
Sleep-Wake Regulation and Its Impact on Working Memory Performance: The Role of Adenosine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sleep-Wake Regulation: Concepts and Empirical Evidence
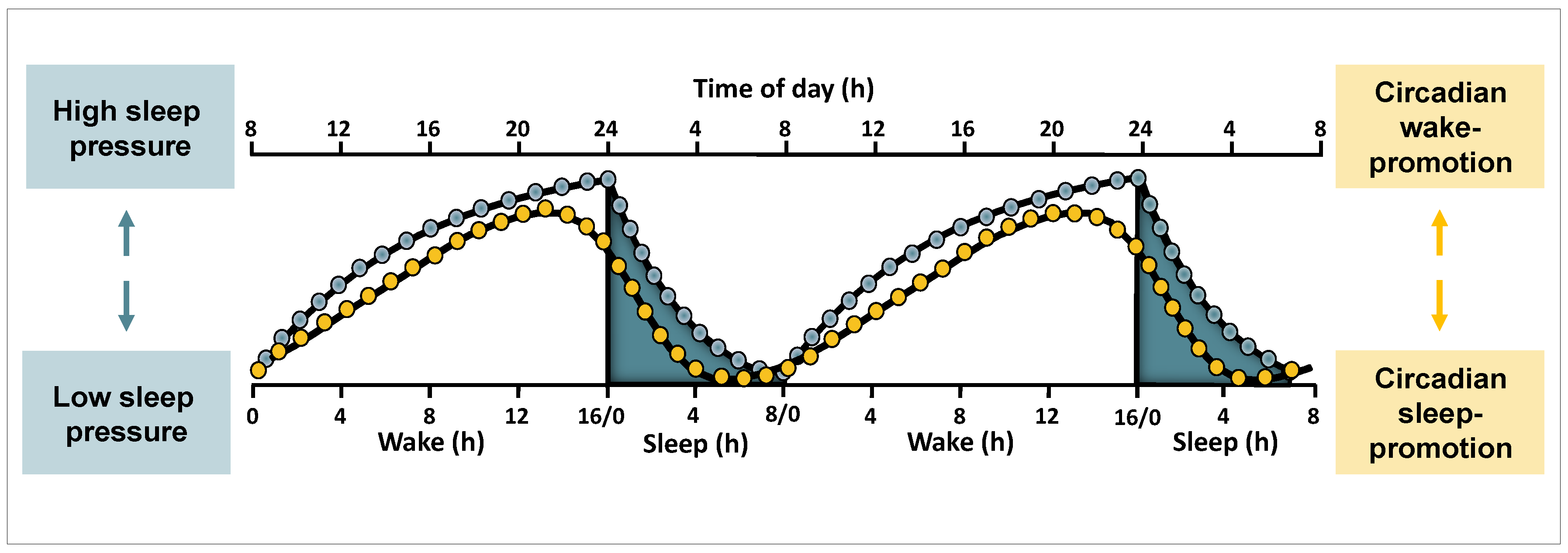
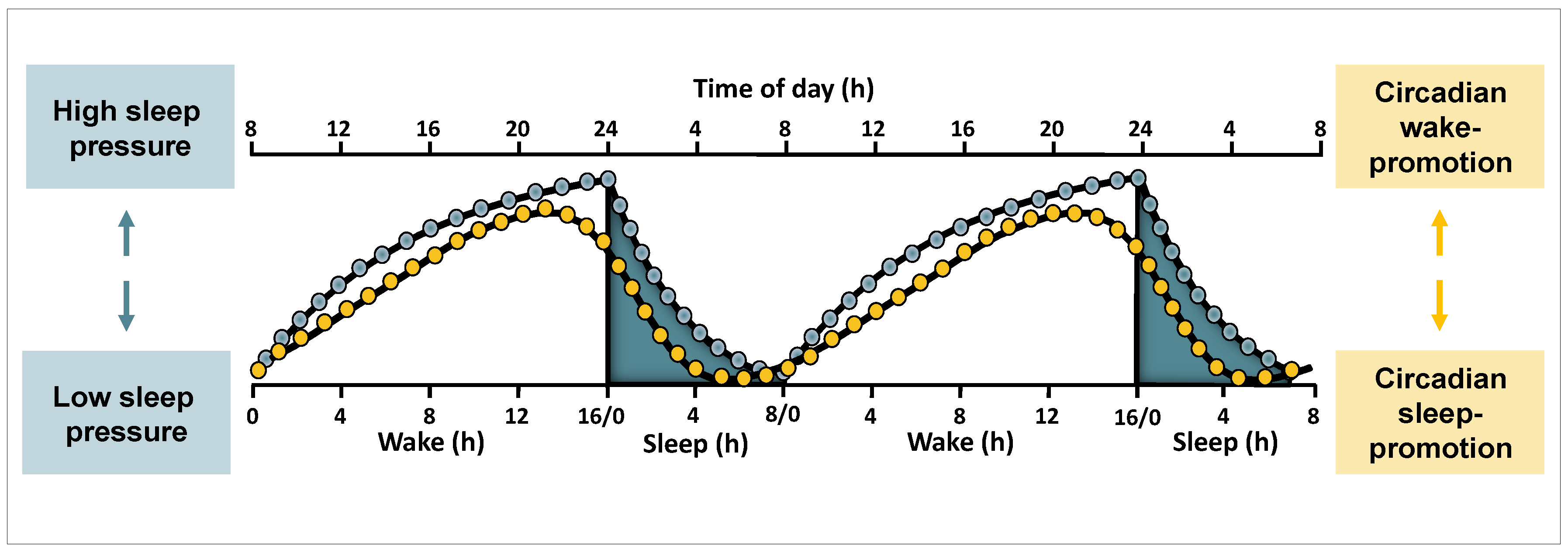
2.1. Sleep-Wake Regulation at a Conceptual Level
2.2. Investigating Circadian and Sleep Homeostatic Mechanisms
