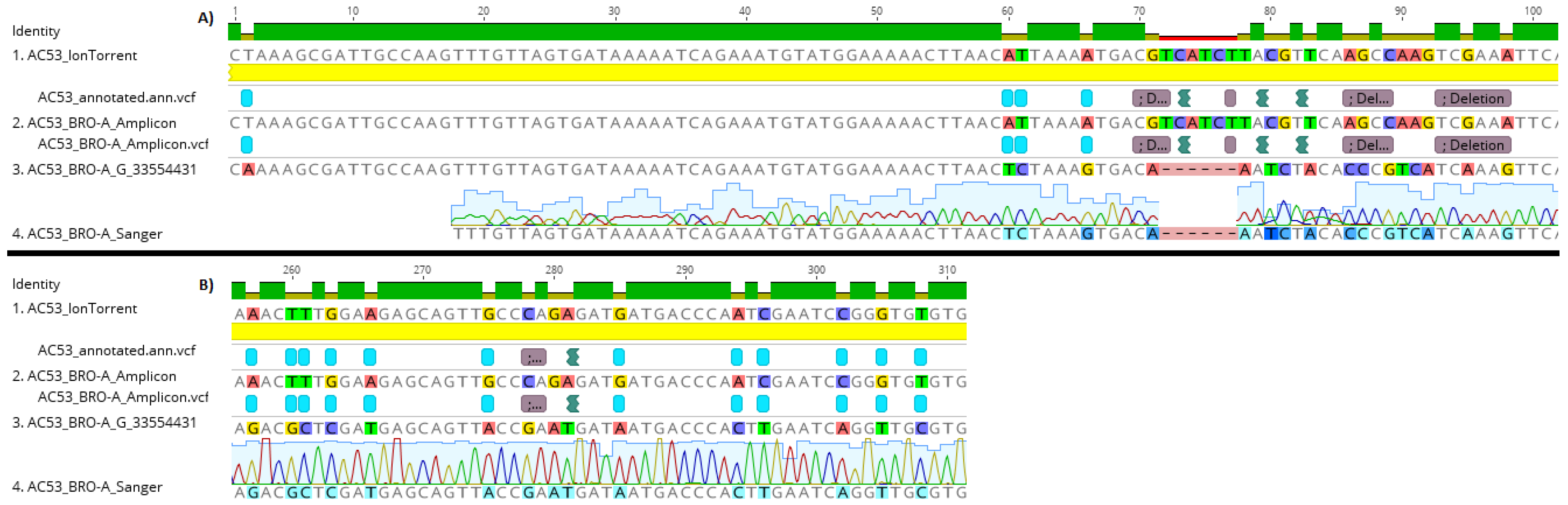
AC53 shotgun sequencing predicted 25 polymorphism in the targeted ‘barcode’ region within BRO-A. All 25 polymorphisms were identified in the amplicon sequences (Figure 3). No polymorphisms were detected in the amplicon data of the DNA polymerase region, as predicted from the shotgun data (Figure S1). A single polymorphism (an ‘A’ substitution at position 293) was detected in the BRO-A amplicon sequences of the derived strain AC53-T2. This polymorphism was confirmed as one of the 25 polymorphisms in AC53 wild-type isolate shotgun and amplicon data.
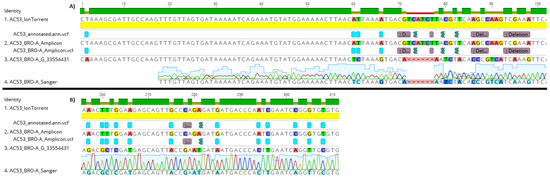
Figure 3.
Fragments A and B of the HaSNPV-AC53 BRO-A target region containing the identified polymorphisms, showing identical alignment of polymorphisms in the amplicon, shotgun and Sanger sequences of the dominant BRO-A variant identified by MetaGaAP within the wild type baculovirus isolate HaSNPV-AC53.
