
MetaGaAP: A Novel Pipeline to Estimate Community Composition and Abundance from Non-Model Sequence Data


Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses
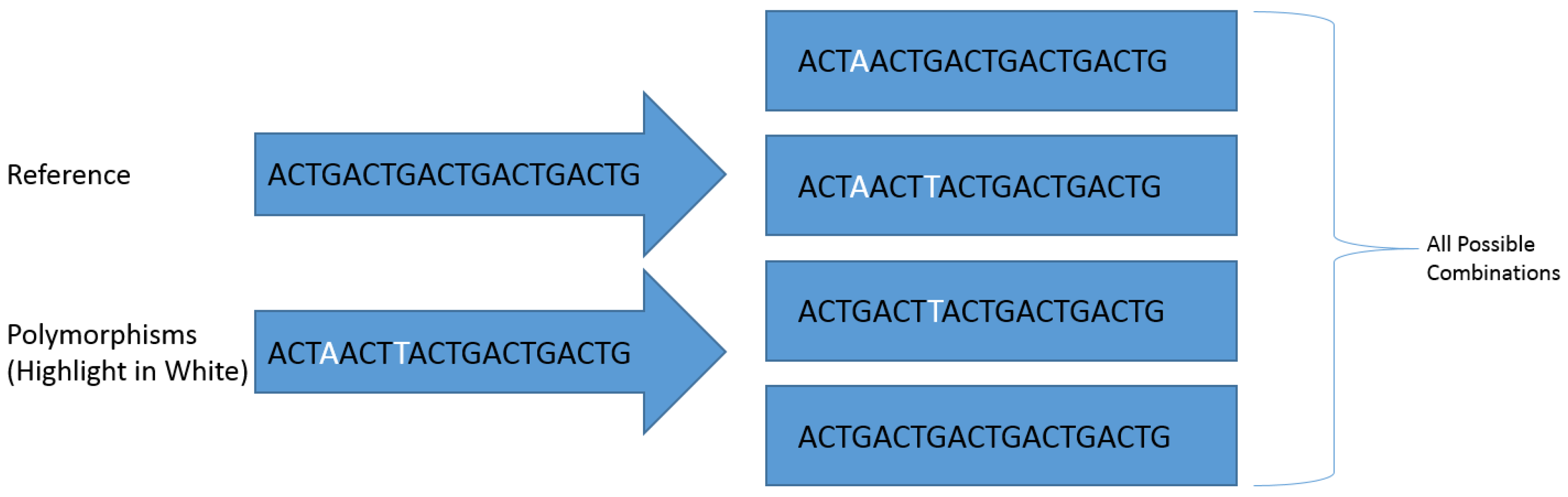
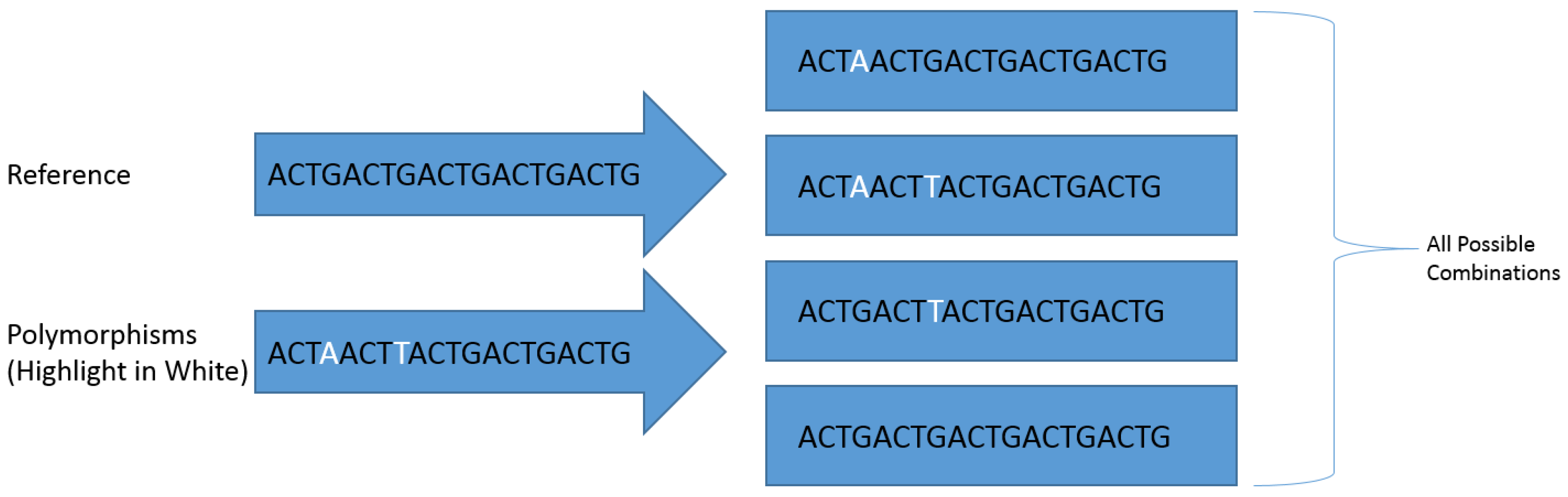
2.2. Identification of High Density Polymorphic Regions in Shotgun Data
2.3. Amplicon Sequencing and Validation of Sequence Polymorphisms
2.4. Sanger Sequencing
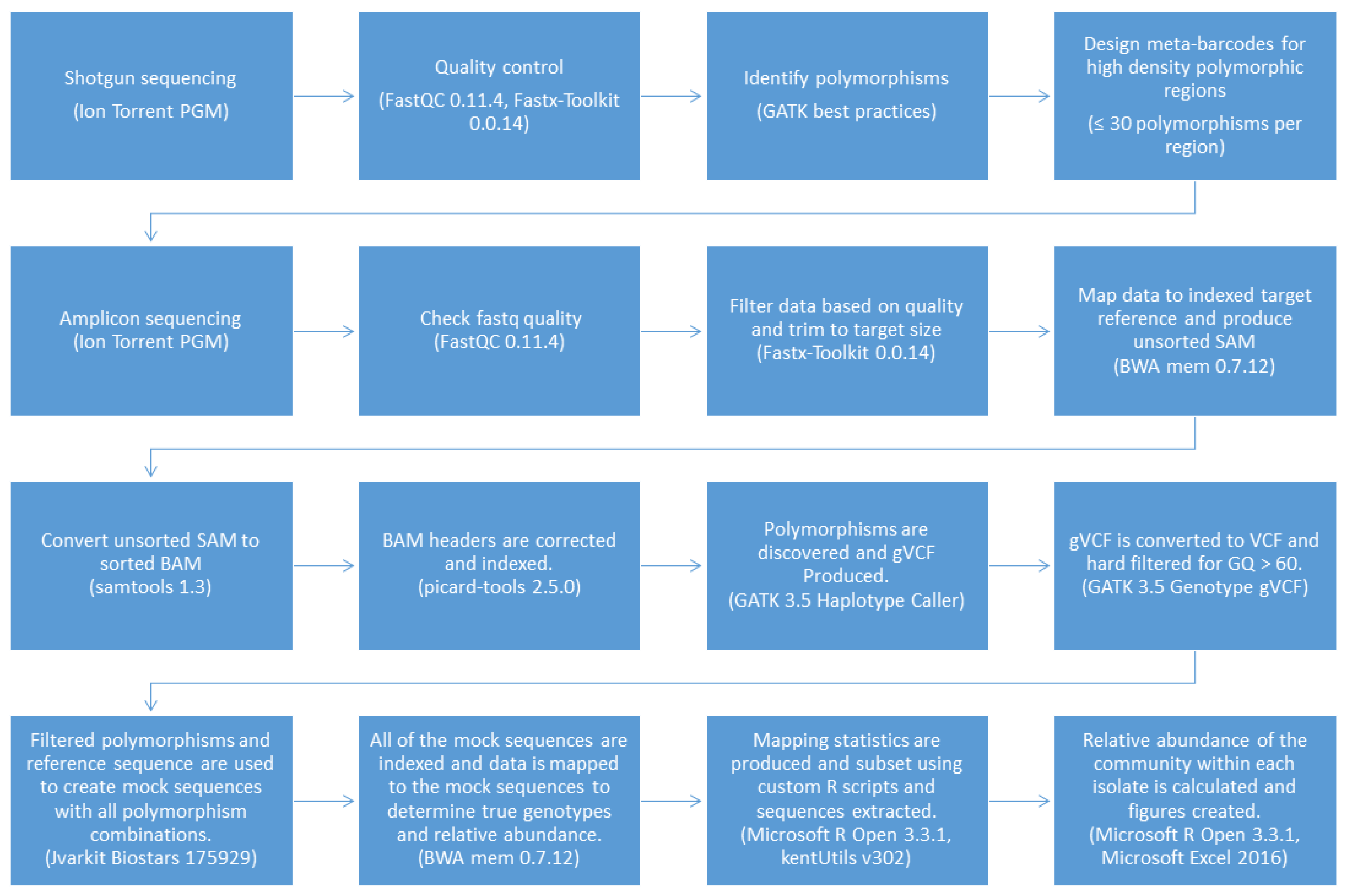
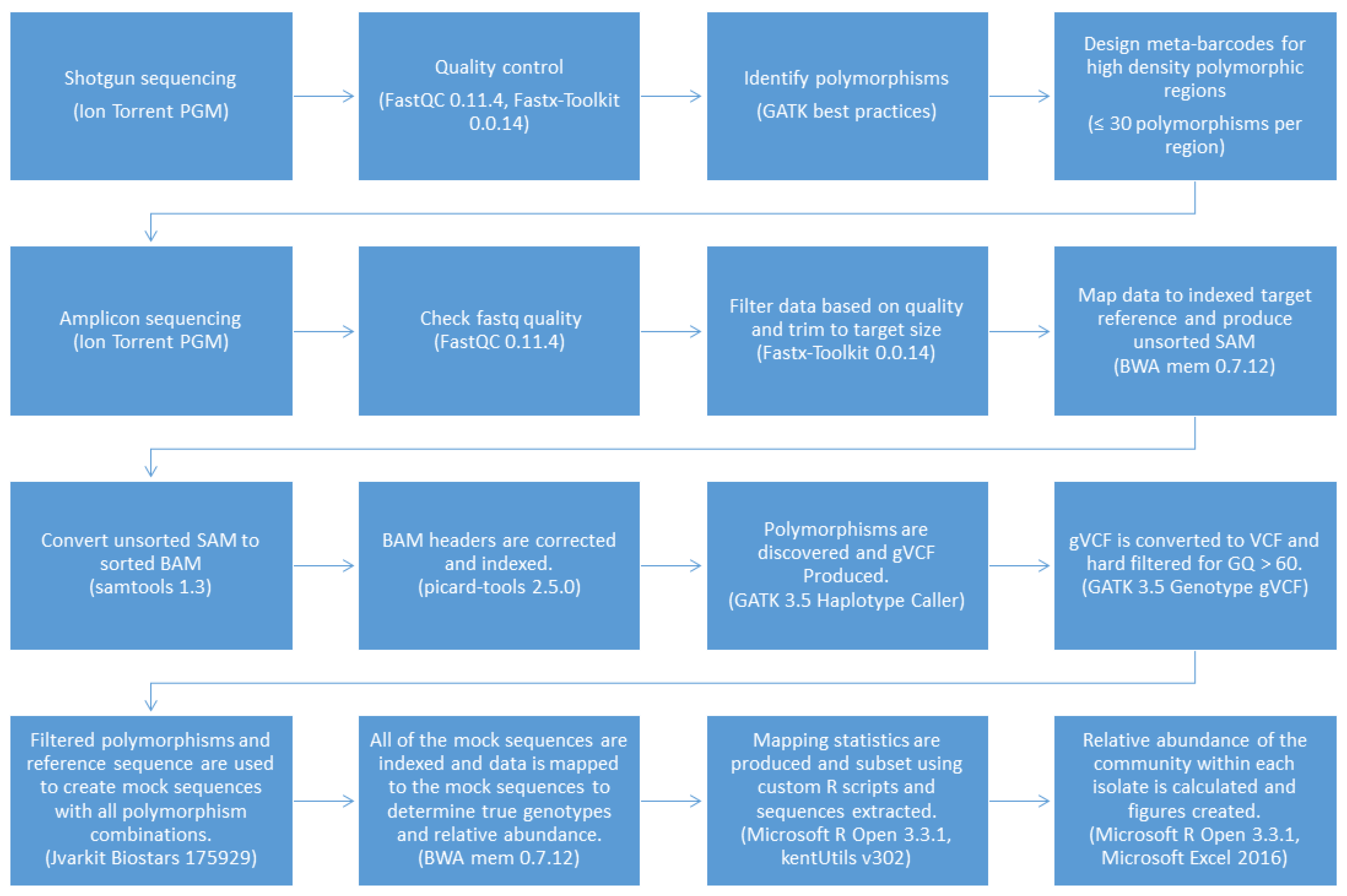
2.5. Genotyping and Abundance Pipeline
2.6. Comparison of amplicon and Sanger sequences
3. Results
3.1. Identification of Polymorphisms in Shotgun Sequence Data
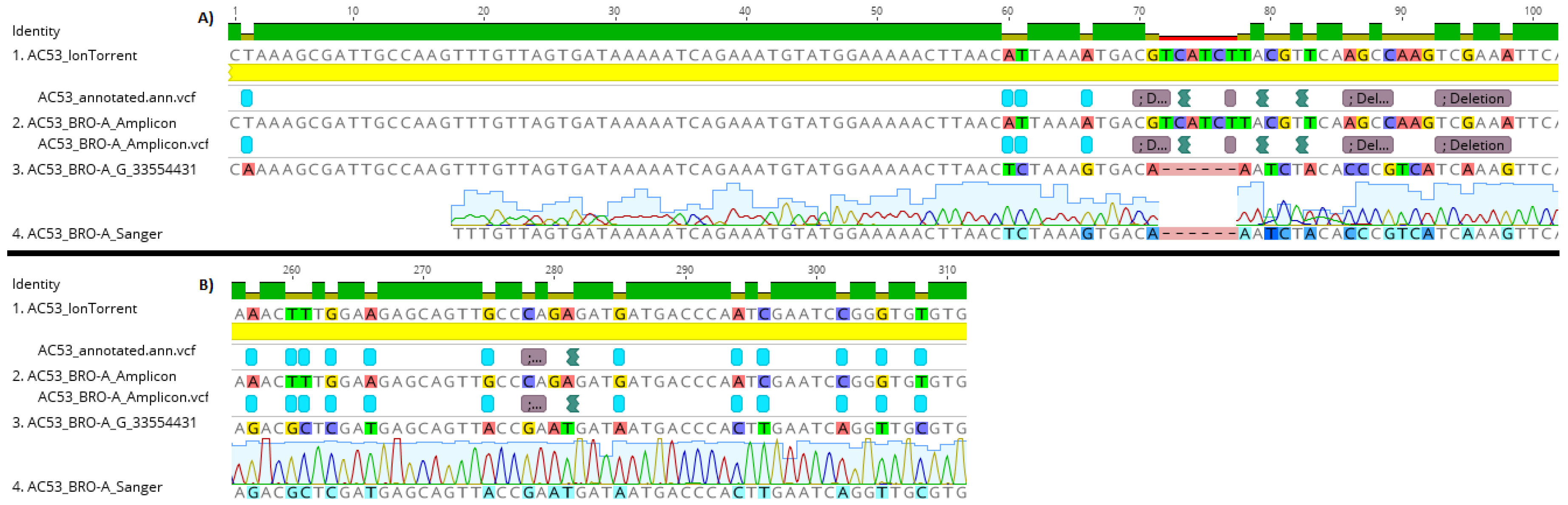
3.2. Validation by Comparison of Amplicon Sequence Variants to Shotgun Sequence Data
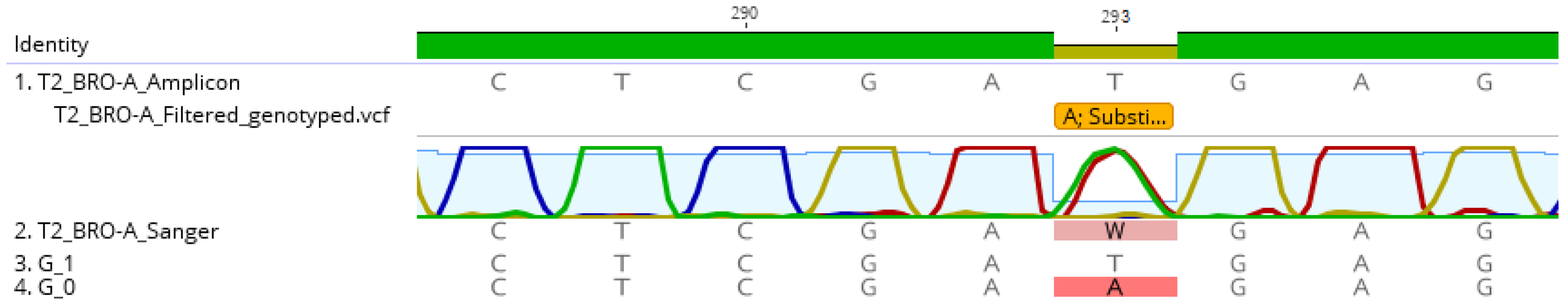
3.3. Genotype Sequence Construction, Abundance Mapping and Validation by Sanger Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
Software and Dataset Availability
References
- Gilbert, J.A.; Dupont, C.L. Microbial metagenomics: Beyond the genome. Annu. Rev. Mar. Sci. 2011, 3, 347–371. [Google Scholar] [CrossRef]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, I. Metagenomics: Tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights 2015, 9, 75–88. [Google Scholar]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.C.; Cram, J.A.; Chen, T.; Fuhrman, J.A.; Sun, F. Accurate genome relative abundance estimation based on shotgun metagenomic reads. PLoS ONE 2011, 6, e27992. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Z.; Bushman, F.D.; Li, H. A model-based approach for species abundance quantification based on shotgun metagenomic data. Stat. Biosci. 2016. [Google Scholar] [CrossRef]