2.3. Circadian and Sleep Homeostatic Regulation of Sleep and Waking Functions
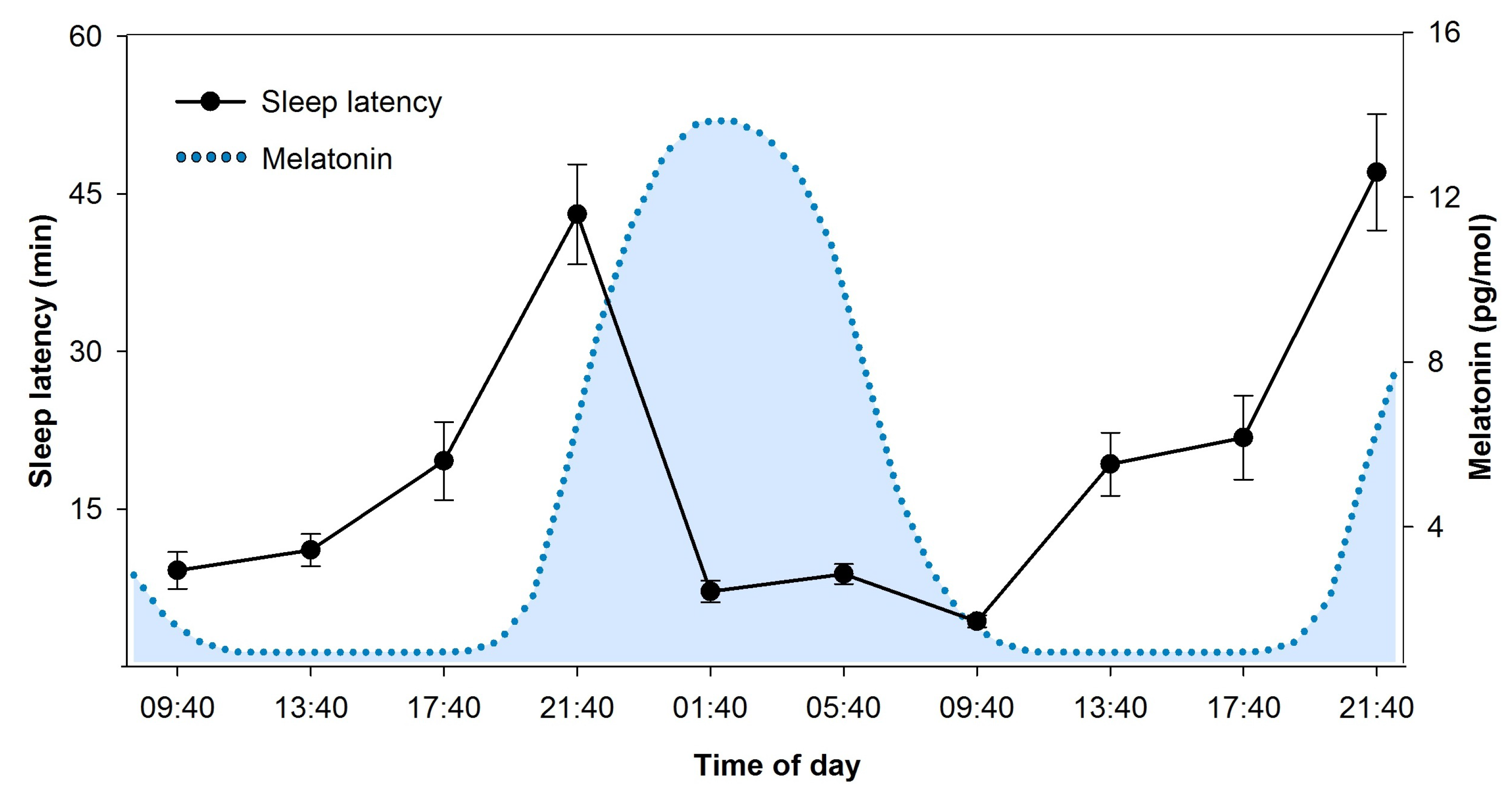
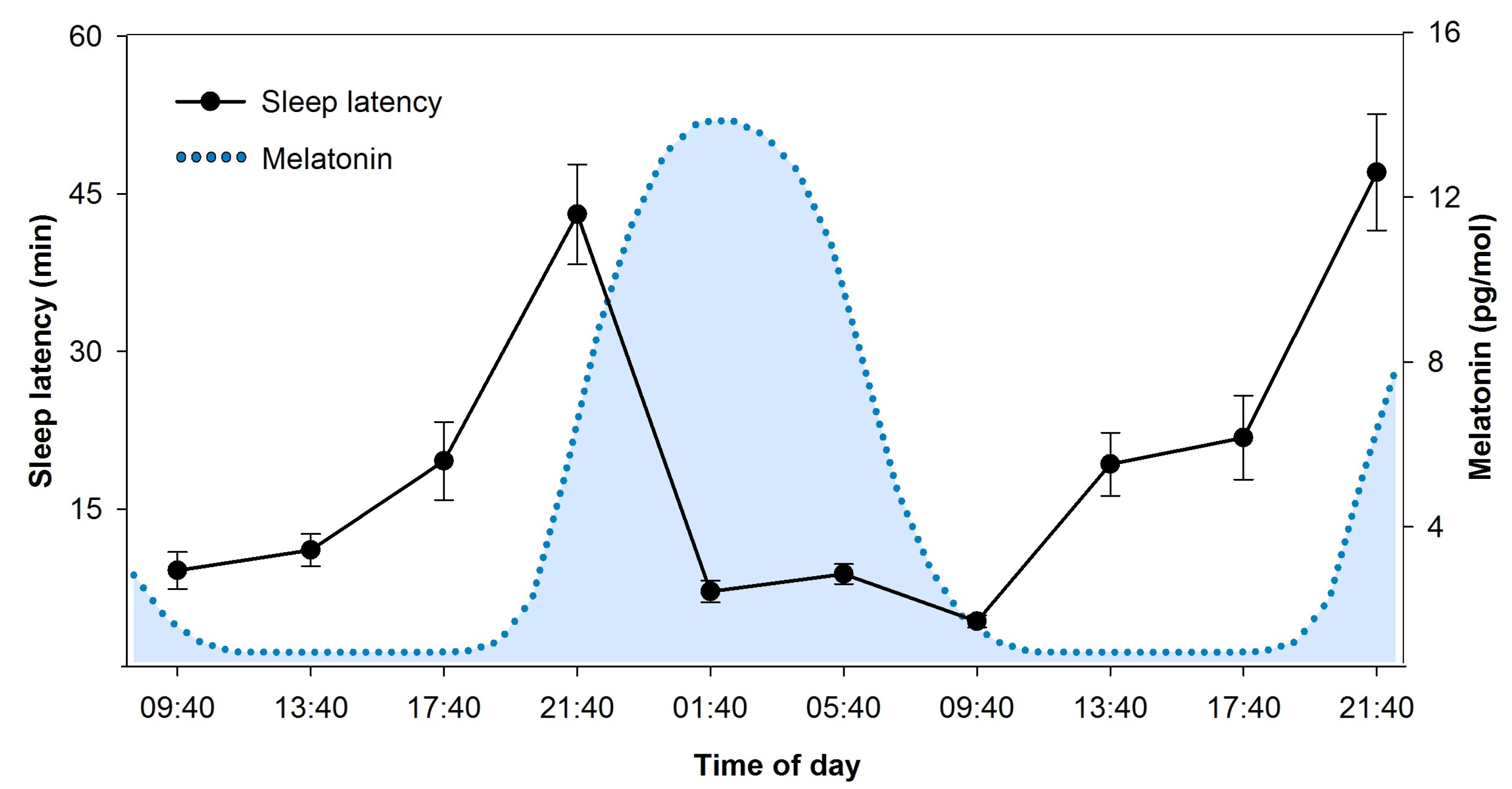
2.3.1. Circadian and Homeostatic Regulation of Sleep Features
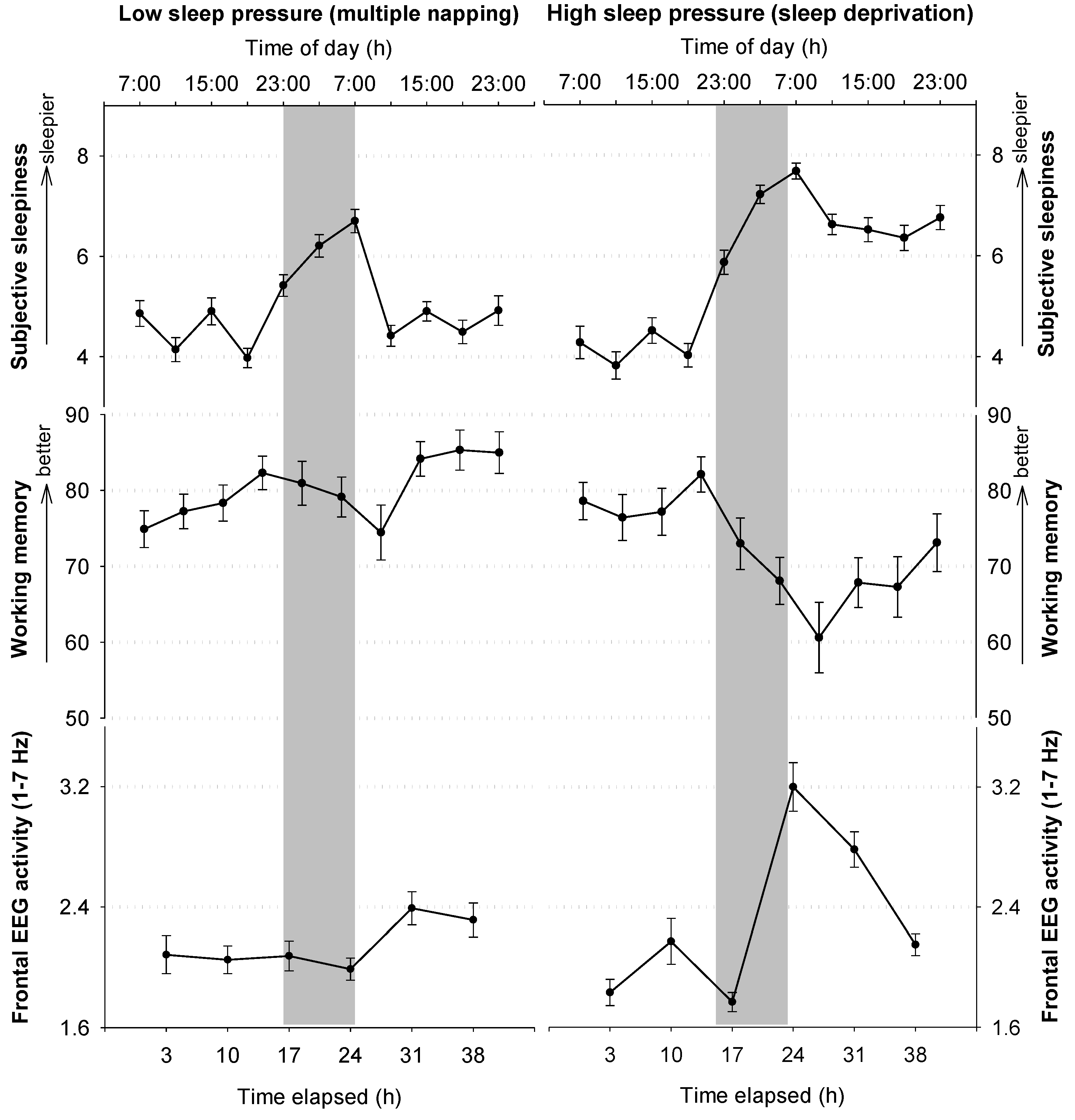
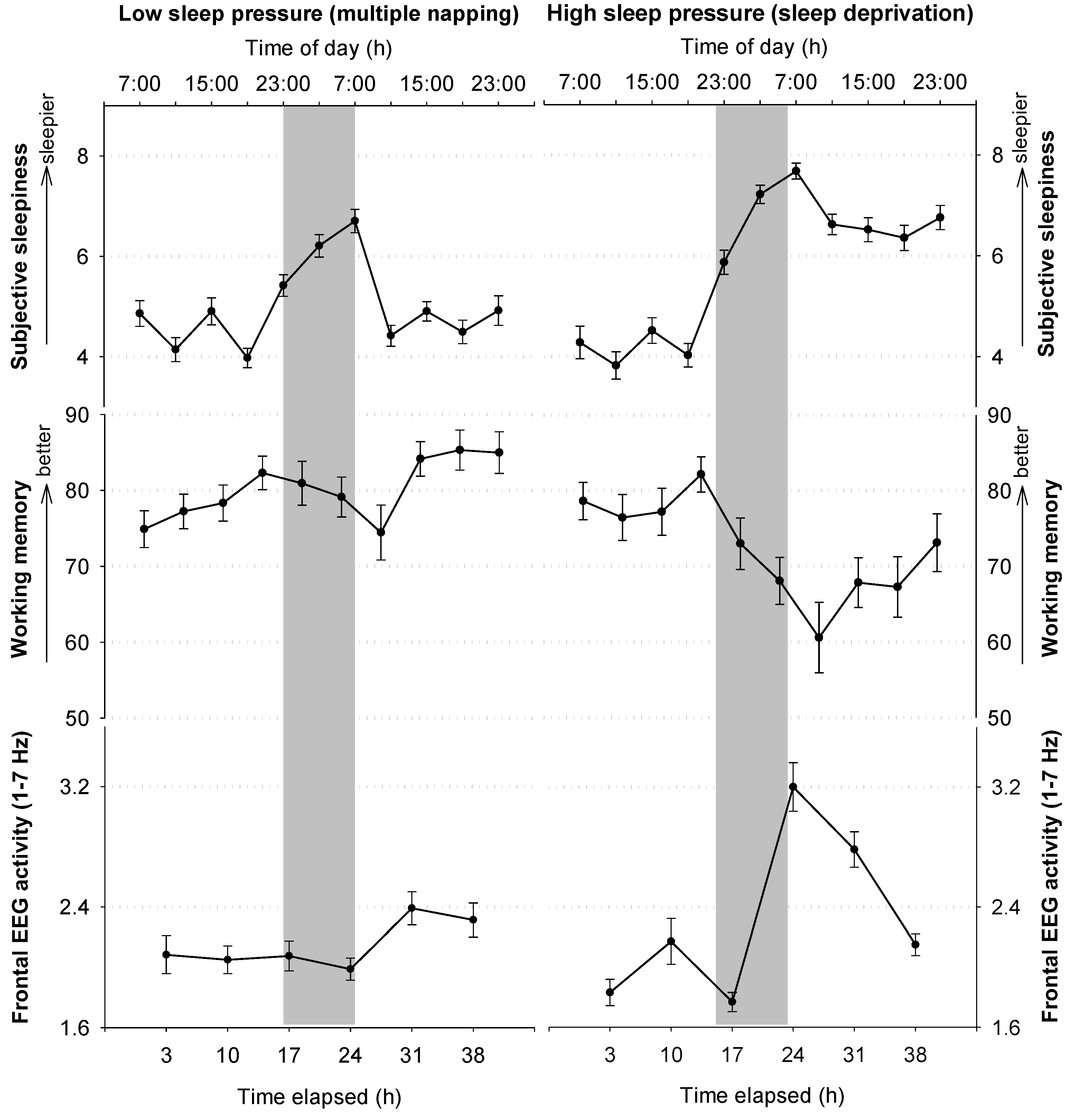
2.3.2. Circadian and Homeostatic Regulation of Waking Functions
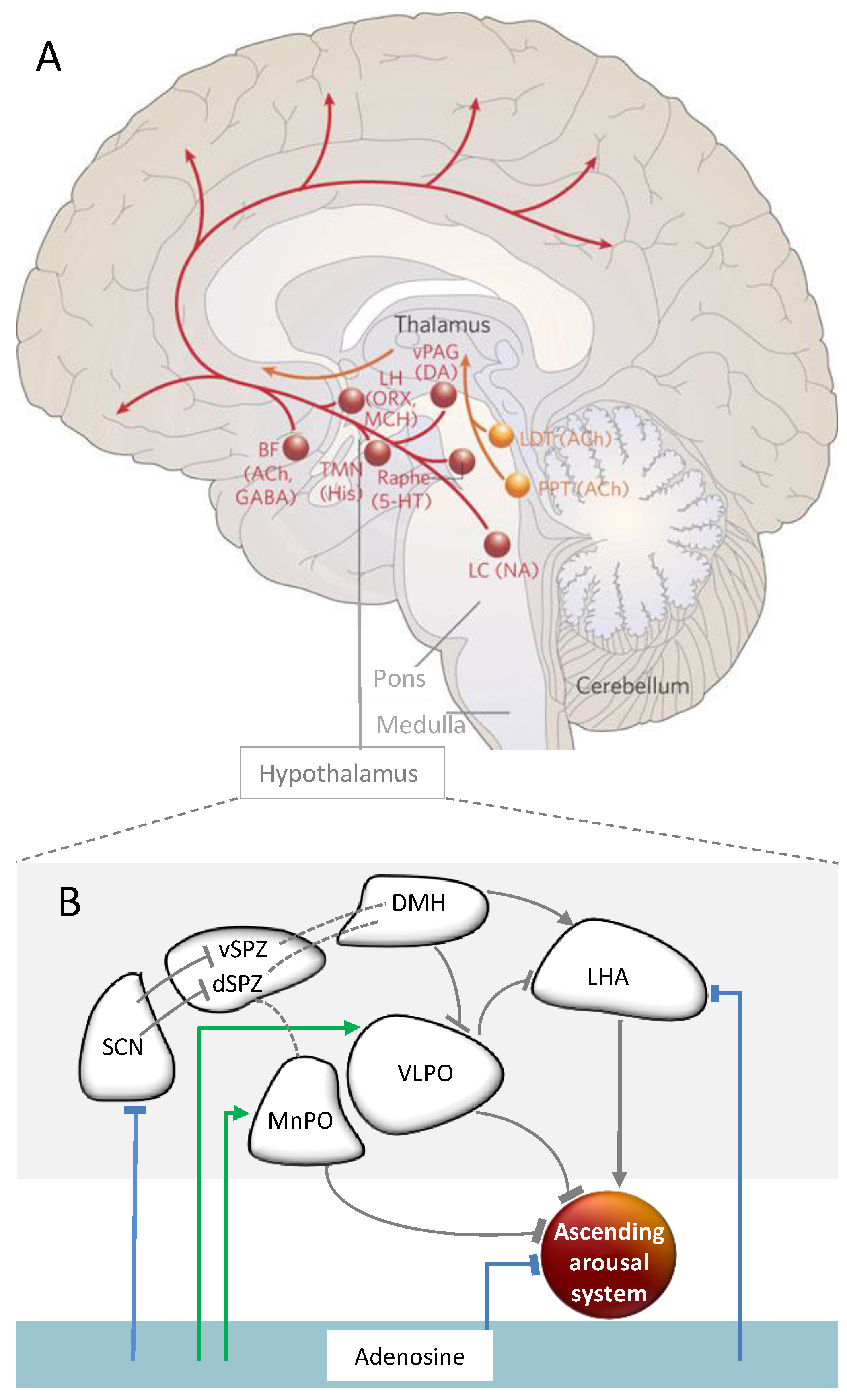
