
The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis

Abstract
:1. Introduction
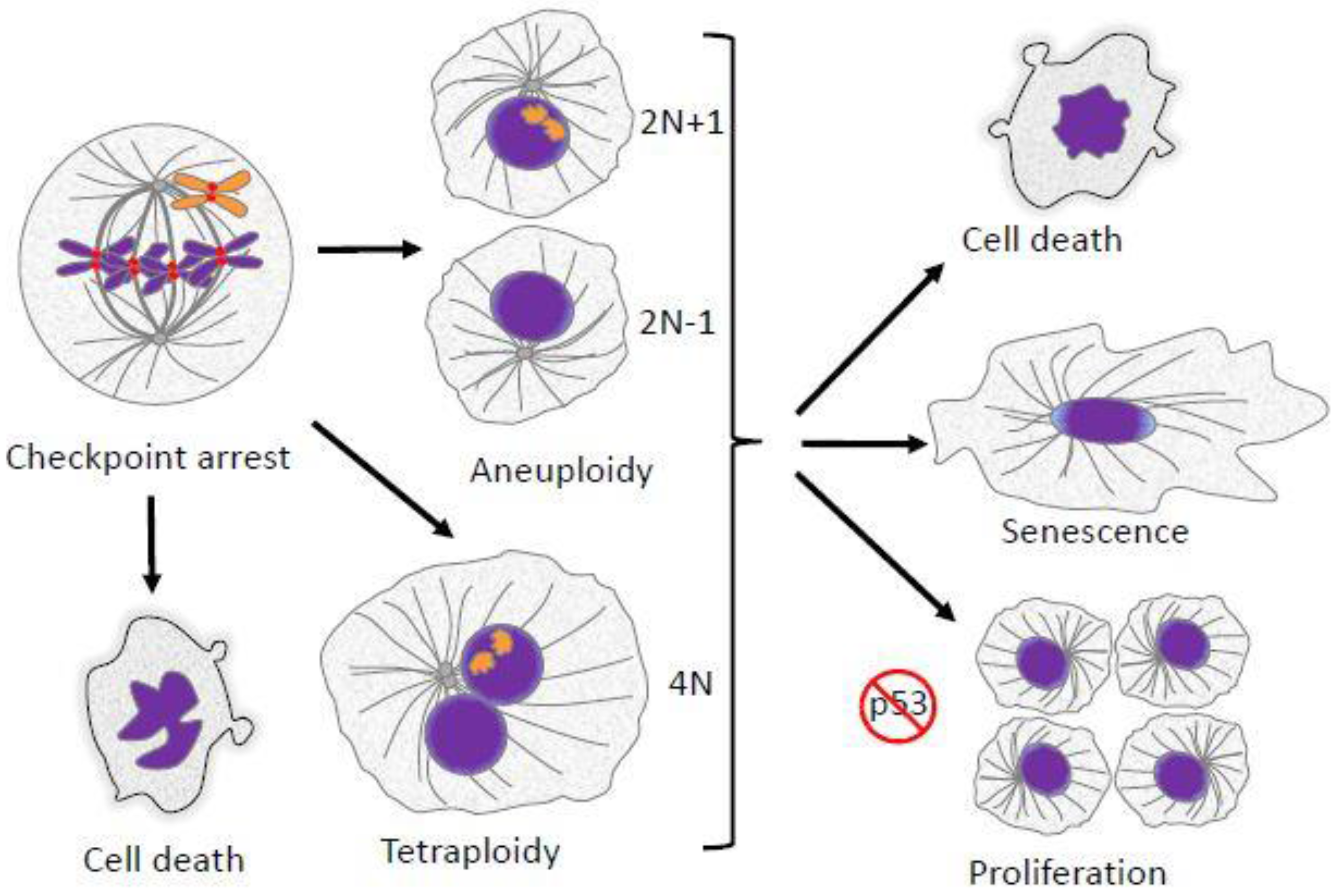
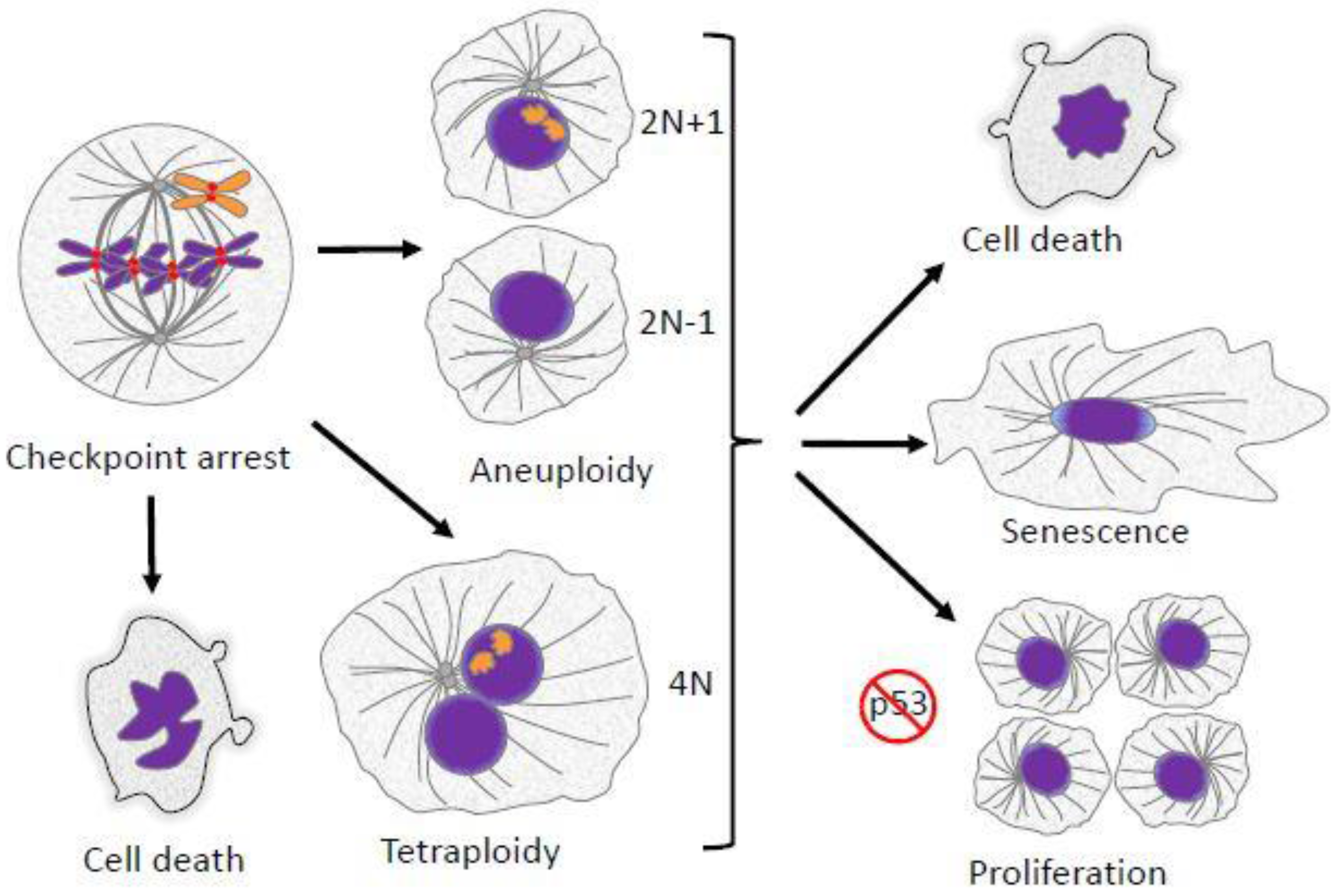
2. Aneuploidy in Mitosis
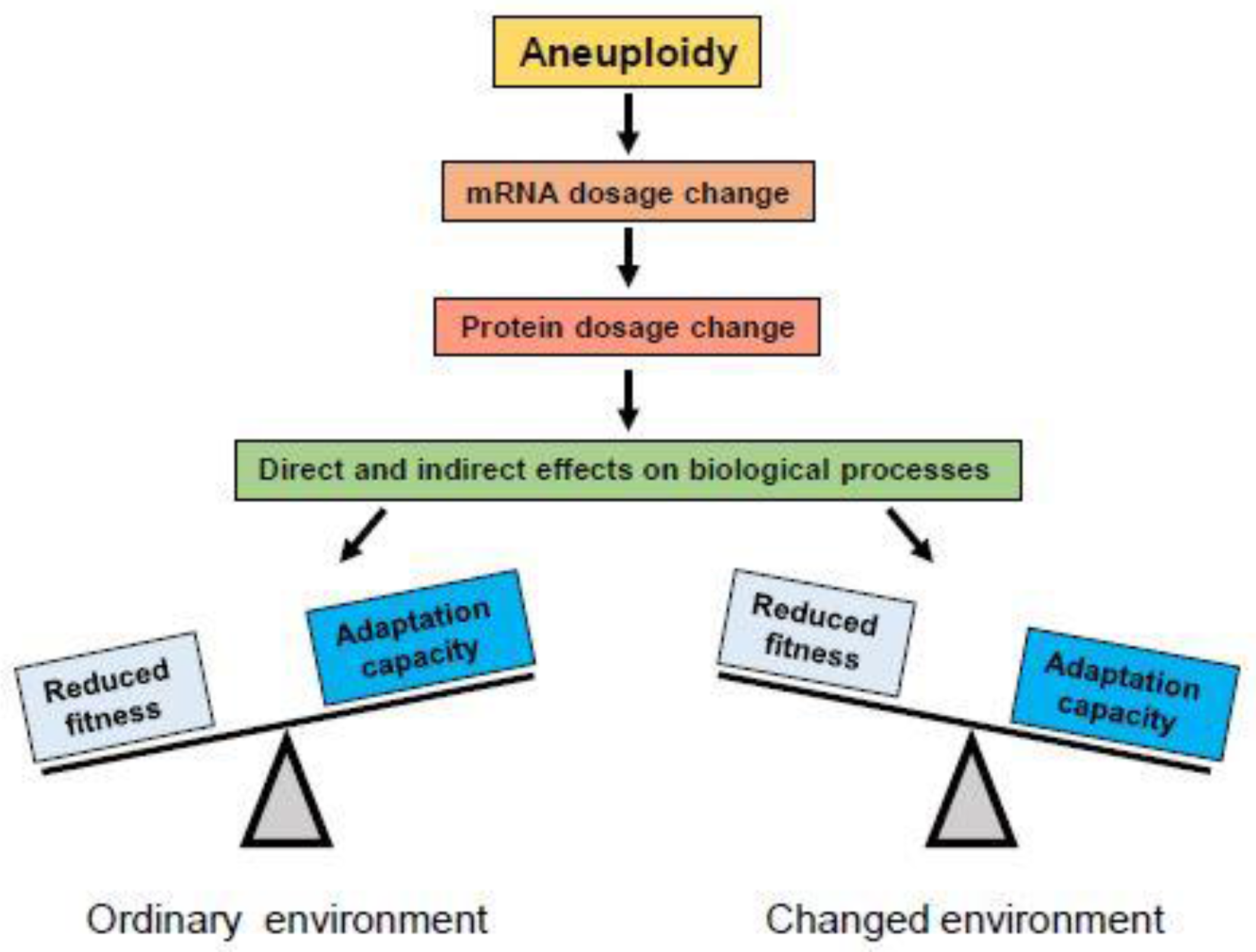
2.1. Effects of Aneuploidy on Gene Dosage
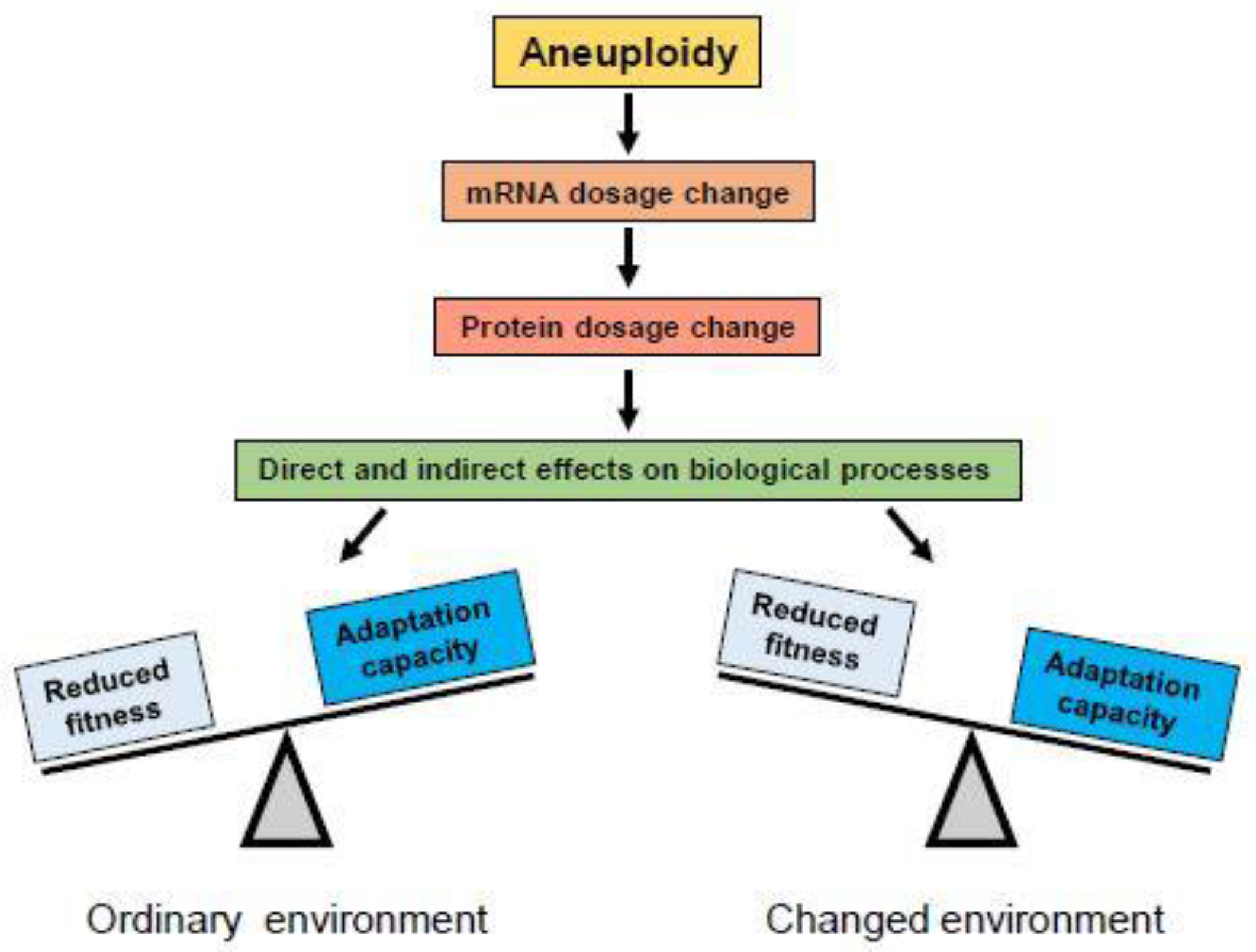
2.2. Effects of Aneuploidy on Cellular Fitness
2.3. Aneuploidy in Fungi
2.4. Aneuploidy in Mammalian Cells
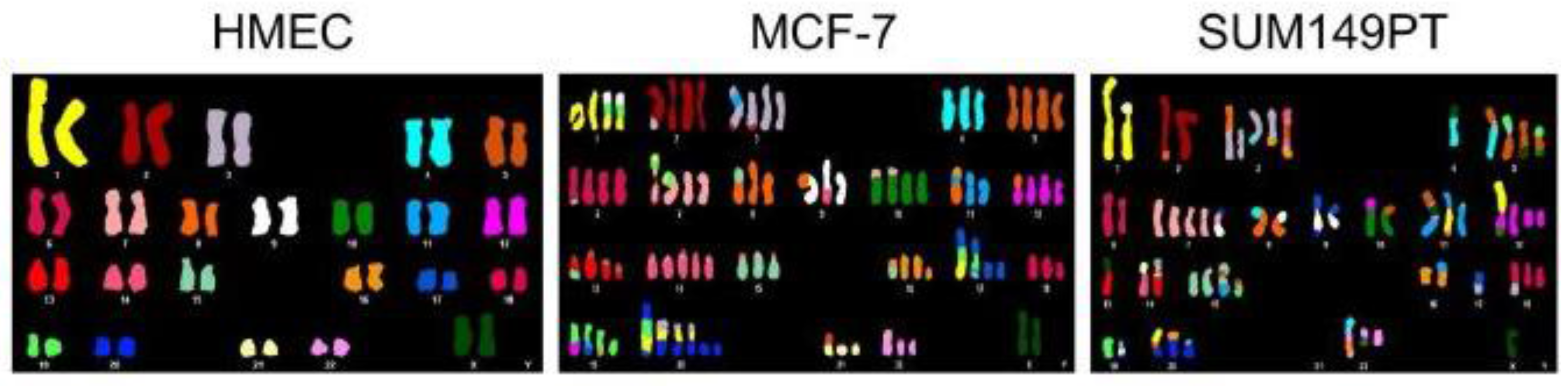
2.5. Aneuploidy as a Driver for Genomic and Chromosomal Instability
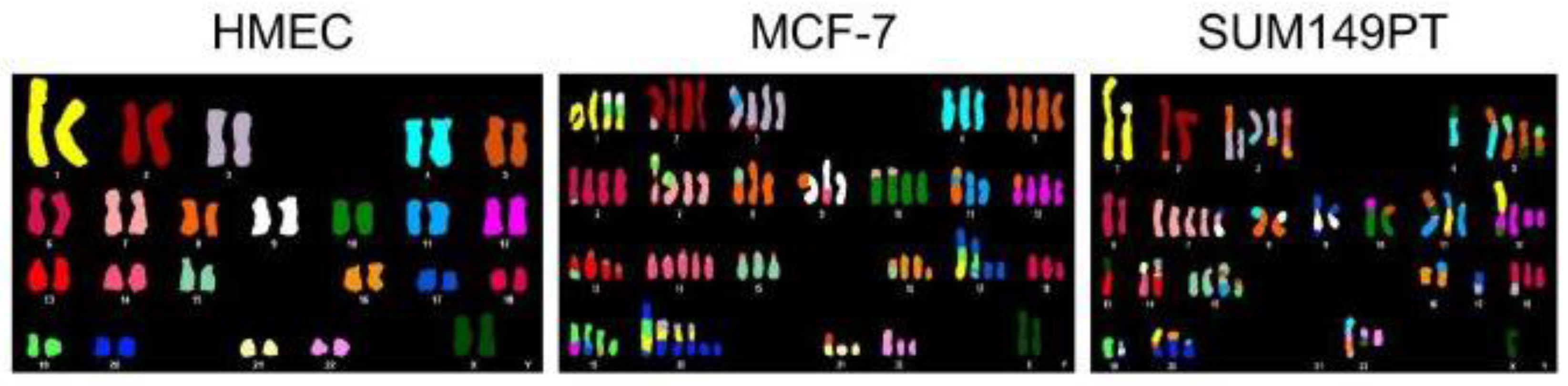
2.6. Aneuploidy and Cancer
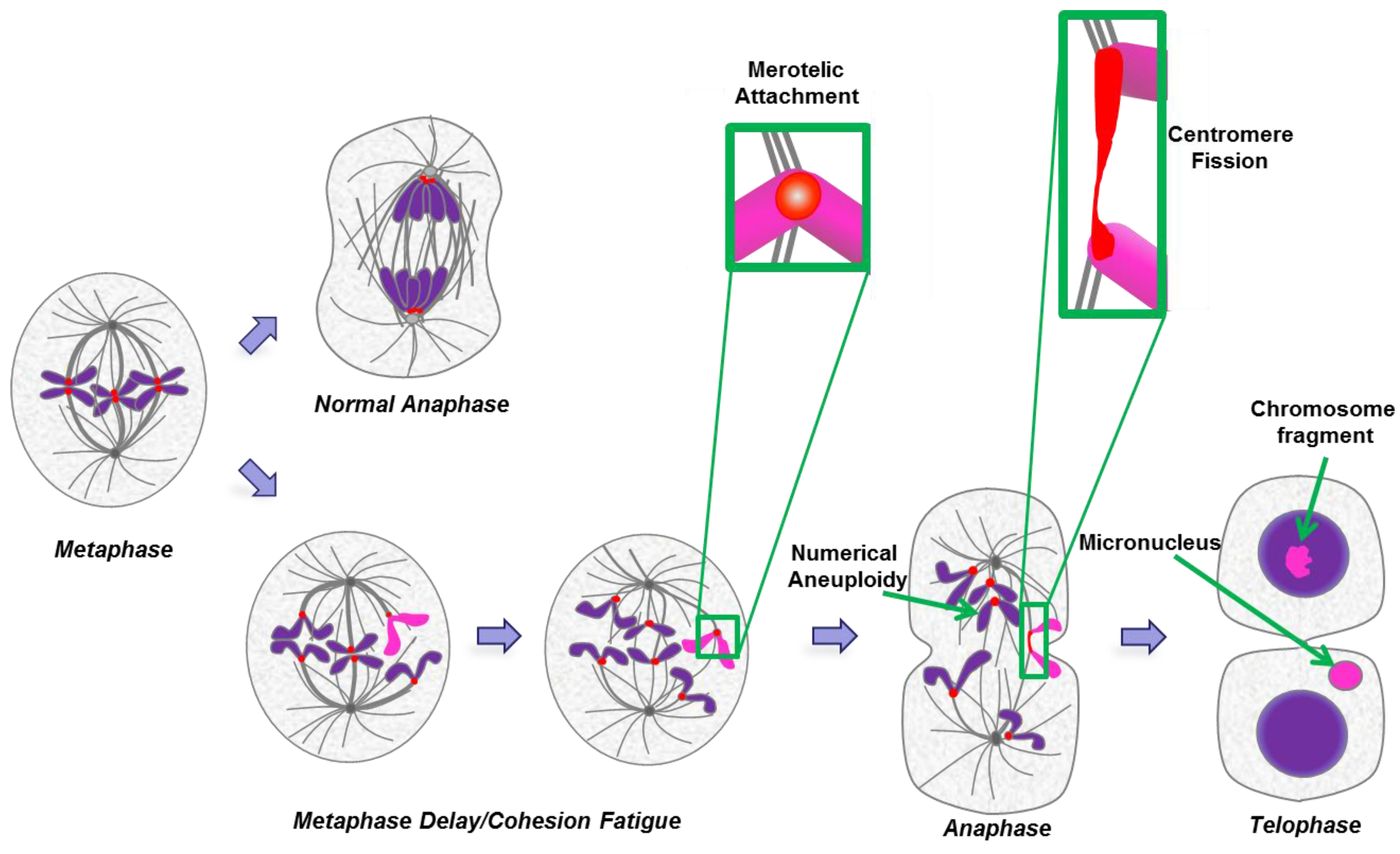
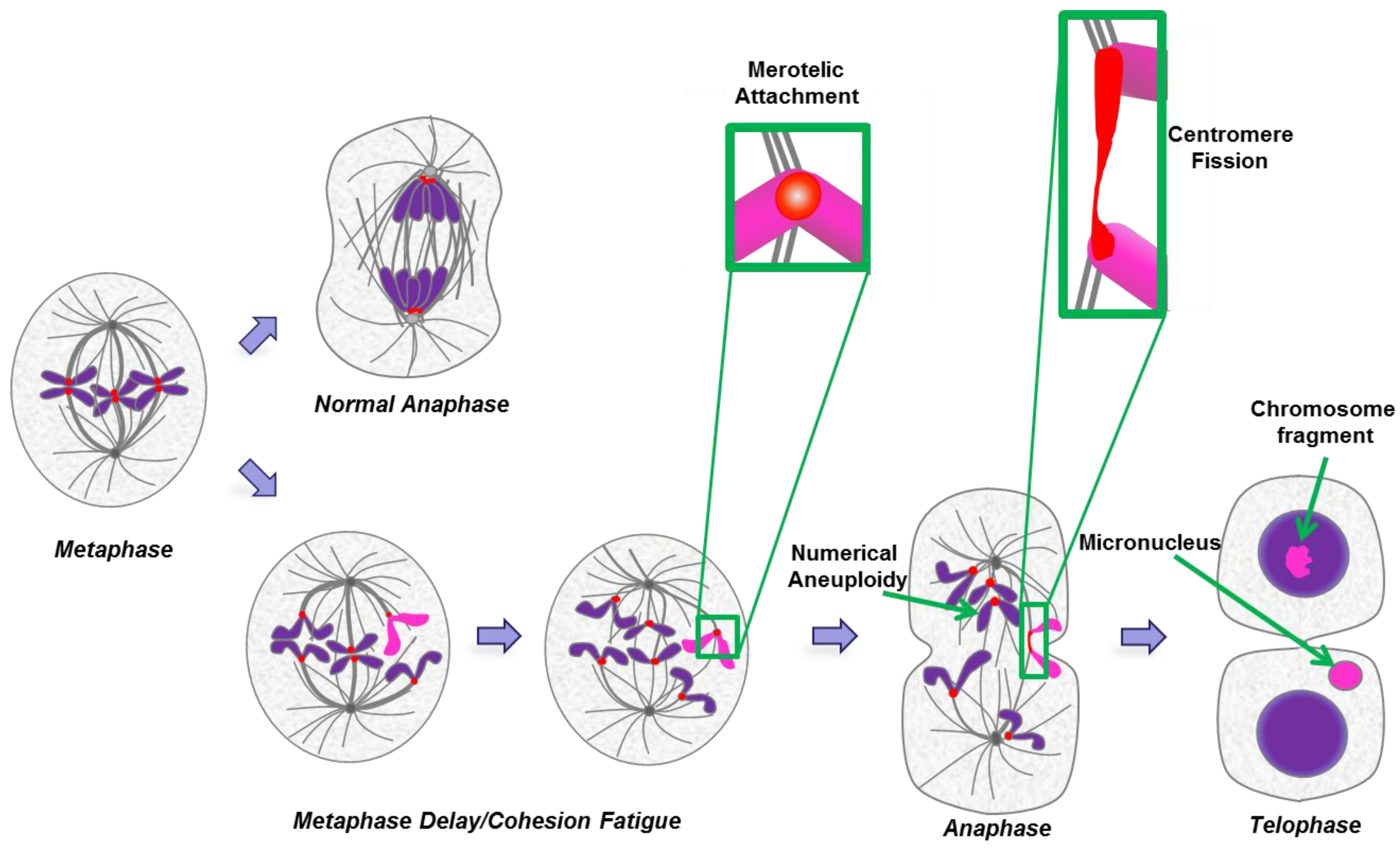
2.7. Cohesion Fatigue and Centromere Fission
2.8. Modern Analysis and Implications of Cancer Aneuploidy
2.9. Aneuploidy and Drug Resistance
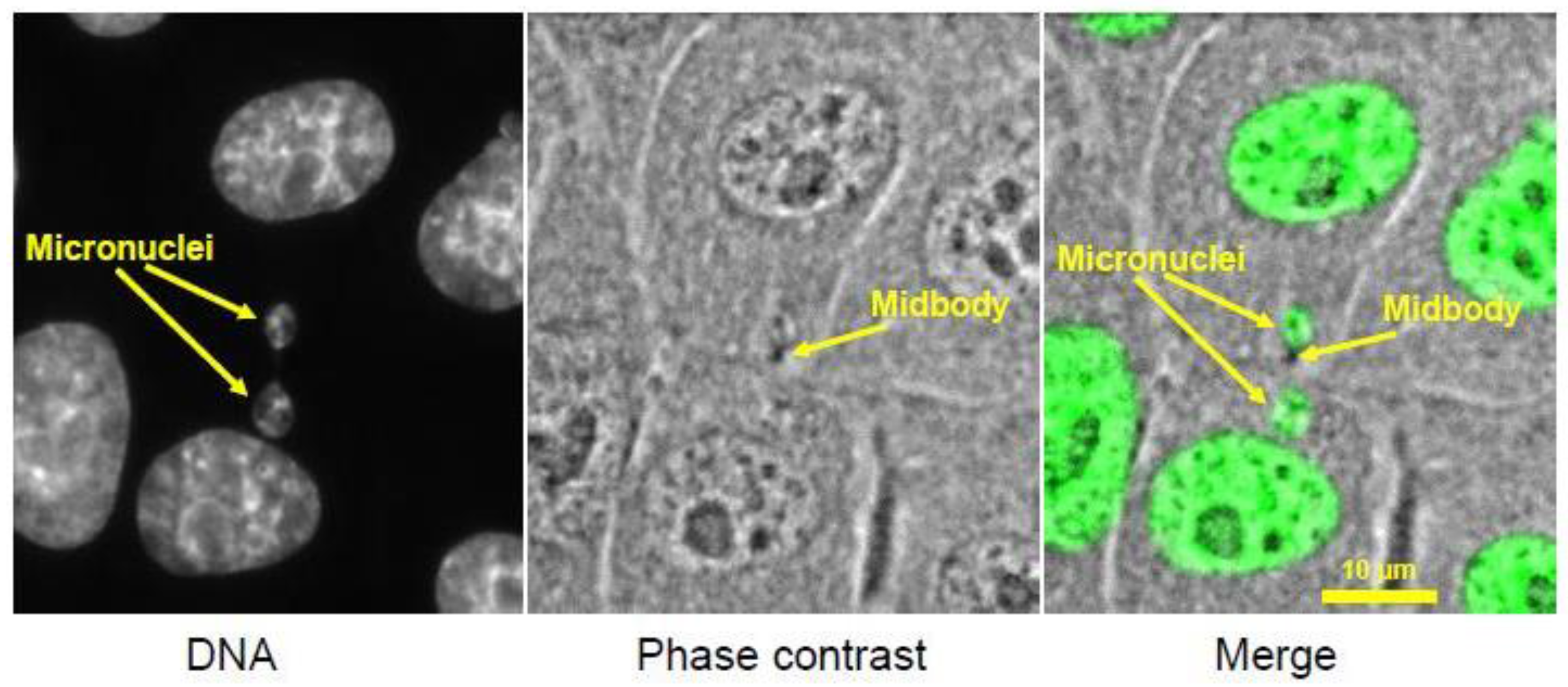
3. Micronuclei
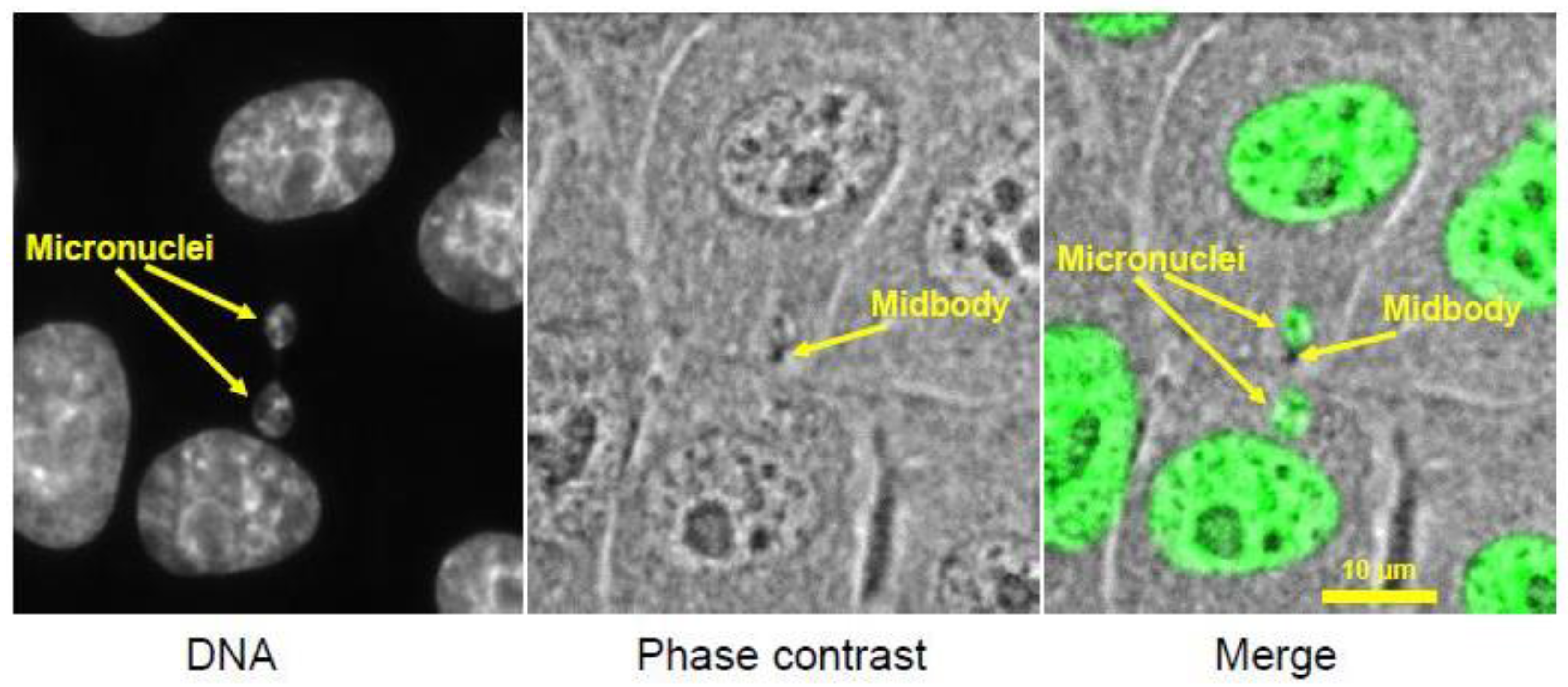
3.1. Footprint of Mitotic Error
3.2. Causes and Consequences of Chromosome Entrapment in Micronuclei
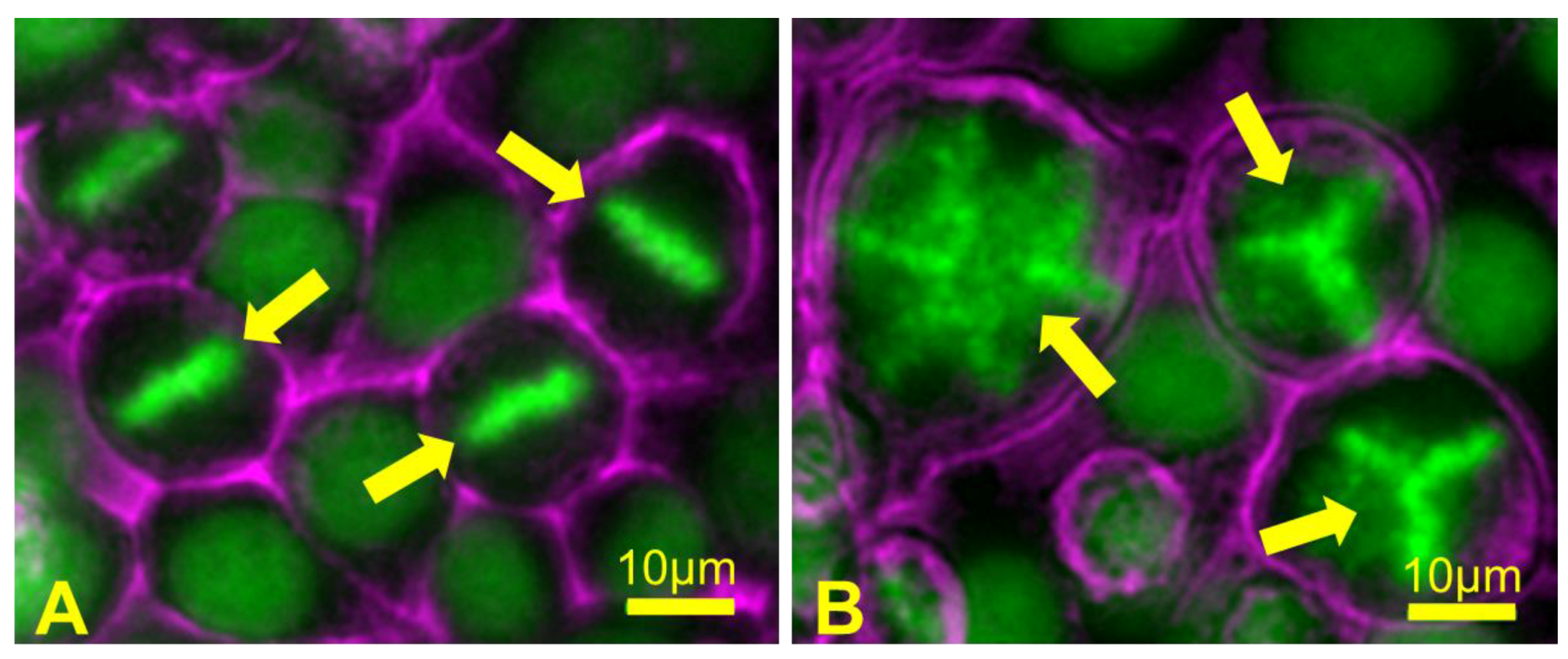
4. Aneuploidy in Meiosis
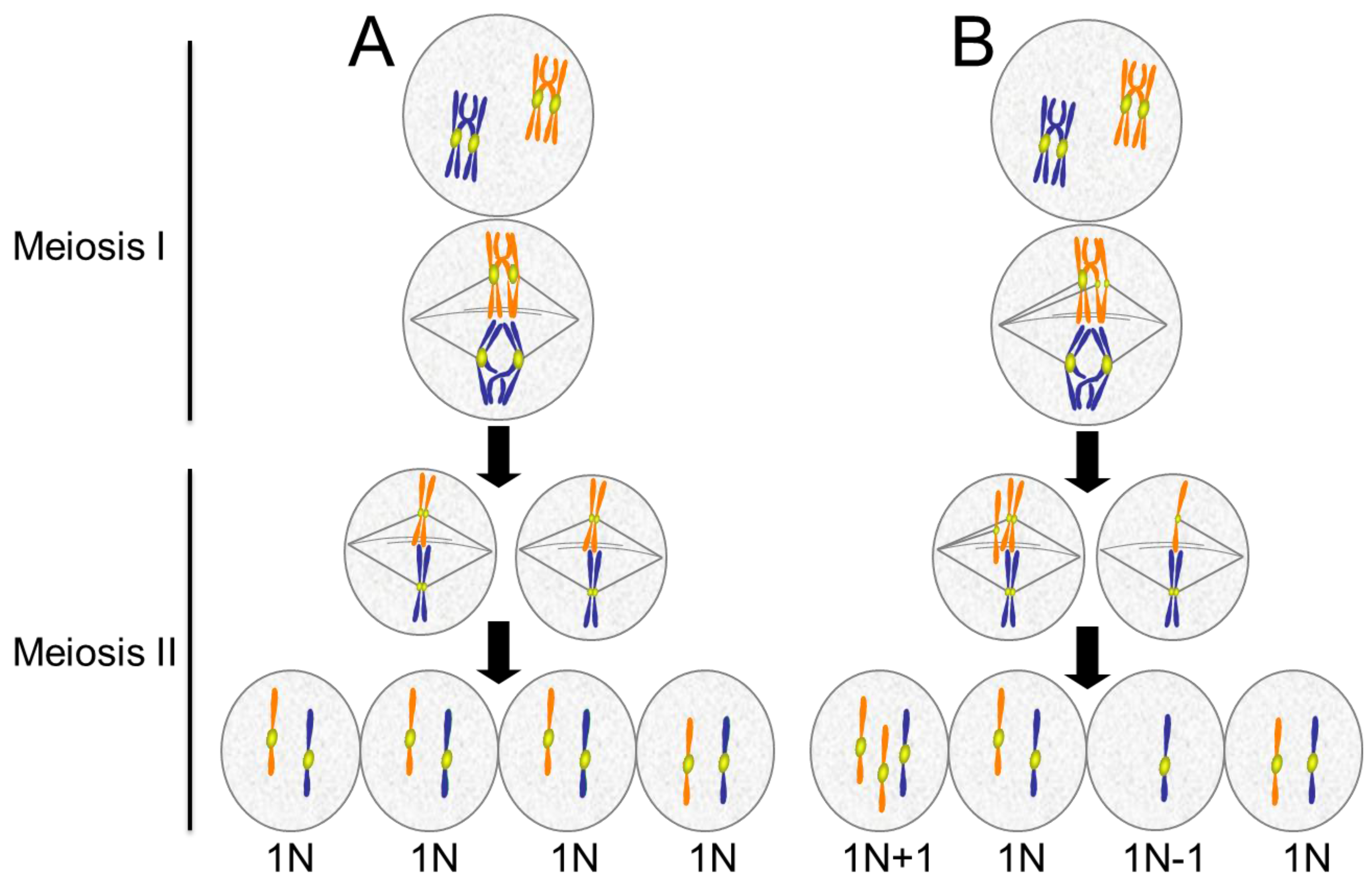
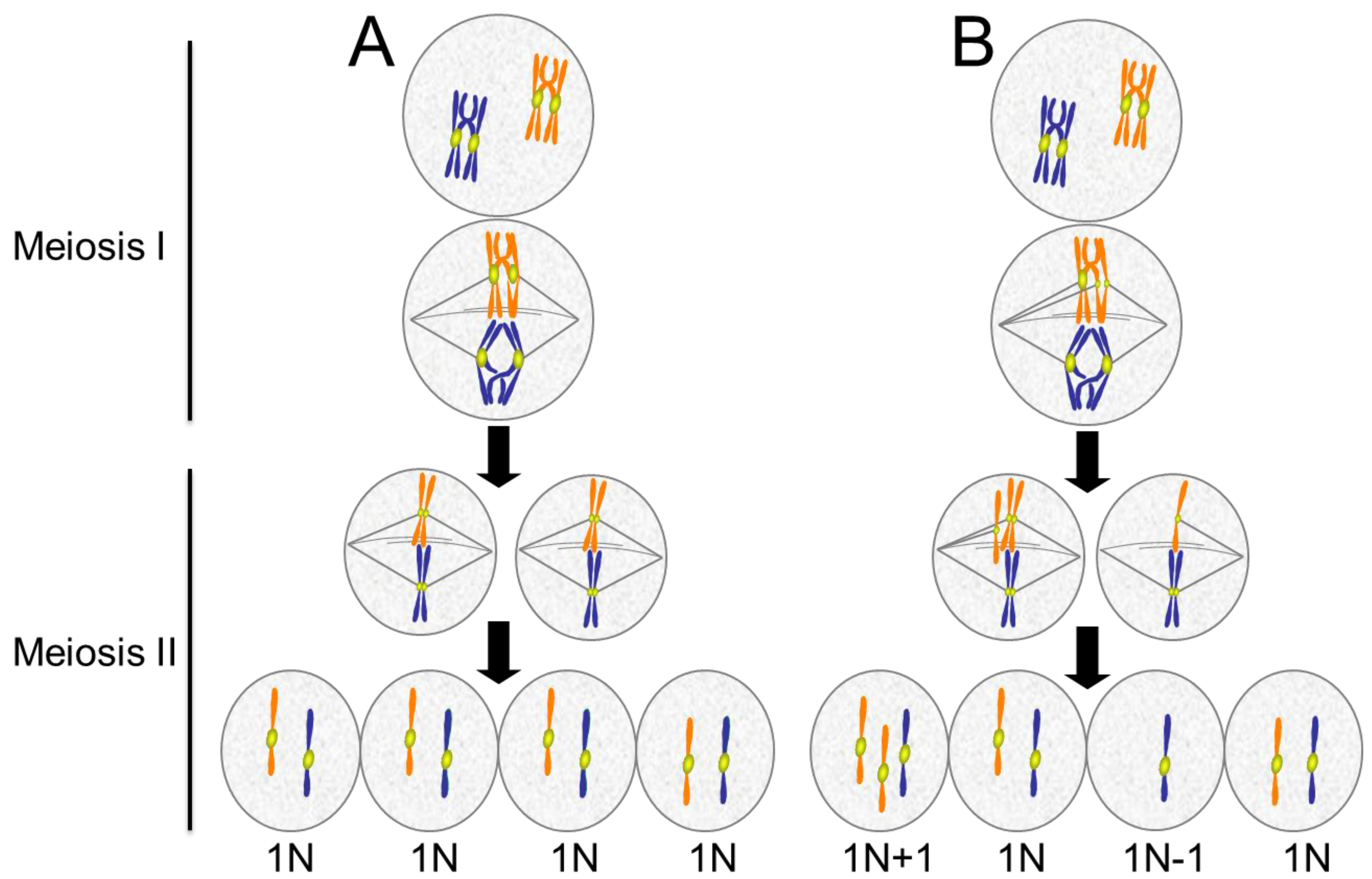
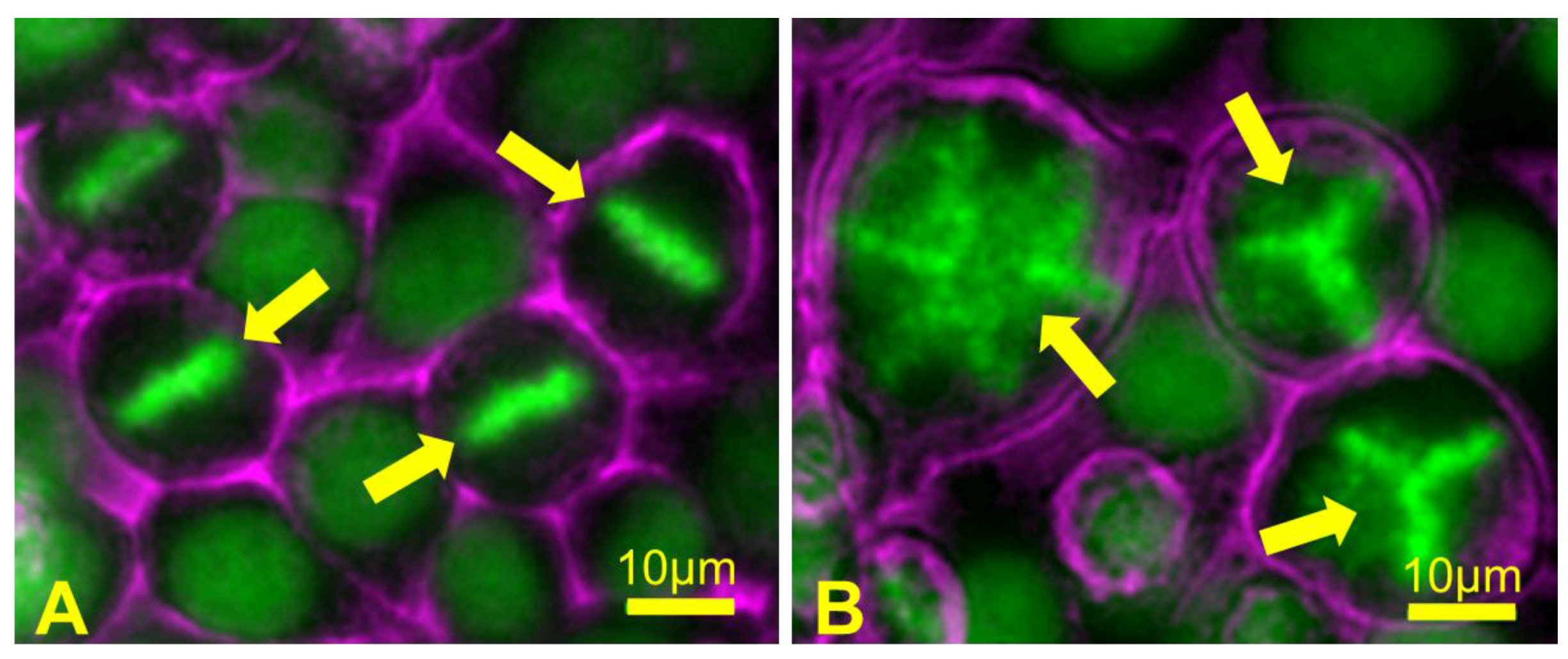
4.1. Causes of Aneuploidy in Meiosis
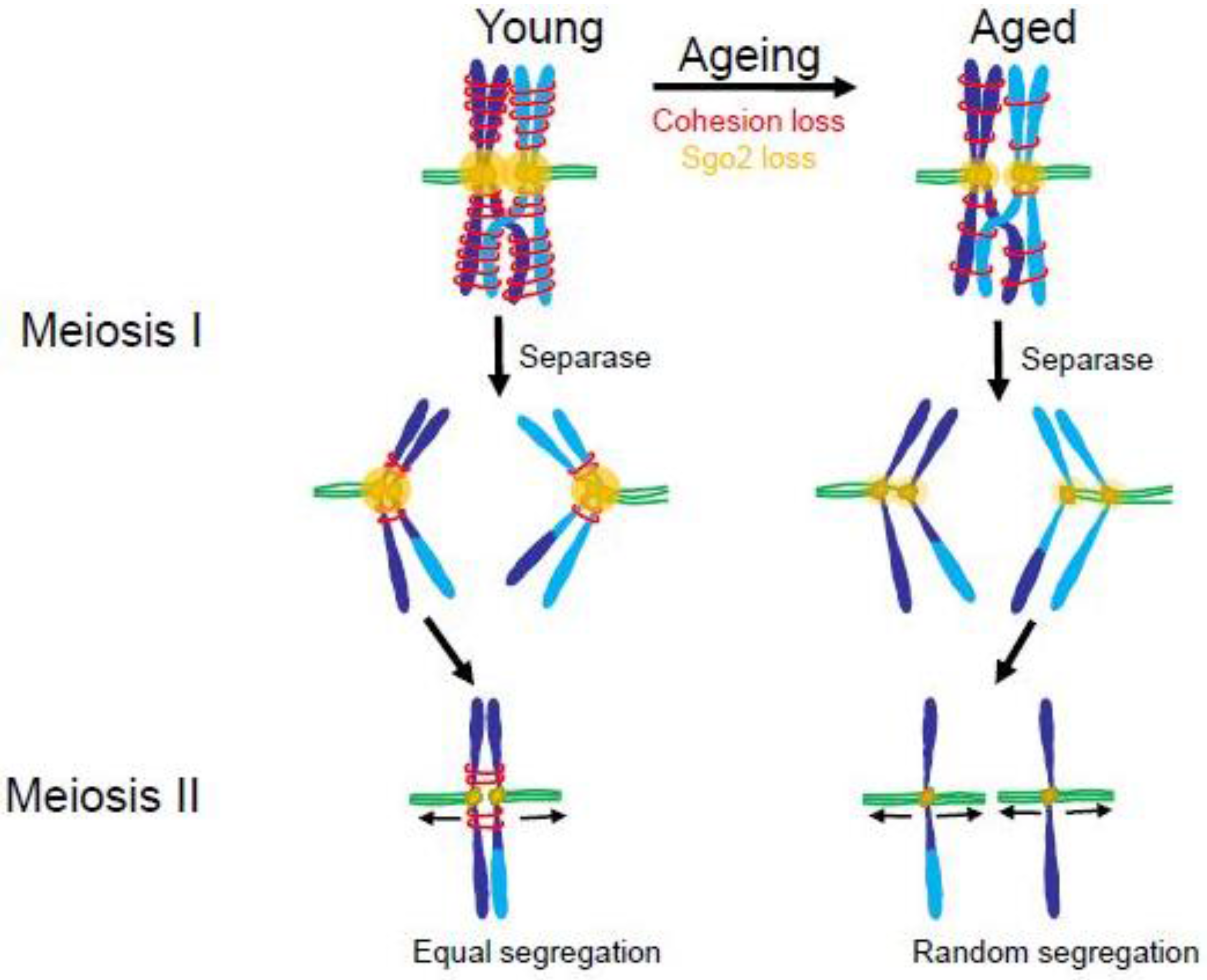
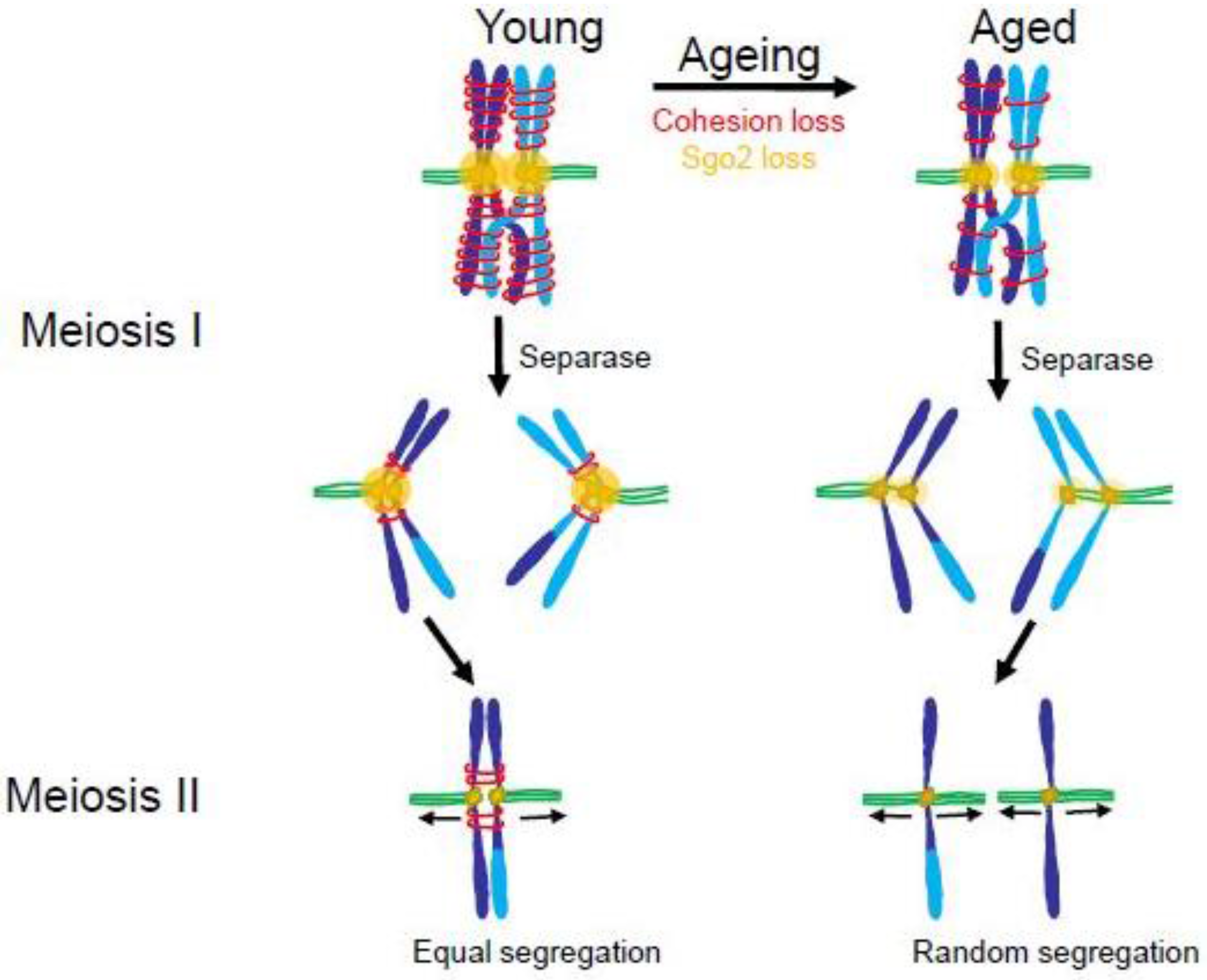
4.2. The Maternal Age Effect
4.3. Consequences of Aneuploidy in Meiosis
4.4. Meiotic Aneuploidy and Cancer
5. Polyploidy
5.1. Sources for Polyploidy
5.2. Polyploidy in Fungi
5.3. Polyploidy in Animals and Plants
5.4. Polyploidy Can Lead to Aneuploidy
5.5. Polyploidy in Cancer
6. Ploidy Aberrations and P53
6.1. Ideas in Evolution and Cancer
6.2. Concepts for Activation and Function
7. Conclusions and Perspectives for Human Health
Acknowledgments
Conflicts of interest
References
- Gascoigne, K.E.; Taylor, S.S. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. P53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Duensing, S. Guilt by association? P53 and the development of aneuploidy in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Dalton, W.B.; Nandan, M.O.; Moore, R.T.; Yang, V.W. Human cancer cells commonly acquire DNA damage during mitotic arrest. Cancer Res. 2007, 67, 11487–11492. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Cesare, A.J.; Fitzpatrick, J.A.; Lazzerini-Denchi, E.; Karlseder, J.A. Telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol. 2012, 19, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Bekier, M.E.; Fischbach, R.; Lee, J.; Taylor, W.R. Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol. Cancer. Ther. 2009, 8, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Chu, R.; Terrano, D.T.; Chambers, T.C. Cdk1/cyclin B plays a key role in mitotic arrest-induced apoptosis by phosphorylation of Mcl-1, promoting its degradation and freeing Bak from sequestration. Biochem. Pharmacol. 2012, 83, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Sakurikar, N.; Eichhorn, J.M.; Chambers, T.C. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J. Biol. Chem. 2012, 287, 39193–39204. [Google Scholar] [CrossRef] [PubMed]
- Sloss, O.; Topham, C.; Diez, M.; Taylor, S. Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget 2016, 7, 5176–5192. [Google Scholar] [CrossRef]
- Orth, J.D.; Loewer, A.; Lahav, G.; Mitchison, T.J. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell 2012, 23, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Uetake, Y.; Sluder, G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr. Biol. 2010, 20, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, N.; Rancati, G.; Zhu, J.; Bradford, W.D.; Saraf, A.; Florens, L.; Sanderson, B.W.; Hattem, G.L.; Li, R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 2010, 468, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Stingele, S.; Stoehr, G.; Peplowska, K.; Cox, J.; Mann, M.; Storchova, Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 2012, 8, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Upender, M.B.; Habermann, J.K.; McShane, L.M.; Korn, E.L.; Barrett, J.C.; Difilippantonio, M.J.; Ried, T. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res. 2004, 64, 6941–6949. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.M.; Dephoure, N.; Panneerselvam, A.; Tucker, C.M.; Whittaker, C.A.; Gygi, S.P.; Dunham, M.J.; Amon, A. Identification of aneuploidy-tolerating mutations. Cell 2010, 143, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Rancati, G.; Pavelka, N.; Fleharty, B.; Noll, A.; Trimble, R.; Walton, K.; Perera, A.; Staehling-Hampton, K.; Seidel, C.W.; Li, R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 2008, 135, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Dephoure, N.; Hwang, S.; O’Sullivan, C.; Dodgson, S.E.; Gygi, S.P.; Amon, A.; Torres, E.M. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. Elife 2014, 3, e03023. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 2008, 322, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.J.; Amon, A. New insights into the troubles of aneuploidy. Annu. Rev. Cell Dev. Biol. 2012, 28, 189–214. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.Y.; Boselli, M.; Dunham, M.J.; Amon, A. Effects of aneuploidy on cellular physiology and Cell Div.ision in haploid yeast. Science 2007, 317, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Gallone, B.; Steensels, J.; Prahl, T.; Soriaga, L.; Saels, V.; Herrera-Malaver, B.; Merlevede, A.; Roncoroni, M.; Voordeckers, K.; Miraglia, L.; et al. Domestication and divergence of saccharomyces cerevisiae beer yeasts. Cell 2016, 166, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, A.; Simpson, J.T.; Salinas, F.; Barre, B.; Parts, L.; Zia, A.; Nguyen Ba, A.N.; Moses, A.M.; Louis, E.J.; Mustonen, V.; et al. A high-definition view of functional genetic variation from natural yeast genomes. Mol. Biol. Evol. 2014, 31, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Dunham, M.J.; Badrane, H.; Ferea, T.; Adams, J.; Brown, P.O.; Rosenzweig, F.; Botstein, D. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2002, 99, 16144–16149. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.; Richter, C.; Kvitek, D.J.; Pugh, T.; Sherlock, G. Analysis of the Saccharomyces cerevisiae pan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments. Genome Res. 2012, 22, 908–924. [Google Scholar] [CrossRef] [PubMed]