- Kunin, V.; He, S.; Warnecke, F.; Peterson, S.B.; Martin, H.G.; Haynes, M.; Ivanova, N.; Blackall, L.L.; Breitbart, M.; Rohwer, F. A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res. 2008, 18, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Sanschagrin, S.; Yergeau, E. Next-generation sequencing of 16S ribosomal RNA gene amplicons. J. Vis. Exp. 2014, 29, e51709. [Google Scholar] [CrossRef] [PubMed]
- Brittnacher, M.J.; Heltshe, S.L.; Hayden, H.S.; Radey, M.C.; Weiss, E.J.; Damman, C.J.; Zisman, T.L.; Suskind, D.L.; Miller, S.I. Gutss: An alignment-free sequence comparison method for use in human intestinal microbiome and fecal microbiota transplantation analysis. PLoS ONE 2016, 11, e0158897. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.W.; Ji, Y.; Emerson, B.C.; Wang, X.; Ye, C.; Yang, C.; Ding, Z. Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 2012, 3, 613–623. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Põlme, S.; Riit, T.; Liiv, I.; Kõljalg, U.; Kisand, V.; Nilsson, H.; Hildebrand, F. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Barbe, V.; Herniou, E.A. Ultra deep sequencing of a baculovirus population reveals widespread genomic variations. Viruses 2015, 7, 3625–3646. [Google Scholar] [PubMed]
- Sipos, R.; Székely, A.; Révész, S.; Márialigeti, K. Addressing PCR biases in environmental microbiology studies. Bioremediat. Methods Protoc. 2010, 599, 37–58. [Google Scholar]
- McElroy, K.; Thomas, T.; Luciani, F. Deep sequencing of evolving pathogen populations: Applications, errors, and bioinformatic solutions. Microb. Inform. Exp. 2014, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G. Introduction to the Baculoviruses and Their Taxonomy. In Baculovirus Molecular Biology; National Center for Biotechnology Information: Bethesda, MD, USA, 2011. [Google Scholar]