2.4. Neuronal Underpinnings of Sleep and Wakefulness and the Role of Adenosine
2.4.1. Adenosinergic Regulation of Sleep Homeostasis
A Role of Adenosine in Sleep Homeostasis—Implicated Brain Regions
Why Does Adenosine Increase with Time Spent Awake? Contributions of Its Metabolization
2.4.2. Pathways of Circadian Arousal Promotion
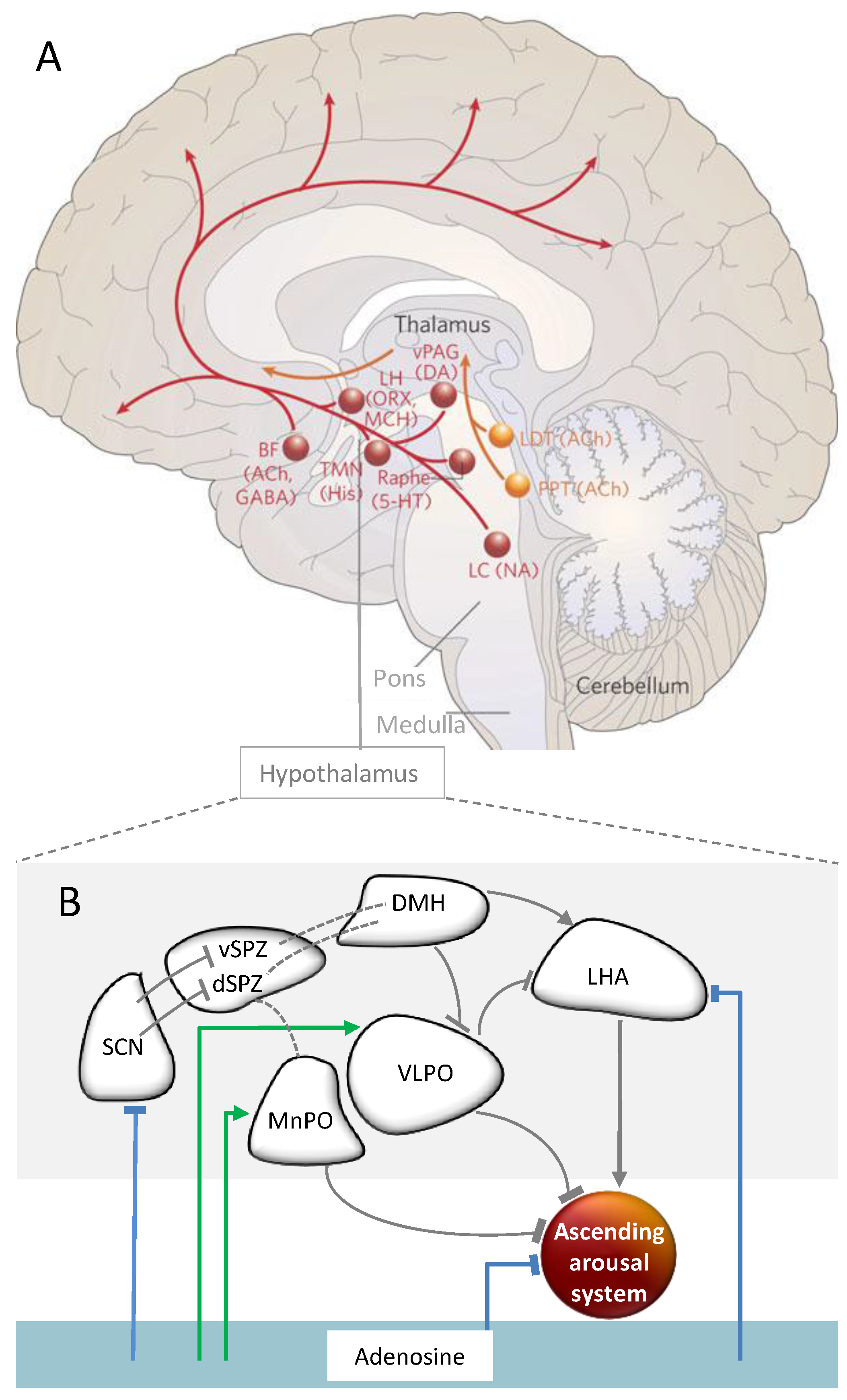
2.5. Interactions of Circadian and Sleep Homeostatic Mechanisms