- Voordeckers, K.; Kominek, J.; Das, A.; Espinosa-Cantu, A.; De Maeyer, D.; Arslan, A.; Van Pee, M.; Van Der Zande, E.; Meert, W.; Yang, Y.; et al. Adaptation to high ethanol reveals complex evolutionary pathways. PLoS Genet. 2015, 11, e1005635. [Google Scholar] [CrossRef] [PubMed]
- Kvitek, D.J.; Will, J.L.; and Gasch, A.P. Variations in stress sensitivity and genomic expression in diverse S. cerevisiae isolates. PLoS Genet. 2008, 4, e1000223. [Google Scholar]
- Borneman, A.R.; Desany, B.A.; Riches, D.; Affourtit, J.P.; Forgan, A.H.; Pretorius, I.S.; Egholm, M.; Chambers, P.J. Whole-genome comparison reveals novel genetic elements that characterize the genome of industrial strains of Saccharomyces cerevisiae. PLoS Genet. 2011, 7, e1001287. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.R.; Roberts, C.J.; Dai, H.; Jones, A.R.; Meyer, M.R.; Slade, D.; Burchard, J.; Dow, S.; Ward, T.R.; Kidd, M.J.; et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat. Genet. 2000, 25, 333–337. [Google Scholar] [PubMed]
- Gresham, D.; Desai, M.M.; Tucker, C.M.; Jenq, H.T.; Pai, D.A.; Ward, A.; DeSevo, C.G.; Botstein, D.; Dunham, M.J. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008, 4, e1000303. [Google Scholar] [CrossRef] [PubMed]
- Fries, B.C.; Casadevall, A. Serial isolates of Cryptococcus neoformans from patients with AIDS differ in virulence for mice. J. Infect. Dis. 1998, 178, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Selmecki, A.; Gerami-Nejad, M.; Paulson, C.; Forche, A.; Berman, J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 2008, 68, 624–641. [Google Scholar] [CrossRef] [PubMed]
- Selmecki, A.M.; Dulmage, K.; Cowen, L.E.; Anderson, J.B.; Berman, J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet. 2009, 5, e1000705. [Google Scholar] [CrossRef] [PubMed]
- Selmecki, A.; Forche, A.; Berman, J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 2006, 313, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, A.C.; Fu, M.S.; Mukaremera, L.; Li, Z.; Ormerod, K.L.; Fraser, J.A.; Berman, J.; Nielsen, K. Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. mBio 2015, 6, e01340-15. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 2014, 5, 4825–4835. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Baker, D.; Zhang, J.; Andrews, P.W.; Barbaric, I. Aneuploidy in pluripotent stem cells and implications for cancerous transformation. Protein Cell 2014, 5, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Werbowetski-Ogilvie, T.E.; Bosse, M.; Stewart, M.; Schnerch, A.; Ramos-Mejia, V.; Rouleau, A.; Wynder, T.; Smith, M.J.; Dingwall, S.; Carter, T.; et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009, 27, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cheng, L.; Jia, Y.; Liu, G.; Li, C.; Song, S.; Bradley, A.; Huang, Y. Aneuploid embryonic stem cells exhibit impaired differentiation and increased neoplastic potential. EMBO J. 2016, 35, 2285–2300. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.A.; Seidel, C.W.; Box, A.C.; Rancati, G.; Li, R. Transcriptome analysis of tetraploid cells identifies Cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol. Biol. Cell 2016, 27, 3065–3084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Arena, S.; Gallicchio, M.; Zecchin, D.; Martini, M.; Flonta, S.E.; Stella, G.M.; Lamba, S.; Cancelliere, C.; Russo, M.; et al. Replacement of normal with mutant alleles in the genome of normal human cells unveils mutation-specific drug responses. Proc. Natl. Acad. Sci. USA 2008, 105, 20864–20869. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Eskiocak, U.; Kim, S.B.; Roig, A.I.; Hight, S.K.; Lulla, D.R.; Zou, Y.S.; Batten, K.; Wright, W.E.; Shay, J.W. Characterization of aneuploid populations with trisomy 7 and 20 derived from diploid human colonic epithelial cells. Neoplasia 2011, 13, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, S.D.; Douglas, T.A.; Nicholson, J.M.; Vila-Casadesus, M.; Kantzler, C.L.; Wangsa, D.; Barroso-Vilares, M.; Kale, S.D.; Logarinho, E.; Cimini, D. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci. Rep. 2016, 6, 22828–22839. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.A.; Zhu, J.; Li, R. Aneuploidy and chromosomal instability: A vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013, 32, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, E.L.; Dorer, R.K.; Murray, A.W.; Schuyler, S.C. Reduced Mad2 expression keeps relaxed kinetochores from arresting budding yeast in mitosis. Mol. Biol. Cell 2011, 22, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Pavelka, N.; Bradford, W.D.; Rancati, G.; Li, R. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet. 2012, 8, e1002719. [Google Scholar] [CrossRef] [PubMed]
- Sheltzer, J.M.; Blank, H.M.; Pfau, S.J.; Tange, Y.; George, B.M.; Humpton, T.J.; Brito, I.L.; Hiraoka, Y.; Niwa, O.; Amon, A. Aneuploidy drives genomic instability in yeast. Science 2011, 333, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.M.; Macedo, J.C.; Mattingly, A.J.; Wangsa, D.; Camps, J.; Lima, V.; Gomes, A.M.; Doria, S.; Ried, T.; Logarinho, E.; et al. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. Elife 2015, 4, e05068. [Google Scholar] [CrossRef] [PubMed]
- Passerini, V.; Ozeri-Galai, E.; de Pagter, M.S.; Donnelly, N.; Schmalbrock, S.; Kloosterman, W.P.; Kerem, B.; Storchova, Z. The presence of extra chromosomes leads to genomic instability. Nat. Commun. 2016, 7, 10754–10765. [Google Scholar] [CrossRef] [PubMed]
- Hansemann, D. Ueber asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung. Virchows Arch. 1890, 119, 299–326. [Google Scholar] [CrossRef]
- Ribbert, H. Zur frage der entstehung maligner tumoren. Naturwissenschaften 1914, 2, 676–679. (In German) [Google Scholar] [CrossRef]