3.3. Genotype Sequence Construction, Abundance Mapping and Validation by Sanger Sequencing
The 25 polymorphisms identified in the BRO-A amplicon ‘barcode’ region of isolate AC53 generated 3.4 × 107 possible combinations of polymorphisms in reference sequences in the custom database. Mapping of the amplicon sequences data to this database identified 329 of these possible sequences were present in the amplicon sequencing, with a minimum of 1 read mapping to them. Of these, 28 amplicon sequences with between 21× and 258,084× coverage were identified (Table 2). Genotype abundance was estimated from the number of reads mapping to each of these 28 hypothetical variants.
Table 2.
Relative abundance of the identified AC53 BRO-A community composition that were above the 20x coverage threshold with G_33554431 identified as the dominant strain in the population.
Genotype G_33554431 accounted for 97% of the reads and was thus predicted to be the dominant genotype, while the second most abundant genotype (G_33554303) accounted for 0.62% of the reads (Table 2). The dominant genotype G_33554431 was confirmed by Sanger sequencing, with 100% sequence similarity (Figure 3).
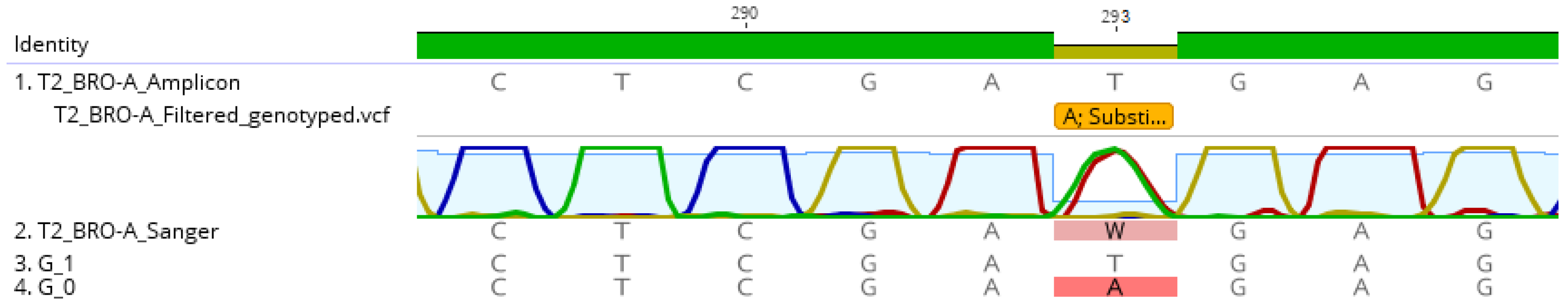
The single polymorphism detected in the BRO-A amplicon sequences of the tissue-culture derived strain AC53-T2 resulted in generation of two reference sequences: with the A substitution or without the substitution (i.e., with a T). Mapping of amplicon sequence data to the two reference sequences showed that both were present in similar abundance: the T genotype accounted for 54% of reads and the A genotype for 46% of reads (Table 3). The dominant T genotype of the derived strain AC53-T2 had 100% sequence similarity with the dominant G_33554431 genotype of the AC53 wild type isolate. The minor A genotype of AC53-T2 had 100% homology to genotype G_33553919 of the AC53 wild type isolate, which accounted for only 0.05% of the reads in the wild type isolate (Table 3). The Sanger chromatogram detected both genotypes in strain AC53-T2, with both A and T detected in approximately equal intensity in the chromatogram at position 293 (Figure 4).
Table 3.
Relative abundance of the two BRO-A genotypes within AC53-T2 BRO-A.
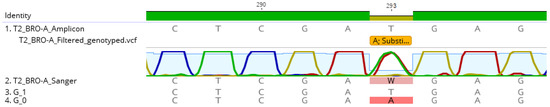
Figure 4.
Comparison of the AC53-T2 reference sequence to the Sanger sequence and the two identified genotypes within HaSNPV-AC53-T2. The Sanger chromatogram at position 293 shows the two competing genotypes which were identified with MetaGaAP and validates relative abundance result.
The single genotype predicted by the amplicon sequencing of DNA polymerase was confirmed to have 100% homology with the Sanger sequencing (Figure S1).