- Rowley, D.L.; Popham, H.J.R.; Harrison, R.L. Genetic variation and virulence of nucleopolyhedroviruses isolated worldwide from the heliothine pests Helicoverpa armigera, Helicoverpa zea, and Heliothis virescens. J. Invertebr. Pathol. 2011, 107, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, M.M.; Vlak, J.M. Baculovirus Genomics. Curr. Drug Targets 2007, 8, 1051–1068. [Google Scholar] [CrossRef]
- Noune, C.; Hauxwell, C. Comparative analysis of HaSNPV-AC53 and derived strains. Viruses 2016, 8, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Cory, J.S.; Green, B.M.; Paul, R.K.; Hunter-Fujita, F. Genotypic and phenotypic diversity of a baculovirus population within an individual insect host. J. Invertebr. Pathol. 2005, 89, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Faulkner, P. A plaque assay for nuclear polyhedrosis viruses using a solid overlay. J. Gen. Virol. 1977, 36, 361–364. [Google Scholar] [CrossRef]
- Graillot, B.; Berling, M.; Blachere-López, C.; Siegwart, M.; Besse, S.; López-Ferber, M. Progressive adaptation of a CpGV isolate to codling moth populations resistant to CpGV-M. Viruses 2014, 6, 5135–5144. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Okano, K.; Rohrmann, G.F. Characterization of the replication of a baculovirus mutant lacking the DNA polymerase gene. Virology 2005, 331, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Redman, E.M.; Wilson, K.; Cory, J.S. Trade-offs and mixed infections in an obligate-killing insect pathogen. J. Anim. Ecol. 2016, 85, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Simon, O.; Palma, L.; Beperet, I.; Munoz, D.; Lopez-Ferber, M.; Caballero, P.; Williams, T. Sequence comparison between three geographically distinct Spodoptera frugiperda multiple nucleopolyhedrovirus isolates: Detecting positively selected genes. J. Invertebr. Pathol. 