- Zasadil, L.M.; Britigan, E.M.; Weaver, B.A. 2n or not 2n: Aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin. Cell Dev. Biol. 2013, 24, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Rasnick, D.; Li, R.; Winters, L.; Rausch, C.; Hehlmann, R. How aneuploidy may cause cancer and genetic instability. Anticancer Res. 1999, 19, 4887–4906. [Google Scholar] [PubMed]
- Zimonjic, D.; Brooks, M.W.; Popescu, N.; Weinberg, R.A.; Hahn, W.C. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res. 2001, 61, 8838–8844. [Google Scholar] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, Y.; Chi, Y.H.; Miyazato, A.; Sheleg, S.; Haller, K.; Peloponese, J.M., Jr.; Li, Y.; Ward, J.M.; Benezra, R.; Jeang, K.T. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007, 67, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, K.; Malureanu, L.; Baker, D.J.; Abraham, S.C.; Van Deursen, J.M. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J. Cell Biol. 2007, 179, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.S.; Liberal, V.; Chatterjee, A.; Kirchwegger, R.; Pasche, B.; Gerald, W.; Dobles, M.; Sorger, P.K.; Murty, V.V.; Benezra, R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001, 409, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Ricke, R.M.; Jeganathan, K.B.; Van Deursen, J.M. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J. Cell Biol. 2011, 193, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Hernando, E.; Diaz-Rodriguez, E.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Lowe, S.W.; Benezra, R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007, 11, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; De Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, Q.; Liu, T.; Swamy, M.; Fang, Y.; Xie, S.; Mahmood, R.; Yang, Y.M.; Xu, M.; Rao, C.V. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004, 64, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, T.; Fang, Y.; Xie, S.; Huang, X.; Mahmood, R.; Ramaswamy, G.; Sakamoto, K.M.; Darzynkiewicz, Z.; Xu, M.; et al. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood 2004, 103, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.K.; Wengenack, T.M.; Poduslo, J.F.; Van Deursen, J.M. Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol. Aging 2007, 28, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Baker, D.J.; D’USCIO, L.V.; Mozammel, G.; Katusic, Z.S.; Van Deursen, J.M. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke 2007, 38, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Rosenberg, M.A.; Jeganathan, K.B.; Hafner, A.V.; Michan, S.; Dai, J.; Baker, D.J.; Cen, Y.; Wu, L.E.; Sauve, A.A.; et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014, 33, 1438–1453. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Dawlaty, M.M.; Wijshake, T.; Jeganathan, K.B.; Malureanu, L.; Van Ree, J.H.; Crespo-Diaz, R.; Reyes, S.; Seaburg, L.; Shapiro, V.; et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat. Cell Biol. 2013, 15, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; Fitzpatrick, D.; Kidd, A.; Mehes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Boceno, M.; Rival, J.M.; Mechinaud, F.; David, A. High risk of malignancy in mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. 2002, 109, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Matsumoto, Y.; Morishima, K.; Izumi, H.; Matsumoto, H.; Ito, E.; Tsutsui, K.; Kobayashi, J.; Tauchi, H.; Kajiwara, Y.; et al. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am. J. Med. Genet. A 2006, 140, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Silk, A.D.; Montagna, C.; Verdier-Pinard, P.; Cleveland, D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.D.; Zasadil, L.M.; Holland, A.J.; Vitre, B.; Cleveland, D.W.; Weaver, B.A. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA 2013, 110, E4134–E4141. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 1993, 90, 9552–9556. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.C.; Chen, R.H.; Murray, A.W.; Salmon, E.D. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 1998, 141, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Komlodi-Pasztor, E.; Sackett, D.; Wilkerson, J.; Fojo, T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.J. The proliferation rate paradox in antimitotic chemotherapy. Mol. Biol. Cell 2012, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Frias, C.; Pampalona, J.; Genesca, A.; Tusell, L. Telomere dysfunction and genome instability. Front. Biosci. 2012, 17, 2181–2196. [Google Scholar] [CrossRef]
- Gimenez-Abian, J.F.; Sumara, I.; Hirota, T.; Hauf, S.; Gerlich, D.; De La Torre, C.; Ellenberg, J.; Peters, J.M. Regulation of sister chromatid cohesion between chromosome arms. Curr. Biol. 2004, 14, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Gorbsky, G.J. The mitotic spindle checkpoint. Curr. Biol. 2001, 11, R1001–R1004. [Google Scholar] [CrossRef]
- Bompard, G.; Rabeharivelo, G.; Cau, J.; Abrieu, A.; Delsert, C.; Morin, N. P21-activated kinase 4 (PAK4) is required for metaphase spindle positioning and anchoring. Oncogene 2013, 32, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Duijf, P.H.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cong, B.; Yuan, H.; Chen, L.; Lv, Y.; Bai, C.; Nan, X.; Shi, S.; Yue, W.; Pei, X. Overexpression of spindlin1 induces metaphase arrest and chromosomal instability. J. Cell. Physiol. 2008, 217, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Yazinski, S.A.; Nicolay, B.; Bryll, A.; Zou, L.; Dyson, N.J. Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol. Cell 2014, 53, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Longworth, M.S.; Dyson, N.J. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes. Dev. 2010, 24, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Daum, J.R.; Potapova, T.A.; Sivakumar, S.; Daniel, J.J.; Flynn, J.N.; Rankin, S.; Gorbsky, G.J. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr. Biol. 2011, 21, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Taylor, S.S. Cohesion fatigue explains why pharmacological inhibition of the APC/C induces a spindle checkpoint-dependent mitotic arrest. PLoS ONE 2012, 7, e49041. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.; Gassmann, R.; Oegema, K.; Desai, A. Uncoordinated loss of chromatid cohesion is a common outcome of extended metaphase arrest. PLoS ONE 2011, 6, e22969. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Brinkley, B.R.; Zinkowski, R.P.; Mollon, W.L.; Davis, F.M.; Pisegna, M.A.; Pershouse, M.; Rao, P.N. Movement and segregation of kinetochores experimentally detached from mammalian chromosomes. Nature 1988, 336, 251–254. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, C.B.; Loncarek, J.; Hergert, P.; Kourtidis, A.; Conklin, D.S.; Khodjakov, A. The spindle assembly checkpoint is satisfied in the absence of interkinetochore tension during mitosis with unreplicated genomes. J. Cell Biol. 2008, 183, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl. Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Cameron, L.A.; Salmon, E.D. Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Curr. Biol. 2004, 14, 2149–2155. [Google Scholar] [CrossRef] [PubMed]