4. Discussion
Shotgun sequencing and identification of polymorphisms was used to identify of custom ‘barcode’ regions in the viral metapopulation of the wild type baculovirus isolate HaSNPV-AC53. Hr and non-coding regions were excluded to reduce possible primer bias and sequencing errors. The highest number of polymorphisms was identified in an AT-rich region of the BRO-A ORF. The high abundance of mutations in BRO-A has been previously described in published whole-genome sequences of SNPV isolates from Helicoverpa spp. [19,60,61] and a polymorphism rich ‘custom barcode’ region was selected within the ORF [62,63,64].
In contrast, only five polymorphisms were identified across the entire 3 kbp DNA polymerase ORF. A previous study using a different HaSNPV isolate identified 60 polymorphisms in the DNA polymerase ORF using 454 pyrosequencing with only 30× coverage, but the authors had expected DNA polymerase to be much more highly conserved [33]. Our results support this expectation and we suggest that the low coverage of the 454 pyrosequencing may have led to overestimation of polymorphisms in that study [65,66,67].
Comparison of the amplicon sequence data identified the same 25 polymorphisms in BRO-A and absence of polymorphisms in DNA polymerase, as predicted from the shotgun sequence data of HaSNPV-AC53. In contrast, a single polymorphism was predicted in the amplicon data of the tissue culture derived strain AC53-T2, which was also confirmed as one of the 25 polymorphisms predicted from the shotgun data of the parent isolate. This validates the use of amplicon and shotgun sequence to compare polymorphisms using the GATK best practices guidelines [38,39,40,68].
Comparison of amplicon data with the database of all possible combinations of polymorphisms using MetaGaAP identified 28 variants within the HaSNPV-AC53 wild type viral metapopulation at the level of 20× read coverage. A dominant variant within the wild type HaSNPV-AC53 accounted for 97% of the population. In contrast, two variants of approximately equal abundance were identified in the derived strain AC53-T2. The slightly more abundant variant in AC53-T2 had 100% sequence similarity to the dominant variant in the parent isolate, and the other variant had 100% sequence similarity to a minor variant accounting for 0.05% of abundance in the parent isolate. This supports the sensitivity of MetaGaAP to detect and identify minor variants as low as 129× coverage. We suggest including strains with a minimum 20× coverage threshold (to eliminate ‘false positives’ due to sequencing error). However, the presence of minor genotypes with coverage below 129× would require confirmation by, for example, detection in multiple deep sequencing of the isolate during different stages of infection, or large scale sequence or virus cloning and characterisation.
Sanger sequencing is the ‘gold-standard’ for validation of NGS datasets and has the lowest error rates [69]. Sanger sequencing confirmed the identification of the predicted dominant variant in both the BRO-A and DNA Polymerase amplicons of the HaSNPV wild type metapopulation. Furthermore, Sanger sequencing detected both the predicted variants within the derived strain AC53-T2 in the approximately equal proportions calculated by MetaGaAP. This confirmed the validity both of the identification of variants and the calculation of their relative abundance by MetaGaAP.
Current tools for 16S based taxonomic classification of clinical isolates use either pairwise or non-pairwise alignments to a very limited set of sequences from culture collections. Most meta-barcode analyses of microbial communities use partial regions of 16S and 18S ribosomal RNA and, to a lesser degree, the ITS region of fungi, while ‘barcodes’ for viruses are limited to a few significant virus types such as small RNA viruses [1,2,3,6,7,15,70,71]. These approaches are primarily used for taxonomic classification and rely on either phylogenetic clustering or alignment scores in comparison to sequences in reference databases such as Greengenes for 16S [9,72,73,74,75,76] However, these approaches are limited by errors such as submission of misannotated sequences or identification based on short or partial sequences, in addition to the limited sequence availability for non-model organisms [77,78,79].
5. Conclusions
MetaGaAP accurately identified and estimated abundance of variants in a virus metapopulation by generating a custom database from sequence data and comparison with ultra-deep sequencing of amplicons of novel, polymorphism-rich ‘barcode’ regions in the viral metagenome. However, the computer data handling and processing time increases as the number of polymorphisms increases and the number of possible combinations generated in the database increases by 2y, where y = number of polymorphisms. The application is thus practically limited regions with 30 or fewer polymorphisms.