2.5.1. The Genetic Clockwork and Sleep Homeostasis
2.5.2. Brain Regions and Substances Mediating the Interaction
3. Working Memory
3.1. Working Memory at a Conceptual Level
3.2. Neuronal Underpinnings
3.2.1. Brain Activity Patterns
3.2.2. Neurotransmitters and Neuromodulators
3.3. Impact of Sleep Loss
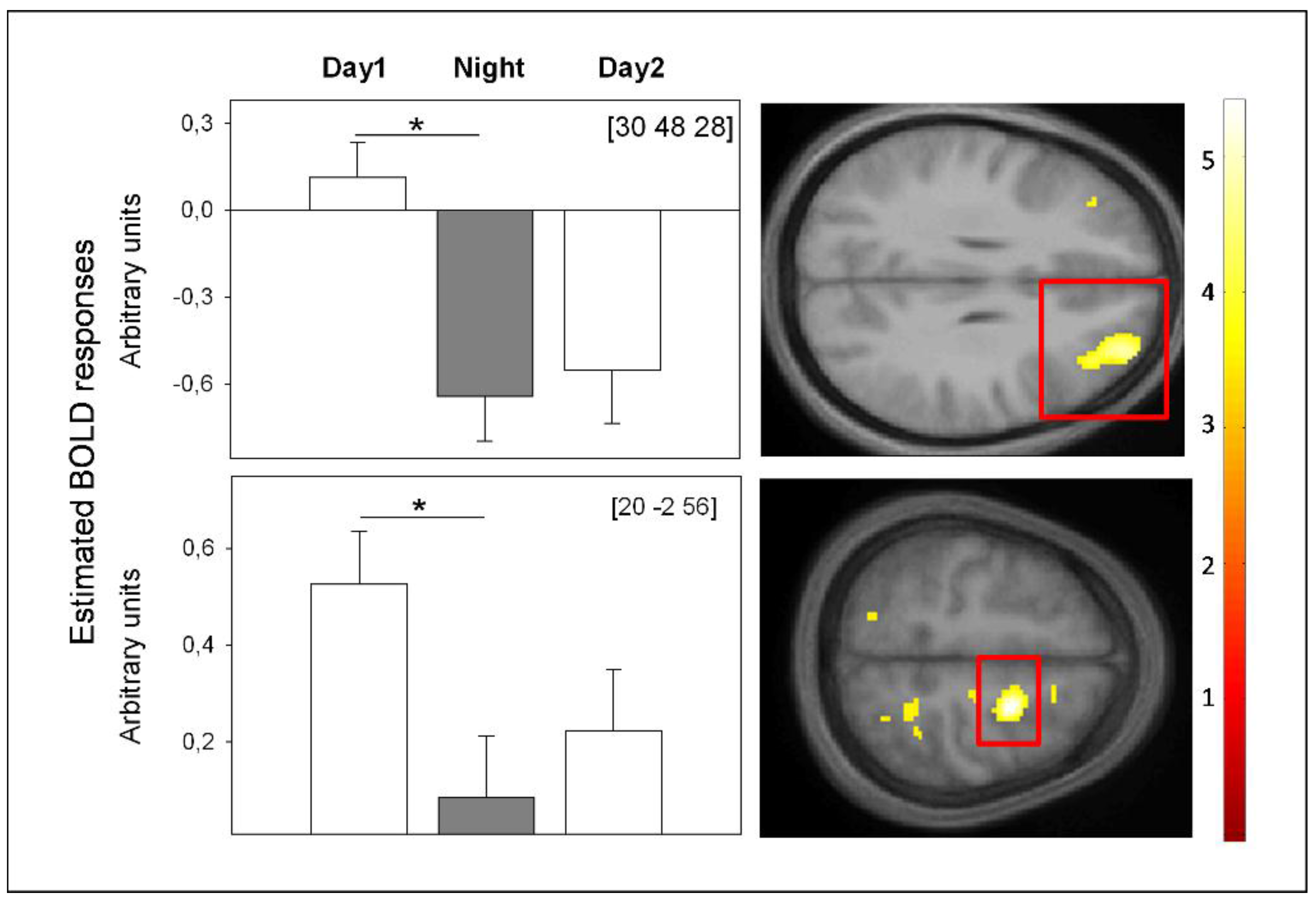
Do Adenosinergic Mechanisms Play a Role in Sleep-Pressure-Related WM Deficits?