- Daum, J.R.; Wren, J.D.; Daniel, J.J.; Sivakumar, S.; McAvoy, J.N.; Potapova, T.A.; Gorbsky, G.J. Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Curr. Biol. 2009, 19, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Opyrchal, M.; Salisbury, J.L.; Iankov, I.; Goetz, M.P.; McCubrey, J.; Gambino, M.W.; Malatino, L.; Puccia, G.; Ingle, J.N.; Galanis, E.; et al. Inhibition of Cdk2 kinase activity selectively targets the CD44+/CD24-/Low stem-like subpopulation and restores chemosensitivity of SUM149PT triple-negative breast cancer cells. Int. J. Oncol. 2014, 45, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.A.; Gamero, M.C.; Trachana, V.; Futterer, A.; Pacios-Bras, C.; Diaz-Concha, N.P.; Cigudosa, J.C.; Martinez, A.C.; Van Wely, K.H. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc. Natl. Acad. Sci. USA 2010, 107, 4159–4164. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.C.; Van Wely, K.H. Centromere fission, not telomere erosion, triggers chromosomal instability in human carcinomas. Carcinogenesis 2011, 32, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 2001, 153, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [PubMed]
- Lee, J.K.; Choi, Y.L.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. 2016, 11, 283–312. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- How, C.; Bruce, J.; So, J.; Pintilie, M.; Haibe-Kains, B.; Hui, A.; Clarke, B.A.; Hedley, D.W.; Hill, R.P.; Milosevic, M.; et al. Chromosomal instability as a prognostic marker in cervical cancer. BMC Cancer 2015, 15, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Bjork, J.; Hoglund, M.; Mertens, F.; Dal Cin, P.; Akerman, M.; Mandahl, N. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol. 2001, 158, 199–206. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Krasnitz, A.; Rodgers, L.; Cook, K.; Meth, J.; Kendall, J.; Riggs, M.; Eberling, Y.; Troge, J.; Grubor, V.; et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010, 20, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. Cancer genomics: One cell at a time. Genome Biol. 2014, 15, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015, 25, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Bradford, W.D.; Seidel, C.W.; Li, R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 2012, 482, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Mulla, W.A.; Kucharavy, A.; Tsai, H.J.; Rubinstein, B.; Conkright, J.; McCroskey, S.; Bradford, W.D.; Weems, L.; Haug, J.S.; et al. Targeting the adaptability of heterogeneous aneuploids. Cell 2015, 160, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.D.; Berman, J.; Brand, A.C. High frame-rate resolution of cell division during Candida albicans filamentation. Fungal Genet. Biol. 2016, 88, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, A.C.; Berman, J. Shift and adapt: The costs and benefits of karyotype variations. Curr. Opin. Microbiol. 2015, 26, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Stindl, R.; Hehlmann, R. Explaining the high mutation rates of cancer cells to drug and multidrug resistance by chromosome reassortments that are catalyzed by aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 97, 14295–14300. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Nicke, B.; Schuett, M.; Eklund, A.C.; Ng, C.; Li, Q.; Hardcastle, T.; Lee, A.; Roy, R.; East, P.; et al. Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. USA 2009, 106, 8671–8676. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, S.; Huang, X.; Li, L.; Zhang, C.; Pan, Q.; Ren, Z.; Zhou, R.; Ren, Y.; Zi, J.; et al. Drug Resistance in colorectal cancer cell lines is partially associated with aneuploidy status in light of profiling gene expression. J. Proteome Res. 2016, 15, 4047–4059. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Stindl, R.; Hehlmann, R. Origin of multidrug resistance in cells with and without multidrug resistance genes: Chromosome reassortments catalyzed by aneuploidy. Proc. Natl. Acad. Sci. USA 2001, 98, 11283–11288. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, N.; Rancati, G.; Li, R. Dr Jekyll and Mr Hyde: Role of aneuploidy in cellular adaptation and cancer. Curr. Opin. Cell Biol. 2010, 22, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Luzhna, L.; Kathiria, P.; Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 2013, 4, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; De Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Hoffelder, D.R.; Luo, L.; Burke, N.A.; Watkins, S.C.; Gollin, S.M.; Saunders, W.S. Resolution of anaphase bridges in cancer cells. Chromosoma 2004, 112, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Stevens, J.B.; Bremer, S.W.; Liu, G.; Abdallah, B.Y.; Ye, C.J. Evolutionary mechanisms and diversity in cancer. Adv. Cancer Res. 2011, 112, 217–253. [Google Scholar] [PubMed]
- Leibowitz, M.L.; Zhang, C.Z.; Pellman, D. Chromothripsis: A new mechanism for rapid karyotype evolution. Annu. Rev. Genet. 2015, 49, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Teitz, L.S.; Kim, D.H.; Shoshani, O.; Skaletsky, H.; Fachinetti, D.; Page, D.C.; Cleveland, D.W. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 2017, 19, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Sansam, C.L.; Pezza, R.J. Connecting by breaking and repairing: mechanisms of DNA strand exchange in meiotic recombination. FEBS J. 2015, 282, 2444–2457. [Google Scholar] [CrossRef] [PubMed]