2011, 107, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L. Genomic sequence analysis of the Illinois strain of the Agrotis ipsilon multiple nucleopolyhedrovirus. Virus Genes 2009, 38, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Christian, P.D.; Gibb, N.; Kasprzak, A.B.; Richards, A. A rapid method for the identification and differentiation of Helicoverpa nucleopolyhedroviruses (NPV Baculoviridae) isolated from the environment. J. Virol. Methods 2001, 96, 51–65. [Google Scholar] [CrossRef]
- Lightner, D.V.; Redman, R.M.; Bell, T.A. Observations on the geographic distribution, pathogenesis and morphology of the baculovirus from Penaeus monodon Fabricius. Aquaculture 1983, 32, 209–233. [Google Scholar] [CrossRef]
- Crawford, A.M.; Zelazny, B.; Alfiler, A.R. Genotypic variation in geographical isolates of oryctes baculovirus. J. Gen. Virol. 1986, 67, 949–952. [Google Scholar] [CrossRef]
- Gettig, R.R.; McCarthy, W.J. Genotypic variation among wild isolates of Heliothis spp nuclear polyhedrosis viruses from different geographical regions. Virology 1982, 117, 245–252. [Google Scholar] [CrossRef]
- Baillie, V.L.; Bouwer, G. High levels of genetic variation within Helicoverpa armigera nucleopolyhedrovirus populations in individual host insects. Arch. Virol. 2012, 157, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Baillie, V.L.; Bouwer, G. High levels of genetic variation within core Helicoverpa armigera nucleopolyhedrovirus genes. Virus Genes 2012, 44, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Baillie, V.L.; Bouwer, G. Development of highly sensitive assays for detection of genetic variation in key Helicoverpa armigera nucleopolyhedrovirus genes. J. Virol. Methods 