3.4. Circadian Modulation
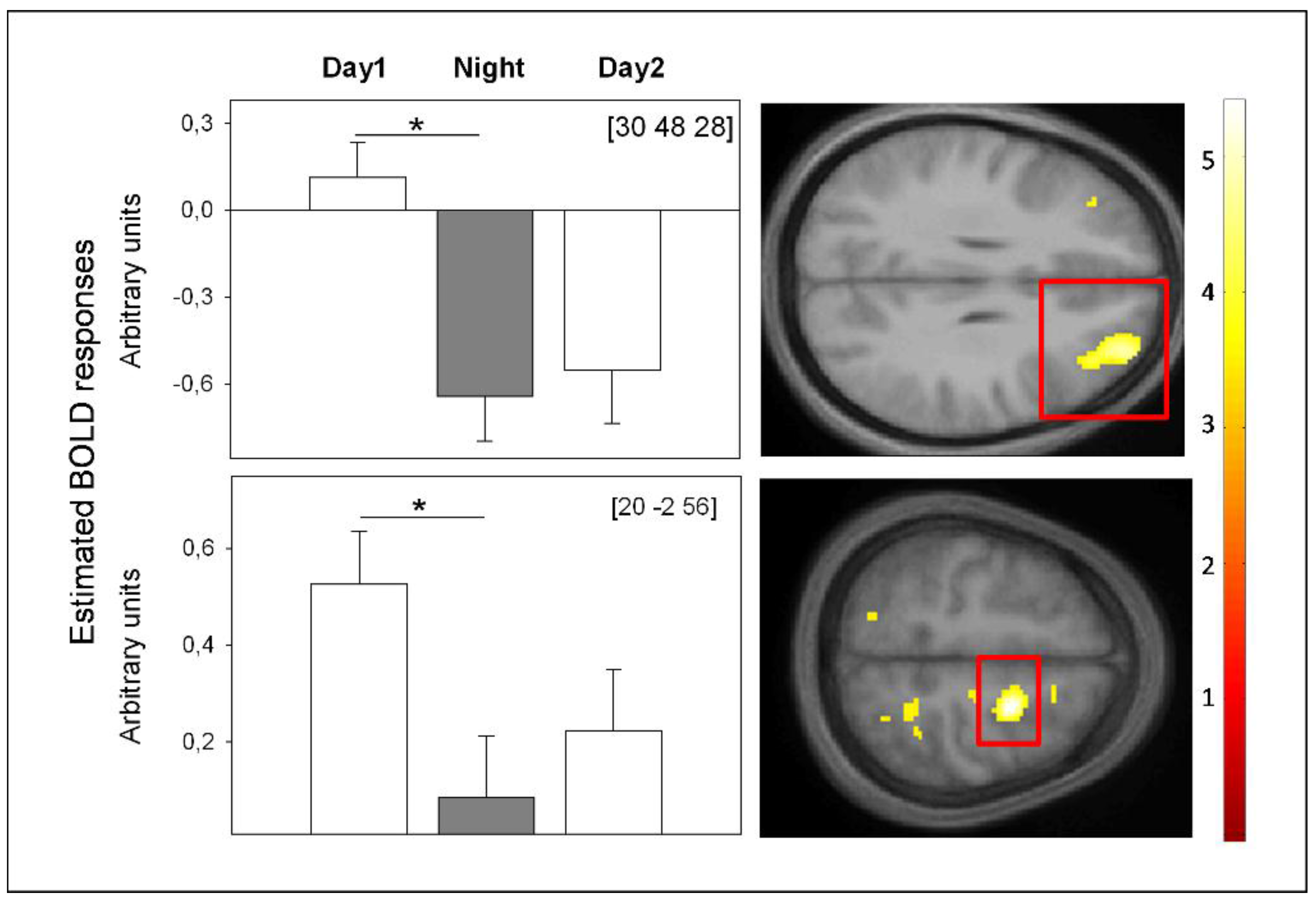
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Borbely, A.A. A two process model of sleep regulation. Hum. Neurobiol. 1982, 1, 195–204. [Google Scholar] [PubMed]
- Borbély, A.A.; Acherman, P. Sleep homeostasis and models of sleep regulation. In Principles and Practices of Sleep Medicine; Kryger, M.H., Roth, T., Dement, W.C., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2005; pp. 405–418. [Google Scholar]
- Franken, P. A role for clock genes in sleep homeostasis. Curr. Opin. Neurobiol. 2013, 23, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Deboer, T.; Detari, L.; Meijer, J.H. Long term effects of sleep deprivation on the mammalian circadian pacemaker. Sleep 2007, 30, 257–262. [Google Scholar] [PubMed]
- Schmidt, C.; Collette, F.; Leclercq, Y.; Sterpenich, V.; Vandewalle, G.; Berthomier, P.; Berthomier, C.; Phillips, C.; Tinguely, G.; Darsaud, A.; et al. Homeostatic sleep pressure and responses to sustained attention in the suprachiasmatic area. Science 2009, 324, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Dijk, D.J.; Franken, P. Interaction of sleep homeostatis and circadian rhythmicity: Dependent or independent systems? In Principles and Practice of Sleep Medicine; Kryger, M.H., Roth, T., Dement, W.C., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2005; pp. 418–435. [Google Scholar]
- Landolt, H.P. Sleep homeostasis: A role for adenosine in humans? Biochem. Pharmacol. 2008, 75, 2070–2079. [Google Scholar] [CrossRef] [PubMed]
- Porkka-Heiskanen, T.; Kalinchuk, A.V. Adenosine, energy metabolism and sleep homeostasis. Sleep Med. Rev. 2011, 15, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Porkka-Heiskanen, T. Sleep homeostasis. Curr. Opin. Neurobiol. 2013, 23, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Chee, M.W.; Thomas, R.J. Functional neuroimaging of sleep deprivation. In Neuroimaging of Sleep and Sleep Disorders; Nofzinger, E., Maquet, P., Thorpy, M.J., Eds.; Cambrindge University Press: New York, NY, USA, 2013; pp. 129–136. [Google Scholar]
- Vandewalle, G.; Archer, S.N.; Wuillaume, C.; Balteau, E.; Degueldre, C.; Luxen, A.; Maquet, P.; Dijk, D.J. Functional magnetic resonance imaging-assessed brain responses during an executive task depend on interaction of sleep homeostasis, circadian phase, and per3 genotype. J. Neurosci. 2009, 29, 7948–7956. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.F.; Maire, M.; Gabel, V.; Viola, A.U.; Kolodyazhniy, V.; Strobel, W.; Gotz, T.; Bachmann, V.; Landolt, H.P.; Cajochen, C.; et al. Insights into behavioral vulnerability to differential sleep pressure and circadian phase from a functional ada polymorphism. J. Biol. Rhythms. 2014, 29, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.C.; Groeger, J.A.; Santhi, N.; Arbon, E.L.; Lazar, A.S.; Hasan, S.; von Schantz, M.; Archer, S.N.; Dijk, D.J. Effects of partial and acute total sleep deprivation on performance across cognitive domains, individuals and circadian phase. PLoS ONE 2012, 7, e45987. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, V.; Klein, C.; Bodenmann, S.; Schafer, N.; Berger, W.; Brugger, P.; Landolt, H.P. The bdnf val66met polymorphism modulates sleep intensity: Eeg frequency- and state-specificity. Sleep 2012, 35, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Scullin, M.K.; Bliwise, D.L. Sleep, cognition, and normal aging: Integrating a half century of multidisciplinary research. Perspect Psychol. Sci. 2015, 10, 97–137. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, J.K.; Ritz-De Cecco, A.; Czeisler, C.A.; Dijk, D.J. Circadian temperature and melatonin rhythms, sleep, and neurobehavioral function in humans living on a 20-h day. Am. J. Physiol. 1999, 277, R1152–R1163. [Google Scholar] [PubMed]
- Dijk, D.J.; Duffy, J.F.; Czeisler, C.A. Circadian and sleep/wake dependent aspects of subjective alertness and cognitive performance. J. Sleep Res. 1992, 1, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.J.; Wang, W.; Ronda, J.M.; Wyatt, J.K.; Duffy, J.F. Circadian and wake-dependent influences on subjective sleepiness, cognitive throughput, and reaction time performance in older and young adults. Sleep 2010, 33, 481–490. [Google Scholar] [PubMed]
- Lee, J.H.; Wang, W.; Silva, E.J.; Chang, A.M.; Scheuermaier, K.D.; Cain, S.W.; Duffy, J.F. Neurobehavioral performance in young adults living on a 28-h day for 6 weeks. Sleep 2009, 32, 905–913. [Google Scholar] [PubMed]
- Grady, S.; Aeschbach, D.; Wright, K.P., Jr.; Czeisler, C.A. Effect of modafinil on impairments in neurobehavioral performance and learning associated with extended wakefulness and circadian misalignment. Neuropsychopharmacol 2010, 35, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, J.K.; Cajochen, C.; Ritz-De Cecco, A.; Czeisler, C.A.; Dijk, D.J. Low-dose repeated caffeine administration for circadian-phase-dependent performance degradation during extended wakefulness. Sleep 2004, 27, 374–381. [Google Scholar] [PubMed]
- Sagaspe, P.; Taillard, J.; Amieva, H.; Beck, A.; Rascol, O.; Dartigues, J.F.; Capelli, A.; Philip, P. Influence of age, circadian and homeostatic processes on inhibitory motor control: A go/nogo task study. PLoS ONE 2012, 7, e39410. [Google Scholar] [CrossRef] [PubMed]
- Blatter, K.; Opwis, K.; Munch, M.; Wirz-Justice, A.; Cajochen, C. Sleep loss-related decrements in planning performance in healthy elderly depend on task difficulty. J. Sleep Res. 2005, 14, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Cajochen, C.; Khalsa, S.B.; Wyatt, J.K.; Czeisler, C.A.; Dijk, D.J. Eeg and ocular correlates of circadian melatonin phase and human performance decrements during sleep loss. Am. J. Physiol. 1999, 277, R640–649. [Google Scholar] [PubMed]
- Borbely, A.A.; Achermann, P. Sleep homeostasis and models of sleep regulation. J. Biol. Rhythms. 1999, 14, 557–568. [Google Scholar] [PubMed]
- Tononi, G.; Cirelli, C. Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 2014, 81, 12–34. [Google Scholar] [CrossRef] [PubMed]