- Bomblies, K.; Jones, G.; Franklin, C.; Zickler, D.; Kleckner, N. The challenge of evolving stable polyploidy: Could an increase in “crossover interference distance” play a central role? Chromosoma 2016, 125, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Eaker, S.; Pyle, A.; Cobb, J.; Handel, M.A. Evidence for meiotic spindle checkpoint from analysis of spermatocytes from Robertsonian-chromosome heterozygous mice. J. Cell Sci. 2001, 114, 2953–2965. [Google Scholar] [PubMed]
- Vrooman, L.A.; Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Evidence for paternal age-related alterations in meiotic chromosome dynamics in the mouse. Genetics 2014, 196, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Brunet, S.; Pahlavan, G.; Taylor, S.; Maro, B. Functionality of the spindle checkpoint during the first meiotic division of mammalian oocytes. Reproduction 2003, 126, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Homer, H.A.; McDougall, A.; Levasseur, M.; Murdoch, A.P.; Herbert, M. Mad2 is required for inhibiting securin and cyclin B degradation following spindle depolymerisation in meiosis I mouse oocytes. Reproduction 2005, 130, 829–843. [Google Scholar] [CrossRef] [PubMed]
- LeMaire-Adkins, R.; Radke, K.; Hunt, P.A. Lack of checkpoint control at the metaphase/anaphase transition: A mechanism of meiotic nondisjunction in mammalian females. J. Cell Biol. 1997, 139, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Maro, B.; Kubiak, J.Z.; Polanski, Z. A single bivalent efficiently inhibits cyclin B1 degradation and polar body extrusion in mouse oocytes indicating robust SAC during female meiosis I. PLoS ONE 2011, 6, e27143. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Li, R.; Ma, C.; Chen, E.; Liu, X.J. Xenopus oocyte meiosis lacks spindle assembly checkpoint control. J. Cell Biol. 2013, 201, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.W.; Kirschner, M.W. Cyclin synthesis drives the early embryonic cell cycle. Nature 1989, 339, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Minshull, J.; Sun, H.; Tonks, N.K.; Murray, A.W. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell 1994, 79, 475–486. [Google Scholar] [CrossRef]
- Kuliev, A.; Zlatopolsky, Z.; Kirillova, I.; Spivakova, J.; Cieslak Janzen, J. Meiosis errors in over 20,000 oocytes studied in the practice of preimplantation aneuploidy testing. Reprod. Biomed. Online 2011, 22, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Alfarawati, S.; Goodall, N.N.; Sanchez-Garcia, J.F.; Colls, P.; Wells, D. The cytogenetics of polar bodies: Insights into female meiosis and the diagnosis of aneuploidy. Mol. Hum. Reprod. 2011, 17, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Steuerwald, N.M.; Bermudez, M.G.; Wells, D.; Munne, S.; Cohen, J. Maternal age-related differential global expression profiles observed in human oocytes. Reprod. Biomed. Online 2007, 14, 700–708. [Google Scholar] [CrossRef]
- Yun, Y.; Holt, J.E.; Lane, S.I.; McLaughlin, E.A.; Merriman, J.A.; Jones, K.T. Reduced ability to recover from spindle disruption and loss of kinetochore spindle assembly checkpoint proteins in oocytes from aged mice. Cell Cycle 2014, 13, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Ma, P.; Zhu, W.; Schultz, R.M. Age-associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev. Biol. 2008, 316, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Duncan, F.E.; Hornick, J.E.; Lampson, M.A.; Schultz, R.M.; Shea, L.D.; Woodruff, T.K. Chromosome cohesion decreases in human eggs with advanced maternal age. Aging Cell 2012, 11, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Lane, S.I.; Jones, K.T. Premature dyad separation in meiosis II is the major segregation error with maternal age in mouse oocytes. Development 2014, 141, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Rankin, S.; Dawson, D.S. Recent advances in cohesin biology. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Revenkova, E.; Herrmann, K.; Adelfalk, C.; Jessberger, R. Oocyte cohesin expression restricted to predictyate stages provides full fertility and prevents aneuploidy. Curr. Biol. 2010, 20, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Tachibana-Konwalski, K.; Godwin, J.; Van Der Weyden, L.; Champion, L.; Kudo, N.R.; Adams, D.J.; Nasmyth, K. Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev. 2010, 24, 2505–2516. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.; Schultz, R.M.; Lampson, M.A. Age-dependent susceptibility of chromosome cohesion to premature separase activation in mouse oocytes. Biol. Reprod. 2011, 85, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Lister, L.M.; Kouznetsova, A.; Hyslop, L.A.; Kalleas, D.; Pace, S.L.; Barel, J.C.; Nathan, A.; Floros, V.; Adelfalk, C.; Watanabe, Y.; et al. Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr. Biol. 2010, 20, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Keefe, D.L. Defective cohesin is associated with age-dependent misaligned chromosomes in oocytes. Reprod. Biomed. Online 2008, 16, 103–112. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Fujiwara, R.; Nishizawa, H.; Ito, M.; Kogo, H.; Inagaki, H.; Ohye, T.; Kato, T.; Fujii, T.; Kurahashi, H. Age-related decrease of meiotic cohesins in human oocytes. PLoS ONE 2014, 9, e96710. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, C.S.; Newnham, L.J.; Capalbo, A.; Natesan, S.A.; Joshi, H.A.; Cimadomo, D.; Griffin, D.K.; Sage, K.; Summers, M.C.; Thornhill, A.R.; et al. Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat. Genet. 2015, 47, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Ljunger, E.; Cnattingius, S.; Lundin, C.; Anneren, G. Chromosomal anomalies in first-trimester miscarriages. Acta Obstet. Gynecol. Scand. 