2011, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.W.; Jordan, F.L.; Maier, R.M. Analysis of artifacts suggests DGGE should not be used for quantitative diversity analysis. J. Microbiol. Methods 2013, 92, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Lueders, T.; Friedrich, M.W. Evaluation of PCR amplification bias by terminal restriction fragment length polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Appl. Environ. Microbiol. 2003, 69, 320–326. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013. [Google Scholar] [CrossRef]
- Yu, X.; Sun, S. Comparing a few SNP calling algorithms using low-coverage sequencing data. BMC Bioinform. 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Noune, C.; Hauxwell, C. Complete genome sequences of seven helicoverpa armigera SNPV-AC53-Derived strains. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Noune, C.; Hauxwell, C. Complete genome sequences of helicoverpa armigera single nucleopolyhedrovirus strains AC53 and H25EA1 from Australia. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 September 2014).
- Gordon, A.; Hannon, G.J. Fastx-toolkit. FASTQ/A short-reads pre-processing tools. 2010; unpublished work. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. Available online: https://arxiv.org/abs/1303.3997 (accessed on 26 May 2013).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Institute, B. Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 9 September 2016).
- Pierre, L. JVarkit: Java Utilities for Bioinformatics. Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.822.1547&rep=rep1&type=pdf (accessed on 26 May 2015).
- Bushnell, B. BBMap Short Read Aligner. Available online: http://sourceforge.net/projects/bbmap (accessed on 18 September 2016).