- Dijk, D.J.; Czeisler, C.A. Paradoxical timing of the circadian rhythm of sleep propensity serves to consolidate sleep and wakefulness in humans. Neurosci. Lett. 1994, 166, 63–68. [Google Scholar] [CrossRef]
- Edgar, D.M.; Dement, W.C.; Fuller, C.A. Effect of scn lesions on sleep in squirrel monkeys: Evidence for opponent processes in sleep-wake regulation. J. Neurosci. 1993, 13, 1065–1079. [Google Scholar] [PubMed]
- Saper, C.B. The central circadian timing system. Curr. Opin. Neurobiol. 2013, 23, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Cajochen, C.; Chellappa, S.; Schmidt, C. What keeps us awake? The role of clocks and hourglasses, light, and melatonin. Int. Rev. Neurobiol. 2010, 93, 57–90. [Google Scholar] [PubMed]
- Hut, R.A.; Beersma, D.G. Evolution of time-keeping mechanisms: Early emergence and adaptation to photoperiod. Philos. Trans. R. Soc. Lond. B 2011, 366, 2141–2154. [Google Scholar] [CrossRef] [PubMed]
- Lavie, P. Ultrashort sleep-waking schedule. III. 'Gates' and 'forbidden zones' for sleep. Electroencephalogr. Clin. Neurophysiol. 1986, 63, 414–425. [Google Scholar] [CrossRef]
- Minors, D.S.; Waterhouse, J.M. Does 'anchor sleep' entrain circadian rhythms? Evidence from constant routine studies. J. Physiol. 1983, 345, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Cajochen, C.; Jewett, M.E.; Dijk, D.J. Human circadian melatonin rhythm phase delay during a fixed sleep-wake schedule interspersed with nights of sleep deprivation. J. Pineal. Res. 2003, 35, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Birchler-Pedross, A.; Schroder, C.M.; Munch, M.; Knoblauch, V.; Blatter, K.; Schnitzler-Sack, C.; Wirz-Justice, A.; Cajochen, C. Subjective well-being is modulated by circadian phase, sleep pressure, age, and gender. J. Biol. Rhythms. 2009, 24, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Cajochen, C.; Knoblauch, V.; Krauchi, K.; Renz, C.; Wirz-Justice, A. Dynamics of frontal eeg activity, sleepiness and body temperature under high and low sleep pressure. Neuroreport 2001, 12, 2277–2281. [Google Scholar] [CrossRef] [PubMed]
- Graw, P.; Krauchi, K.; Knoblauch, V.; Wirz-Justice, A.; Cajochen, C. Circadian and wake-dependent modulation of fastest and slowest reaction times during the psychomotor vigilance task. Physiol. Behav. 2004, 80, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Knoblauch, V.; Martens, W.; Wirz-Justice, A.; Krauchi, K.; Cajochen, C. Regional differences in the circadian modulation of human sleep spindle characteristics. Eur. J. Neurosci. 2003, 18, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Munch, M.; Knoblauch, V.; Blatter, K.; Schroder, C.; Schnitzler, C.; Krauchi, K.; Wirz-Justice, A.; Cajochen, C. Age-related attenuation of the evening circadian arousal signal in humans. Neurobiol. Aging. 2005, 26, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Maire, M.; Reichert, C.; Schmidt, C. Sleep-wake rhythms and cognition. J. Cogn. Behav. Psychother. 2013, 13, 133–170. [Google Scholar]
- Dijk, D.J.; Czeisler, C.A. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J. Neurosci. 1995, 15, 3526–3538. [Google Scholar] [PubMed]
- Dijk, D.J.; Shanahan, T.L.; Duffy, J.F.; Ronda, J.M.; Czeisler, C.A. Variation of electroencephalographic activity during non-rapid eye movement and rapid eye movement sleep with phase of circadian melatonin rhythm in humans. J. Physiol. 1997, 505, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Cajochen, C.; Foy, R.; Dijk, D.J. Frontal predominance of a relative increase in sleep delta and theta eeg activity after sleep loss in humans. Sleep Res. Online 1999, 2, 65–69. [Google Scholar] [PubMed]
- Strogatz, S.H.; Kronauer, R.E.; Czeisler, C.A. Circadian pacemaker interferes with sleep onset at specific times each day: Role in insomnia. Am. J. Physiol. 1987, 253, R172–R178. [Google Scholar] [PubMed]
- Dijk, D.J.; Edgar, D.M. Circadian and homoestatic control of wakefulness and sleep. In Regulation of Sleep and Wakefulness; Turek, F.W., Zee, P.C., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1999; pp. 111–147. [Google Scholar]
- Borbely, A.A.; Baumann, F.; Brandeis, D.; Strauch, I.; Lehmann, D. Sleep deprivation: Effect on sleep stages and eeg power density in man. Electroencephalogr. Clin. Neurophysiol. 1981, 51, 483–495. [Google Scholar] [CrossRef]
- Knoblauch, V.; Krauchi, K.; Renz, C.; Wirz-Justice, A.; Cajochen, C. Homeostatic control of slow-wave and spindle frequency activity during human sleep: Effect of differential sleep pressure and brain topography. Cereb Cortex 2002, 12, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.S.; Lazar, Z.I.; Dijk, D.J. Circadian regulation of slow waves in human sleep: Topographical aspects. Neuroimage 2015, 116, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.F.; Maire, M.; Gabel, V.; Hofstetter, M.; Viola, A.U.; Kolodyazhniy, V.; Strobel, W.; Goetz, T.; Bachmann, V.; Landolt, H.P.; et al. The circadian regulation of sleep: Impact of a functional ada-polymorphism and its association to working memory improvements. PLoS ONE 2014, 9, e113734. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kolodyazhniy, V.; Späti, J.; Frey, S.; Gotz, T.; Wirz-Justice, A.; Krauchi, K.; Cajochen, C.; Wilhelm, F.H. An improved method for estimating human circadian phase derived from multichannel ambulatory monitoring and artificial neural networks. Chronobiol. Int. 2012, 29, 1078–1097. [Google Scholar] [CrossRef] [PubMed]
- Cajochen, C.; Wyatt, J.K.; Czeisler, C.A.; Dijk, D.J. Separation of circadian and wake duration-dependent modulation of eeg activation during wakefulness. Neuroscience 2002, 114, 1047–1060. [Google Scholar] [CrossRef]
- Maire, M.; Reichert, C.F.; Gabel, V.; Viola, A.U.; Strobel, W.; Krebs, J.; Landolt, H.P.; Bachmann, V.; Cajochen, C.; Schmidt, C. Sleep ability mediates individual differences in the vulnerability to sleep loss: Evidence from a per3 polymorphism. Cortex 2014, 52, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Akerstedt, T.; Gillberg, M. Subjective and objective sleepiness in the active individual. Int. J. Neurosci. 1990, 52, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Cajochen, C.; Blatter, K.; Wallach, D. Circadian and sleep-wake dependent impact on neurobehavioral function. Psychol. Belg. 2004, 44, 59–80. [Google Scholar]