2005, 84, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.; Sanchez, A.; Bruguera, J.; Margarit, E.; Borrell, A.; Borobio, V.; Soler, A. Cytogenetic study of spontaneous abortions using semi-direct analysis of chorionic villi samples detects the broadest spectrum of chromosome abnormalities. Am. J. Med. Genet. A 2008, 146A, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fritz, B.; Hallermann, C.; Olert, J.; Fuchs, B.; Bruns, M.; Aslan, M.; Schmidt, S.; Coerdt, W.; Muntefering, H.; Rehder, H. Cytogenetic analyses of culture failures by comparative genomic hybridisation (CGH)-Re-evaluation of chromosome aberration rates in early spontaneous abortions. Eur. J. Hum. Genet. 2001, 9, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Bittles, A.H.; Bower, C.; Hussain, R.; Glasson, E.J. The four ages of Down syndrome. Eur. J. Public Health 2007, 17, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Sybert, V.P.; McCauley, E. Turner’s syndrome. N. Engl. J. Med. 2004, 351, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.K.; Disteche, C.M. Dosage compensation of the active X chromosome in mammals. Nat. Genet. 2006, 38, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Burgoyne, P.S.; Ojarikre, A.; Bauer, J.; Sargent, C.A.; Amorim, A.; Affara, N.A. Transcriptional changes in response to X chromosome dosage in the mouse: Implications for X inactivation and the molecular basis of Turner Syndrome. BMC Genom. 2010, 11, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Otter, M.; Schrander-Stumpel, C.T.; Curfs, L.M. Triple X syndrome: A review of the literature. Eur. J. Hum. Genet. 2010, 18, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Visootsak, J.; Graham, J.M., Jr. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J. Rare Dis. 2006, 1, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Abramowitz, L.K.; Olivier-Van Stichelen, S.; Hanover, J.A. Chromosome imbalance as a driver of sex disparity in disease. J. Genom. 2014, 2, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Malinge, S.; Crispino, J. Myeloid leukemia in Down syndrome. Crit. Rev. Oncog. 2011, 16, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Hama, A.; Muramatsu, H.; Makishima, H.; Sugimoto, Y.; Szpurka, H.; Jasek, M.; O’Keefe, C.; Takahashi, Y.; Sakaguchi, H.; Doisaki, S.; et al. Molecular lesions in childhood and adult acute megakaryoblastic leukaemia. Br. J. Haematol. 2012, 156, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Rainis, L.; Toki, T.; Pimanda, J.E.; Rosenthal, E.; Machol, K.; Strehl, S.; Gottgens, B.; Ito, E.; Izraeli, S. The proto-oncogene ERG in megakaryoblastic leukemias. Cancer Res. 2005, 65, 7596–7602. [Google Scholar] [PubMed]
- Stankiewicz, M.J.; Crispino, J.D. ETS2 and ERG promote megakaryopoiesis and synergize with alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood 2009, 113, 3337–3347. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.H.; Stiller, C.A.; Walker, L.; Rahman, N. Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J. Med. Genet. 2006, 43, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, D.N. Binucleation in mammalian liver. Studies on the control of cytokinesis in vivo. Exp. Cell Res. 1972, 74, 455–465. [Google Scholar] [CrossRef]
- Eggert, U.S.; Mitchison, T.J.; Field, C.M. Animal cytokinesis: From parts list to mechanisms. Annu. Rev. Biochem. 2006, 75, 543–566. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, M. The molecular requirements for cytokinesis. Science 2005, 307, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009, 136, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Mullins, J.M.; Biesele, J.J. Terminal phase of cytokinesis in D-98s cells. J. Cell Biol. 1977, 73, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.A.; Daum, J.R.; Pittman, B.D.; Hudson, J.R.; Jones, T.N.; Satinover, D.L.; Stukenberg, P.T.; Gorbsky, G.J. The reversibility of mitotic exit in vertebrate cells. Nature 2006, 440, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Budirahardja, Y.; Klompmaker, R.; Medema, R.H. Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep. 2005, 6, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, S.; Tighe, A.; D’Alise, A.M.; Taylor, S.S.; Musacchio, A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 2010, 190, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Muro, Y.; Todokoro, K. Thrombopoietin-induced polyploidization of bone marrow megakaryocytes is due to a unique regulatory mechanism in late mitosis. J. Cell Biol. 1997, 139, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Vitrat, N.; Cohen-Solal, K.; Pique, C.; Le Couedic, J.P.; Norol, F.; Larsen, A.K.; Katz, A.; Vainchenker, W.; Debili, N. Endomitosis of human megakaryocytes are due to abortive mitosis. Blood 1998, 91, 3711–3723. [Google Scholar] [PubMed]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: polyploidy with purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [PubMed]
- Zhimulev, I.F.; Belyaeva, E.S.; Semeshin, V.F.; Koryakov, D.E.; Demakov, S.A.; Demakova, O.V.; Pokholkova, G.V.; Andreyeva, E.N. Polytene chromosomes: 70 years of genetic research. Int. Rev. Cytol. 2004, 241, 203–275. [Google Scholar] [PubMed]
- Zybina, E.V.; Zybina, T.G. Polytene chromosomes in mammalian cells. Int. Rev. Cytol. 1996, 165, 53–119. [Google Scholar] [PubMed]
- Hu, D.; Cross, J.C. Development and function of trophoblast giant cells in the rodent placenta. Int. J. Dev. Biol. 2010, 54, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, F.; Sacco, M.G.; Susani, L.; Montagna, C.; Vezzoni, P. Cell fusion is a physiological process in mouse liver. Hepatology 2008, 48, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Asahina, K.; Fujimori, H.; Ozeki, R.; Shimizu-Saito, K.; Tanaka, Y.; Teramoto, K.; Arii, S.; Takase, K.; Kataoka, M.; et al. Generation of hybrid hepatocytes by cell fusion from monkey embryoid body cells in the injured mouse liver. Histochem. Cell Biol. 2006, 125, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.; Lazebnik, Y. Cell-to-cell fusion as a link between viruses and cancer. Nat. Rev. Cancer 2007, 7, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Okagaki, L.H.; Nielsen, K. Titan cells confer protection from phagocytosis in Cryptococcus neoformans infections. Eukaryot. Cell 2012, 11, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.D.; Hashemi, J.; Bibi, M.; Pulver, R.; Bavli, D.; Nahmias, Y.; Wellington, M.; Sapiro, G.; Berman, J. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol. 2014, 12, e1001815. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.H.; Bickham, J.W.; Honeycutt, R.L.; Ojeda, R.A.; Kohler, N. Discovery of tetraploidy in a mammal. Nature 1999, 401, 341. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.P. The evolutionary consequences of polyploidy. Cell 2007, 131, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Hellsten, U.; Khokha, M.K.; Grammer, T.C.; Harland, R.M.; Richardson, P.; Rokhsar, D.S. Accelerated gene evolution and subfunctionalization in the pseudotetraploid frog Xenopus laevis. BMC Biol. 2007, 5, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Session, A.M.; Uno, Y.; Kwon, T.; Chapman, J.A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, M.H. New insights into triploidy and tetraploidy, from an analysis of model systems for these conditions. Hum. Reprod. 1991, 6, 8–16. [Google Scholar] [PubMed]
- Nakamura, Y.; Takaira, M.; Sato, E.; Kawano, K.; Miyoshi, O.; Niikawa, N. A tetraploid liveborn neonate: Cytogenetic and autopsy findings. Arch. Pathol. Lab. Med. 2003, 127, 1612–1614. [Google Scholar] [PubMed]
- Stefanova, I.; Jenderny, J.; Kaminsky, E.; Mannhardt, A.; Meinecke, P.; Grozdanova, L.; Gillessen-Kaesbach, G. Mosaic and complete tetraploidy in live-born infants: Two new patients and review of the literature. Clin. Dysmorphol. 2010, 19, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.E.; Saxe, D.F.; Muralidharan, K.; Coleman, K.B.; Zacharias, J.F.; Fernhoff, P.M. Unique mosaicism of tetraploidy and trisomy 8: Clinical, cytogenetic, and molecular findings in a live-born infant. Am. J. Med. Genet. 1996, 62, 243–246. [Google Scholar] [CrossRef]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Korthout, H.A.; Caspers, M.P.; Kottenhagen, M.J.; Helmer, Q.; Wang, M. A tormentor in the quest for plant p53-like proteins. FEBS Lett. 2002, 526, 53–57. [Google Scholar] [CrossRef]
- Salman-Minkov, A.; Sabath, N.; Mayrose, I. Whole-genome duplication as a key factor in crop domestication. Nat. Plants 2016, 2, 16115. [Google Scholar] [CrossRef] [PubMed]
- Renny-Byfield, S.; Wendel, J.F. Doubling down on genomes: Polyploidy and crop plants. Am. J. Bot. 2014, 101, 1711–1725. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Quintyne, N.J.; Reing, J.E.; Hoffelder, D.R.; Gollin, S.M.; Saunders, W.S. Spindle multipolarity is prevented by centrosomal clustering. Science 2005, 307, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Hanlon Newell, A.E.; Smith, L.; Wilson, E.M.; Olson, S.B.; Thayer, M.J.; Strom, S.C.; Grompe, M. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 2012, 142, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z.; Breneman, A.; Cande, J.; Dunn, J.; Burbank, K.; O’Toole, E.; Pellman, D. Genome-wide genetic analysis of polyploidy in yeast. Nature 2006, 443, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, P.C.; Cowan, D.S.; Sanchez, C.A.; Barrett, M.T.; Emond, M.J.; Levine, D.S.; Rabinovitch, P.S.; Reid, B.J. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proc. Natl. Acad. Sci. USA 1996, 93, 7081–7084. [Google Scholar] [CrossRef] [PubMed]
- Olaharski, A.J.; Sotelo, R.; Solorza-Luna, G.; Gonsebatt, M.E.; Guzman, P.; Mohar, A.; Eastmond, D.A. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 2006, 27, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Hammer, R.E.; Messing, A.; Palmiter, R.D.; Brinster, R.L. Pancreatic Neoplasia induced by SV40 T-antigen expression in acinar cells of transgenic mice. Science 1987, 238, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Giannoudis, A.; Evans, M.F.; Southern, S.A.; Herrington, C.S. Basal keratinocyte tetrasomy in low-grade squamous intra-epithelial lesions of the cervix is restricted to high and intermediate risk HPV infection but is not type-specific. Br. J. Cancer 2000, 82, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; De Lange, T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Morse, H.C., 3rd; Godfrey, V.L.; Naeem, R.; Justice, M.J. Overexpression of Eg5 causes genomic instability and tumor formation in mice. Cancer Res. 2007, 67, 10138–10147. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Origins and consequences of centrosome aberrations in human cancers. Int. J. Cancer 2006, 119, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.A.; Kwon, M.; Pellman, D. Centrosomes and cancer: How cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009, 28, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Jin, Y.; Lindgren, D.; Persson, J.; Gisselsson, L.; Hanks, S.; Sehic, D.; Mengelbier, L.H.; Ora, I.; Rahman, N.; et al. Generation of trisomies in cancer cells by multipolar mitosis and incomplete cytokinesis. Proc. Natl. Acad. Sci. USA 2010, 107, 20489–20493. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, F.; Thompson, S.L.; Ravi, S.; Compton, D.A.; Dmitrovsky, E. Anaphase catastrophe is a target for cancer therapy. Clin. Cancer Res. 2011, 17, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Leber, B.; Maier, B.; Fuchs, F.; Chi, J.; Riffel, P.; Anderhub, S.; Wagner, L.; Ho, A.D.; Salisbury, J.L.; Boutros, M.; et al. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2010, 2, 33ra38. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.; Durrbaum, M.; Kuffer, C.; Muller, S.; Zaman, G.J.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef] [PubMed]
- Balsas, P.; Galan-Malo, P.; Marzo, I.; Naval, J. Bortezomib resistance in a myeloma cell line is associated to PSMbeta5 overexpression and polyploidy. Leuk. Res. 2012, 36, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Zeng, J.Y.; Zhuang, C.M.; Zhou, Y.Q.; Yao, H.P.; Hu, X.; Zhang, R.; Wang, M.H. Small-molecule inhibitor BMS-777607 induces breast cancer cell polyploidy with increased resistance to cytotoxic chemotherapy agents. Mol. Cancer Ther. 2013, 12, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Hinchcliffe, E.H.; Day, C.A.; Karanjeet, K.B.; Fadness, S.; Langfald, A.; Vaughan, K.T.; Dong, Z. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat. Cell Biol. 2016, 18, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Lentini, L.; Piscitello, D.; Veneziano, L.; Di Leonardo, A. Simultaneous reduction of MAD2 and BUBR1 expression induces mitotic spindle alterations associated with p53 dependent cell cycle arrest and death. Cell Biol. Int. 2014, 38, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Alvarado, A. Cellular hyperproliferation and cancer as evolutionary variables. Curr. Biol. 2012, 22, R772–R778. [Google Scholar] [CrossRef] [PubMed]