Despite this limitation, MetaGaAP has potential application in analysis of community composition where suitable reference sequence databases are not available, complete or accurately assigned, and can be used to identify and quantify strain variants in pathology, ecology and evolutionary studies without the need for viral cloning. MetaGaAP is publicly available for download on GitHub [58].
Supplementary Materials
The following are available online at www.mdpi.com/2079-7737/6/1/14/s1, Figure S1: Comparison of the AC53 DNA polymerase Sanger sequence and the AC53 DNA polymerase reference sequence showing 100% nucleotide identity and no polymorphisms identified., Table S1: Polymorphisms detected within ORFs. BRO-A has the highest number of polymorphisms (30) and HOAR and P74 have the second highest (13).
Acknowledgments
This work was funded by Queensland University of Technology, the Cotton Research Development Corporation and an Australian Government Research Training Program Scholarship. We would like to acknowledge the support of AgBiTech Pty.Ltd in supplying insects and the virus isolate and to thank staff of the Molecular Genetics Research Facility and the Invertebrate & Microbiology Group at QUT for their assistance with sequencing and technical support. Some of the data reported in this paper was obtained at the Central Analytical Research Facility (CARF) operated by the Institute for Future Environments (QUT). Access to CARF is supported by generous funding from the Science and Engineering Faculty (QUT).
Conflicts of Interest
The authors declare a conflict of interest. The Cotton Research Development Corporation has funded the work by C. Noune through a post-graduate student scholarship. AgBiTech Pty Ltd. Provided the sample of HaSNPV-AC53 and previously funded consultancy and research work with C. Hauxwell but did not contribute financially to this study.
Software and Dataset Availability
MetaGaAP is available for download at https://github.com/CNoune/IMG_pipelines. Genotypes described in this paper are available for download at https://researchdatafinder.qut.edu.au/display/n14806.
References
- Gilbert, J.A.; Dupont, C.L. Microbial metagenomics: Beyond the genome. Annu. Rev. Mar. Sci. 2011, 3, 347–371. [Google Scholar] [CrossRef]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, I. Metagenomics: Tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights 2015, 9, 75–88. [Google Scholar]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.C.; Cram, J.A.; Chen, T.; Fuhrman, J.A.; Sun, F. Accurate genome relative abundance estimation based on shotgun metagenomic reads. PLoS ONE 2011, 6, e27992. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Z.; Bushman, F.D.; Li, H. A model-based approach for species abundance quantification based on shotgun metagenomic data. Stat. Biosci. 2016. [Google Scholar] [CrossRef]
- Kunin, V.; He, S.; Warnecke, F.; Peterson, S.B.; Martin, H.G.; Haynes, M.; Ivanova, N.; Blackall, L.L.; Breitbart, M.; Rohwer, F. A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res. 2008, 18, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Sanschagrin, S.; Yergeau, E. Next-generation sequencing of 16S ribosomal RNA gene amplicons. J. Vis. Exp. 2014, 29, e51709. [Google Scholar] [CrossRef] [PubMed]
- Brittnacher, M.J.; Heltshe, S.L.; Hayden, H.S.; Radey, M.C.; Weiss, E.J.; Damman, C.J.; Zisman, T.L.; Suskind, D.L.; Miller, S.I. Gutss: An alignment-free sequence comparison method for use in human intestinal microbiome and fecal microbiota transplantation analysis. PLoS ONE 2016, 11, e0158897. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.W.; Ji, Y.; Emerson, B.C.; Wang, X.; Ye, C.; Yang, C.; Ding, Z. Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 2012, 3, 613–623. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Põlme, S.; Riit, T.; Liiv, I.; Kõljalg, U.; Kisand, V.; Nilsson, H.; Hildebrand, F. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Barbe, V.; Herniou, E.A. Ultra deep sequencing of a baculovirus population reveals widespread genomic variations. Viruses 2015, 7, 3625–3646. [Google Scholar] [PubMed]
- Sipos, R.; Székely, A.; Révész, S.; Márialigeti, K. Addressing PCR biases in environmental microbiology studies. Bioremediat. Methods Protoc. 2010, 599, 37–58. [Google Scholar]
- McElroy, K.; Thomas, T.; Luciani, F. Deep sequencing of evolving pathogen populations: Applications, errors, and bioinformatic solutions. Microb. Inform. Exp. 2014, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G. Introduction to the Baculoviruses and Their Taxonomy. In Baculovirus Molecular Biology; National Center for Biotechnology Information: Bethesda, MD, USA, 2011. [Google Scholar]