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet-next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Microsoft R Open. Available online: https://mran.revolutionanalytics.com/rro/ (accessed on 6 May 2016).
- Kent, J. kentUtils. Available online: https://github.com/ENCODE-DCC/kentUtils (accessed on 12 September 2014).
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Noune, C. The Invertebrates & Microbiology Group Pipelines, GitHub, Queensland University of Technology. Available online: https://github.com/CNoune/IMG_pipelines (accessed on 5 September 2016).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, W.-J.; Wong, J.; Chun, G.; Lu, A.; McCutchen, B.; Presnail, J.; Herrmann, R.; Dolan, M.; Tingey, S.; et al. Comparative analysis of the complete genome sequences of Helicoverpa zea and Helicoverpa armigera single-nucleocapsid nucleopolyhedroviruses. J. Gen. Virol. 2002, 83, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; IJkel, W.F.; Tarchini, R.; Sun, X.; Sandbrink, H.; Wang, H.; Peters, S.; Zuidema, D.; Lankhorst, R.K.; Vlak, J.M. The sequence of the Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus genome. J. Gen. Virol. 2001, 82, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Marnellos, G.; Kammerer, S.; Hoyal, C.R.; Shi, M.M.; Cantor, C.R.; Braun, A. Large-scale validation of single nucleotide polymorphisms in gene regions. Genome Res. 2004, 14, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Piepho, H.-P. Optimal marker density for interval mapping in a backcross population. Heredity 2000, 84, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Beissinger, T.M.; Hirsch, C.N.; Sekhon, R.S.; Foerster, J.M.; Johnson, J.M.; Muttoni, G.; Vaillancourt, B.; Buell, C.R.; Kaeppler, S.M.; De Leon, N. Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics 2013, 193, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Gilles, A.; Meglécz, E.; Pech, N.; Ferreira, S.; Malausa, T.; Martin, J.-F. Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genom. 2011, 12, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J. The effect of sequencing errors on metagenomic gene prediction. BMC Genom. 2009, 10, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, M.C.; Prosperi, L.; Bruselles, A.; Abbate, I.; Rozera, G.; Vincenti, D.; Solmone, M.C.; Capobianchi, M.R.; Ulivi, G. Combinatorial analysis and algorithms for quasispecies reconstruction using next-generation sequencing. BMC Bioinform. 2011, 12, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Puente-Sánchez, F.; Aguirre, J.; Parro, V. A novel conceptual approach to read-filtering in high-throughput amplicon sequencing studies. Nucleic Acids Res. 2016, 44, e40. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2013, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Clarridge, J.E. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [PubMed]
- Mignard, S.; Flandrois, J. 16S rRNA sequencing in routine bacterial identification: A 30-month experiment. J. Microbiol. Methods 2006, 67, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.J.; Koren, O.; Hugenholtz, P.; DeSantis, T.Z.; Walters, W.A.; Caporaso, J.G.; Angenent, L.T.; Knight, R.; Ley, R.E. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012, 6, 94–103. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer | Fragment Size |
|---|---|---|
| BRO-A | * 5′-CATTTGCAAGGATATTGGAGT-3′ # 5′-AAGCTCGTTGGTTATCACAT-3′ | 365 bp |
| DNA Polymerase | * 5′-GTATGACTTATCACGACAATTGC-3′ # 5′-CGGTTTGCATATGTACTCTG-3′ | 325 bp |
| Genotype | Reads | Relative Abundance % |
|---|---|---|