- Leroi, A.M.; Koufopanou, V.; Burt, A. Cancer selection. Nat. Rev. Cancer 2003, 3, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Caulin, A.F.; Maley, C.C. Peto’s Paradox: Evolution’s prescription for cancer prevention. Trends Ecol. Evol. 2011, 26, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Peto, R. Quantitative implications of the approximate irrelevance of mammalian body size and lifespan to lifelong cancer risk. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Sulak, M.; Fong, L.; Mika, K.; Chigurupati, S.; Yon, L.; Mongan, N.P.; Emes, R.D.; Lynch, V.J. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. Elife 2016, 5, e11994. [Google Scholar] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harbor Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. P53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Golomb, L.; Volarevic, S.; Oren, M. P53 and ribosome biogenesis stress: The essentials. FEBS Lett. 2014, 588, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Lambrus, B.G.; Uetake, Y.; Clutario, K.M.; Daggubati, V.; Snyder, M.; Sluder, G.; Holland, A.J. P53 protects against genome instability following centriole duplication failure. J. Cell Biol. 2015, 210, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Cornils, H.; Chiu, S.Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Thery, M.; Camargo, F.D.; Pellman, D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Casenghi, M.; Mangiacasale, R.; Tuynder, M.; Caillet-Fauquet, P.; Elhajouji, A.; Lavia, P.; Mousset, S.; Kirsch-Volders, M.; Cundari, E. P53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp. Cell Res. 1999, 250, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Kurimura, T. Virally transformed cells and cytochalasin B: I. The effect of cytochalasin B on cytokinesis, karyokinesis and DNA synthesis in cells. Exp. Cell Res. 1974, 89, 111–120. [Google Scholar] [CrossRef]
- Incassati, A.; Patel, D.; McCance, D.J. Induction of tetraploidy through loss of p53 and upregulation of Plk1 by human papillomavirus type-16 E6. Oncogene 2006, 25, 2444–2451. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H.; Wahl, G.M. P53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res. 1998, 58, 396–401. [Google Scholar] [PubMed]
- Lanni, J.S.; Jacks, T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell biol. 1998, 18, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Senovilla, L.; Jemaa, M.; Michaud, M.; Galluzzi, L.; Kepp, O.; Nanty, L.; Criollo, A.; Rello-Varona, S.; Manic, G.; et al. Multipolar mitosis of tetraploid cells: Inhibition by p53 and dependency on Mos. EMBO J. 2010, 29, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Krzywicka-Racka, A.; Sluder, G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J. Cell Biol. 2011, 194, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Crockford, A.; Zalmas, L.P.; Gronroos, E.; Dewhurst, S.M.; McGranahan, N.; Cuomo, M.E.; Encheva, V.; Snijders, A.P.; Begum, J.; Purewal, S.; et al. Cyclin D mediates tolerance of genome-doubling in cancers with functional p53. Ann. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Benes, C.; Dyson, N.J. Whole chromosome instability resulting from the synergistic effects of pRB and p53 inactivation. Oncogene 2014, 33, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Rehen, S.K.; Yung, Y.C.; McCreight, M.P.; Kaushal, D.; Yang, A.H.; Almeida, B.S.; Kingsbury, M.A.; Cabral, K.M.; McConnell, M.J.; Anliker, B.; et al. Constitutional aneuploidy in the normal human brain. J. Neurosci. 2005, 25, 2176–2180. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ploidy is the number of sets of chromosomes in a cell or in an organism. |
| Haploid number refers to one set of chromosomes (1N), as in gametes or certain strains of budding yeast. |
| Diploid number refers to two sets of chromosomes (2N) that are homologous (one from each parent). Most animals are diploid. |
| Polyploid denotes a cell with more than two sets of chromosomes (triploid – 3N, tetraploid – 4N, pentaploid – 5N, etc.). |
| Euploid denotes the normal chromosome number in a species, usually an exact multiple of the haploid number (i.e.; human euploid genome contains 46 chromosomes – 2× the haploid number). |
| Chromosomal Instability is the tendency of a cell to gain or lose chromosomes or large segments of chromosomes. It is often abbreviated CIN. |
| Aneuploidy denotes the state of a cell having a chromosome number that deviates from a multiple of the haploid, i.e.; when there are extra or missing single chromosomes. |
| Whole chromosomal aneuploidy is having entire chromosomes gained or lost. |
| Segmental aneuploidy is having large regions of chromosomes deleted, duplicated or translocated from one chromosome to another. Cancer cells often exhibit both whole chromosome aneuploidy and segmental aneuploidy. |
| Trisomy refers to a diploid genome having gained an additional chromosome (2N + 1). Trisomy 21 indicates an extra chromosome 21 in a diploid genome. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potapova, T.; Gorbsky, G.J. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology 2017, 6, 12. https://doi.org/10.3390/biology6010012
Potapova T, Gorbsky GJ. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology. 2017; 6(1):12. https://doi.org/10.3390/biology6010012
Chicago/Turabian StylePotapova, Tamara, and Gary J. Gorbsky. 2017. "The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis" Biology 6, no. 1: 12. https://doi.org/10.3390/biology6010012
APA StylePotapova, T., & Gorbsky, G. J. (2017). The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology, 6(1), 12. https://doi.org/10.3390/biology6010012