- Rowley, D.L.; Popham, H.J.R.; Harrison, R.L. Genetic variation and virulence of nucleopolyhedroviruses isolated worldwide from the heliothine pests Helicoverpa armigera, Helicoverpa zea, and Heliothis virescens. J. Invertebr. Pathol. 2011, 107, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, M.M.; Vlak, J.M. Baculovirus Genomics. Curr. Drug Targets 2007, 8, 1051–1068. [Google Scholar] [CrossRef]
- Noune, C.; Hauxwell, C. Comparative analysis of HaSNPV-AC53 and derived strains. Viruses 2016, 8, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Cory, J.S.; Green, B.M.; Paul, R.K.; Hunter-Fujita, F. Genotypic and phenotypic diversity of a baculovirus population within an individual insect host. J. Invertebr. Pathol. 2005, 89, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Faulkner, P. A plaque assay for nuclear polyhedrosis viruses using a solid overlay. J. Gen. Virol. 1977, 36, 361–364. [Google Scholar] [CrossRef]
- Graillot, B.; Berling, M.; Blachere-López, C.; Siegwart, M.; Besse, S.; López-Ferber, M. Progressive adaptation of a CpGV isolate to codling moth populations resistant to CpGV-M. Viruses 2014, 6, 5135–5144. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Okano, K.; Rohrmann, G.F. Characterization of the replication of a baculovirus mutant lacking the DNA polymerase gene. Virology 2005, 331, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Redman, E.M.; Wilson, K.; Cory, J.S. Trade-offs and mixed infections in an obligate-killing insect pathogen. J. Anim. Ecol. 2016, 85, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Simon, O.; Palma, L.; Beperet, I.; Munoz, D.; Lopez-Ferber, M.; Caballero, P.; Williams, T. Sequence comparison between three geographically distinct Spodoptera frugiperda multiple nucleopolyhedrovirus isolates: Detecting positively selected genes. J. Invertebr. Pathol. 2011, 107, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L. Genomic sequence analysis of the Illinois strain of the Agrotis ipsilon multiple nucleopolyhedrovirus. Virus Genes 2009, 38, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Christian, P.D.; Gibb, N.; Kasprzak, A.B.; Richards, A. A rapid method for the identification and differentiation of Helicoverpa nucleopolyhedroviruses (NPV Baculoviridae) isolated from the environment. J. Virol. Methods 2001, 96, 51–65. [Google Scholar] [CrossRef]
- Lightner, D.V.; Redman, R.M.; Bell, T.A. Observations on the geographic distribution, pathogenesis and morphology of the baculovirus from Penaeus monodon Fabricius. Aquaculture 1983, 32, 209–233. [Google Scholar] [CrossRef]
- Crawford, A.M.; Zelazny, B.; Alfiler, A.R. Genotypic variation in geographical isolates of oryctes baculovirus. J. Gen. Virol. 1986, 67, 949–952. [Google Scholar] [CrossRef]
- Gettig, R.R.; McCarthy, W.J. Genotypic variation among wild isolates of Heliothis spp nuclear polyhedrosis viruses from different geographical regions. Virology 1982, 117, 245–252. [Google Scholar] [CrossRef]
- Baillie, V.L.; Bouwer, G. High levels of genetic variation within Helicoverpa armigera nucleopolyhedrovirus populations in individual host insects. Arch. Virol. 2012, 157, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Baillie, V.L.; Bouwer, G. High levels of genetic variation within core Helicoverpa armigera nucleopolyhedrovirus genes. Virus Genes 2012, 44, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Baillie, V.L.; Bouwer, G. Development of highly sensitive assays for detection of genetic variation in key Helicoverpa armigera nucleopolyhedrovirus genes. J. Virol. Methods 2011, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.W.; Jordan, F.L.; Maier, R.M. Analysis of artifacts suggests DGGE should not be used for quantitative diversity analysis. J. Microbiol. Methods 2013, 92, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Lueders, T.; Friedrich, M.W. Evaluation of PCR amplification bias by terminal restriction fragment length polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Appl. Environ. Microbiol. 2003, 69, 320–326. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013. [Google Scholar] [CrossRef]
- Yu, X.; Sun, S. Comparing a few SNP calling algorithms using low-coverage sequencing data. BMC Bioinform. 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Noune, C.; Hauxwell, C. Complete genome sequences of seven helicoverpa armigera SNPV-AC53-Derived strains. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Noune, C.; Hauxwell, C. Complete genome sequences of helicoverpa armigera single nucleopolyhedrovirus strains AC53 and H25EA1 from Australia. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 September 2014).