| G_33554431 # | 258084 | 97.03 |
| G_33554303 | 1643 | 0.62 |
| G_33552383 | 787 | 0.30 |
| G_16777215 | 666 | 0.25 |
| G_33554423 | 533 | 0.20 |
| G_25165823 | 437 | 0.16 |
| G_33554430 | 437 | 0.16 |
| G_33292287 | 400 | 0.15 |
| G_31457279 | 393 | 0.15 |
| G_33554429 | 261 | 0.10 |
| G_33554399 | 228 | 0.09 |
| G_33554427 | 213 | 0.08 |
| G_33553919 * | 138 | 0.05 |
| G_33554175 | 129 | 0.05 |
| G_33546239 | 123 | 0.05 |
| G_33554367 | 105 | 0.04 |
| G_29360127 | 103 | 0.04 |
| G_33030143 | 103 | 0.04 |
| G_33550335 | 92 | 0.03 |
| G_33552255 | 68 | 0.03 |
| G_33521663 | 62 | 0.02 |
| G_33554415 | 56 | 0.02 |
| G_33554428 | 55 | 0.02 |
| G_20971519 | 52 | 0.02 |
| G_33553407 | 48 | 0.02 |
| G_23068671 | 35 | 0.01 |
| G_33554239 | 28 | 0.01 |
| G_33538047 | 21 | 0.01 |
| Genotype | Reads | Relative Abundance % |
|---|---|---|
| AC53-T2 BRO-A G_1 | 104,065 | 54.27 |
| AC53-T2 BRO-A G_0 | 87,689 | 45.73 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noune, C.; Hauxwell, C. MetaGaAP: A Novel Pipeline to Estimate Community Composition and Abundance from Non-Model Sequence Data. Biology 2017, 6, 14. https://doi.org/10.3390/biology6010014
Noune C, Hauxwell C. MetaGaAP: A Novel Pipeline to Estimate Community Composition and Abundance from Non-Model Sequence Data. Biology. 2017; 6(1):14. https://doi.org/10.3390/biology6010014
Chicago/Turabian StyleNoune, Christopher, and Caroline Hauxwell. 2017. "MetaGaAP: A Novel Pipeline to Estimate Community Composition and Abundance from Non-Model Sequence Data" Biology 6, no. 1: 14. https://doi.org/10.3390/biology6010014
APA StyleNoune, C., & Hauxwell, C. (2017). MetaGaAP: A Novel Pipeline to Estimate Community Composition and Abundance from Non-Model Sequence Data. Biology, 6(1), 14. https://doi.org/10.3390/biology6010014