- Akerstedt, T.; Anund, A.; Axelsson, J.; Kecklund, G. Subjective sleepiness is a sensitive indicator of insufficient sleep and impaired waking function. J. Sleep Res. 2014, 23, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, H.P.; Baynard, M.D.; Maislin, G.; Dinges, D.F. Systematic interindividual differences in neurobehavioral impairment from sleep loss: Evidence of trait-like differential vulnerability. Sleep 2004, 27, 423–433. [Google Scholar] [PubMed]
- Basheer, R.; Strecker, R.E.; Thakkar, M.M.; McCarley, R.W. Adenosine and sleep-wake regulation. Prog. Neurobiol. 2004, 73, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, J.M.; Ferrari, L.L.; Keating, S.A.; Arrigoni, E. Adenosine inhibits glutamatergic input to basal forebrain cholinergic neurons. J. Neurophysiol. 2012, 107, 2769–2781. [Google Scholar] [CrossRef] [PubMed]
- Porkka-Heiskanen, T.; Strecker, R.E.; Thakkar, M.; Bjorkum, A.A.; Greene, R.W.; McCarley, R.W. Adenosine: A mediator of the sleep-inducing effects of prolonged wakefulness. Science 1997, 276, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, M.M.; Delgiacco, R.A.; Strecker, R.E.; McCarley, R.W. Adenosinergic inhibition of basal forebrain wakefulness-active neurons: A simultaneous unit recording and microdialysis study in freely behaving cats. Neuroscience 2003, 122, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Szymusiak, R.; McGinty, D. Hypothalamic regulation of sleep and arousal. Ann. N. Y. Acad. Sci. 2008, 1129, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Latini, S.; Pedata, F. Adenosine in the central nervous system: Release mechanisms and extracellular concentrations. J. Neurochem. 2001, 79, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, M.; Nikonova, E.V.; Zimmerman, J.E.; Galante, R.J.; Zhang, L.; Cater, J.R.; Geiger, J.D.; Pack, A.I. Enzymes of adenosine metabolism in the brain: Diurnal rhythm and the effect of sleep deprivation. J. Neurochem. 2003, 85, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Huang, Z.L.; Fredholm, B.B.; Urade, Y.; Hayaishi, O. Adenosine in the tuberomammillary nucleus inhibits the histaminergic system via a1 receptors and promotes non-rapid eye movement sleep. Proc. Natl. Acad. Sci. USA 2008, 105, 19992–19997. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Mochizuki, T.; Huang, Z.L.; Eguchi, N.; Sugita, Y.; Urade, Y.; Hayaishi, O. Dominant localization of adenosine deaminase in leptomeninges and involvement of the enzyme in sleep. Biochem. Biophys Res. Commun. 2003, 312, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Radulovacki, M.; Virus, R.M.; Djuricic-Nedelson, M.; Green, R.D. Hypnotic effects of deoxycorformycin in rats. Brain Res. 1983, 271, 392–395. [Google Scholar] [CrossRef]
- Franken, P.; Chollet, D.; Tafti, M. The homeostatic regulation of sleep need is under genetic control. J. Neurosci. 2001, 21, 2610–2621. [Google Scholar] [PubMed]
- Battistuzzi, G.; Iudicone, P.; Santolamazza, P.; Petrucci, R. Activity of adenosine deaminase allelic forms in intact erythrocytes and in lymphocytes. Ann. Hum. Genet. 1981, 45, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Riksen, N.P.; Franke, B.; van den Broek, P.; Naber, M.; Smits, P.; Rongen, G.A. The 22g>a polymorphism in the adenosine deaminase gene impairs catalytic function but does not affect reactive hyperaemia in humans in vivo. Pharmacogenet. Genomics 2008, 18, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Retey, J.V.; Adam, M.; Honegger, E.; Khatami, R.; Luhmann, U.F.; Jung, H.H.; Berger, W.; Landolt, H.P. A functional genetic variation of adenosine deaminase affects the duration and intensity of deep sleep in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 15676–15681. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, V.; Klaus, F.; Bodenmann, S.; Schafer, N.; Brugger, P.; Huber, S.; Berger, W.; Landolt, H.P. Functional ada polymorphism increases sleep depth and reduces vigilant attention in humans. Cereb Cortex 2012, 22, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, D.R.; Guindalini, C.; Pellegrino, R.; Barrueco, K.F.; Santos-Silva, R.; Bittencourt, L.R.; Tufik, S. Effects of the adenosine deaminase polymorphism and caffeine intake on sleep parameters in a large population sample. Sleep 2011, 34, 399–402. [Google Scholar] [PubMed]
- Mazzotti, D.R.; Guindalini, C.; de Souza, A.A.; Sato, J.R.; Santos-Silva, R.; Bittencourt, L.R.; Tufik, S. Adenosine deaminase polymorphism affects sleep eeg spectral power in a large epidemiological sample. PLoS ONE 2012, 7, e44154. [Google Scholar] [CrossRef] [PubMed]
- Kuna, S.T.; Maislin, G.; Pack, F.M.; Staley, B.; Hachadoorian, R.; Coccaro, E.F.; Pack, A.I. Heritability of performance deficit accumulation during acute sleep deprivation in twins. Sleep 2012, 35, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Urry, E.; Landolt, H.P. Adenosine, caffeine, and performance: From cognitive neuroscience of sleep to sleep pharmacogenetics. Curr. Top. Behav. Neurosci. 2014. [Google Scholar]
- Davidson, A.J.; Yamazaki, S.; Menaker, M. Scn: Ringmaster of the circadian circus or conductor of the circadian orchestra? Novartis. Found Symp. 2003, 253, 110–125. [Google Scholar] [PubMed]
- Schibler, U.; Sassone-Corsi, P. A web of circadian pacemakers. Cell 2002, 111, 919–922. [Google Scholar] [CrossRef]
- Cohen, R.A.; Albers, H.E. Disruption of human circadian and cognitive regulation following a discrete hypothalamic lesion: A case study. Neurology 1991, 41, 726–729. [Google Scholar] [CrossRef] [PubMed]
- DelRosso, L.M.; Hoque, R.; James, S.; Gonzalez-Toledo, E.; Chesson, A.L., Jr. Sleep-wake pattern following gunshot suprachiasmatic damage. J. Clin. Sleep Med. 2014, 10, 443–445. [Google Scholar] [CrossRef] [PubMed]