- Gordon, A.; Hannon, G.J. Fastx-toolkit. FASTQ/A short-reads pre-processing tools. 2010; unpublished work. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. Available online: https://arxiv.org/abs/1303.3997 (accessed on 26 May 2013).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Institute, B. Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 9 September 2016).
- Pierre, L. JVarkit: Java Utilities for Bioinformatics. Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.822.1547&rep=rep1&type=pdf (accessed on 26 May 2015).
- Bushnell, B. BBMap Short Read Aligner. Available online: http://sourceforge.net/projects/bbmap (accessed on 18 September 2016).
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet-next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Microsoft R Open. Available online: https://mran.revolutionanalytics.com/rro/ (accessed on 6 May 2016).
- Kent, J. kentUtils. Available online: https://github.com/ENCODE-DCC/kentUtils (accessed on 12 September 2014).
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Noune, C. The Invertebrates & Microbiology Group Pipelines, GitHub, Queensland University of Technology. Available online: https://github.com/CNoune/IMG_pipelines (accessed on 5 September 2016).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, W.-J.; Wong, J.; Chun, G.; Lu, A.; McCutchen, B.; Presnail, J.; Herrmann, R.; Dolan, M.; Tingey, S.; et al. Comparative analysis of the complete genome sequences of Helicoverpa zea and Helicoverpa armigera single-nucleocapsid nucleopolyhedroviruses. J. Gen. Virol. 2002, 83, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; IJkel, W.F.; Tarchini, R.; Sun, X.; Sandbrink, H.; Wang, H.; Peters, S.; Zuidema, D.; Lankhorst, R.K.; Vlak, J.M. The sequence of the Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus genome. J. Gen. Virol. 2001, 82, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Marnellos, G.; Kammerer, S.; Hoyal, C.R.; Shi, M.M.; Cantor, C.R.; Braun, A. Large-scale validation of single nucleotide polymorphisms in gene regions. Genome Res. 2004, 14, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Piepho, H.-P. Optimal marker density for interval mapping in a backcross population. Heredity 2000, 84, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Beissinger, T.M.; Hirsch, C.N.; Sekhon, R.S.; Foerster, J.M.; Johnson, J.M.; Muttoni, G.; Vaillancourt, B.; Buell, C.R.; Kaeppler, S.M.; De Leon, N. Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics 2013, 193, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Gilles, A.; Meglécz, E.; Pech, N.; Ferreira, S.; Malausa, T.; Martin, J.-F. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genom. 2011, 12, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J. The effect of sequencing errors on metagenomic gene prediction. BMC Genom. 2009, 10, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, M.C.; Prosperi, L.; Bruselles, A.; Abbate, I.; Rozera, G.; Vincenti, D.; Solmone, M.C.; Capobianchi, M.R.; Ulivi, G. Combinatorial analysis and algorithms for quasispecies reconstruction using next-generation sequencing. BMC Bioinform. 2011, 12, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Puente-Sánchez, F.; Aguirre, J.; Parro, V. A novel conceptual approach to read-filtering in high-throughput amplicon sequencing studies. Nucleic Acids Res. 2016, 44, e40. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2013, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Clarridge, J.E. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [PubMed]
- Mignard, S.; Flandrois, J. 16S rRNA sequencing in routine bacterial identification: A 30-month experiment. J. Microbiol. Methods 2006, 67, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.J.; Koren, O.; Hugenholtz, P.; DeSantis, T.Z.; Walters, W.A.; Caporaso, J.G.; Angenent, L.T.; Knight, R.; Ley, R.E. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012, 6, 94–103. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).