- Mistlberger, R.E. Circadian regulation of sleep in mammals: Role of the suprachiasmatic nucleus. Brain Res. 2005, 49, 429–454. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B.; Scammell, T.E.; Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature 2005, 437, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, C.; Andermann, M.L.; Scammell, T.E. Control of arousal by the orexin neurons. Curr. Opin. Neurobiol. 2013, 23, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Aston-Jones, G. Brain structures and receptors involved in alertness. Sleep Med. 2005, 6, S3–S7. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Chen, S.; Zhu, Y.; Oshinsky, M.L. A neural circuit for circadian regulation of arousal. Nat. Neurosci. 2001, 4, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Gompf, H.S.; Aston-Jones, G. Role of orexin input in the diurnal rhythm of locus coeruleus impulse activity. Brain Res. 2008, 1224, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, C.E.; Brewer, J.M.; Bittman, E.L. Central control of circadian phase in arousal-promoting neurons. PLoS ONE 2013, 8, e67173. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B. The neurobiology of sleep. Continuum (Minneap Minn) 2013, 19, 19–31. [Google Scholar] [PubMed]
- Samuels, E.R.; Szabadi, E. Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function part i: Principles of functional organisation. Curr. Neuropharmacol. 2008, 6, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef] [PubMed]
- Laposky, A.; Easton, A.; Dugovic, C.; Walisser, J.; Bradfield, C.; Turek, F. Deletion of the mammalian circadian clock gene bmal1/mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep 2005, 28, 395–409. [Google Scholar] [PubMed]
- Franken, P.; Dudley, C.A.; Estill, S.J.; Barakat, M.; Thomason, R.; O'Hara, B.F.; McKnight, S.L. Npas2 as a transcriptional regulator of non-rapid eye movement sleep: Genotype and sex interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 7118–7123. [Google Scholar] [CrossRef] [PubMed]
- Kopp, C.; Albrecht, U.; Zheng, B.; Tobler, I. Homeostatic sleep regulation is preserved in mper1 and mper2 mutant mice. Eur. J. Neurosci. 2002, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Wisor, J.P.; O'Hara, B.F.; Terao, A.; Selby, C.P.; Kilduff, T.S.; Sancar, A.; Edgar, D.M.; Franken, P. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Wisor, J.P.; Pasumarthi, R.K.; Gerashchenko, D.; Thompson, C.L.; Pathak, S.; Sancar, A.; Franken, P.; Lein, E.S.; Kilduff, T.S. Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J. Neurosci. 2008, 28, 7193–7201. [Google Scholar] [CrossRef] [PubMed]
- Franken, P.; Dijk, D.J. Circadian clock genes and sleep homeostasis. Eur. J. Neurosci. 2009, 29, 1820–1829. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, S.; Honma, S.; Abe, H.; Ishizaki, K.; Namihira, M.; Ikeda, M.; Honma, K. Clock genes outside the suprachiasmatic nucleus involved in manifestation of locomotor activity rhythm in rats. Eur. J. Neurosci. 2000, 12, 4206–4214. [Google Scholar] [CrossRef]
- Abe, H.; Honma, S.; Namihira, M.; Masubuchi, S.; Honma, K. Behavioural rhythm splitting in the cs mouse is related to clock gene expression outside the suprachiasmatic nucleus. Eur. J. Neurosci. 2001, 14, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, H.; Yoshinobu, Y.; Aida, R.; Moriya, T.; Akiyama, M.; Shibata, S. Restricted-feeding-induced anticipatory activity rhythm is associated with a phase-shift of the expression of mper1 and mper2 mrna in the cerebral cortex and hippocampus but not in the suprachiasmatic nucleus of mice. Eur J Neurosci 2001, 13, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Ruby, C.L.; Vadnie, C.A.; Hinton, D.J.; Abulseoud, O.A.; Walker, D.L.; O'Connor, K.M.; Noterman, M.F.; Choi, D.S. Adenosinergic regulation of striatal clock gene expression and ethanol intake during constant light. Neuropsychopharmacol 2014, 39, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.A.; Yu, Y.V.; Govindaiah, G.; Ye, X.; Artinian, L.; Coleman, T.P.; Sweedler, J.V.; Cox, C.L.; Gillette, M.U. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science 2012, 337, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.; Wang, T.A.; Gillette, M.U. Circadian gating of neuronal functionality: A basis for iterative metaplasticity. Front Syst. Neurosci. 2014, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Roblero, R.; Díaz-Muñoz, M.; Báez-Ruíz, A.; Quinto-Muñoz, D.; Lundkvist, G.; Michel, S. Intracellular calcium as a clock output from scn neurons. In Mechanisms of Circadian Systems in Animals and their Clinical Relevance; Aguilar-Roblero, R., Díaz-Muñoz, M., Fanjul-Moles, M.L., Eds.; Springer: Gewerbestrasse, Switzerland, 2015. [Google Scholar]
- Deboer, T.; Vansteensel, M.J.; Detari, L.; Meijer, J.H. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat. Neurosci. 2003, 6, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Mistlberger, R.E.; Landry, G.J.; Marchant, E.G. Sleep deprivation can attenuate light-induced phase shifts of circadian rhythms in hamsters. Neurosci. Lett. 1997, 238, 5–8. [Google Scholar] [CrossRef]
- van Diepen, H.C.; Lucassen, E.A.; Yasenkov, R.; Groenen, I.; Ijzerman, A.P.; Meijer, J.H.; Deboer, T. Caffeine increases light responsiveness of the mouse circadian pacemaker. Eur. J. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.; Blackburn, M.R.; Wang, S.W.; Ribelayga, C.P.; O'Brien, J. Adenosine and dopamine receptors coregulate photoreceptor coupling via gap junction phosphorylation in mouse retina. J. Neurosci. 2013, 33, 3135–3150. [Google Scholar] [CrossRef] [PubMed]
- Ribelayga, C.; Mangel, S.C. A circadian clock and light/dark adaptation differentially regulate adenosine in the mammalian retina. J. Neurosci. 2005, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Hallworth, R.; Cato, M.; Colbert, C.; Rea, M.A. Presynaptic adenosine a1 receptors regulate retinohypothalamic neurotransmission in the hamster suprachiasmatic nucleus. J. Neurobiol. 2002, 52, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.M.; Markwald, R.R.; McHill, A.W.; Chinoy, E.D.; Snider, J.A.; Bessman, S.C.; Jung, C.M.; O'Neill, J.S.; Wright, K.P., Jr. Effects of caffeine on the human circadian clock in vivo and in vitro. Sci. Trans. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.P., Jr.; Badia, P.; Myers, B.L.; Plenzler, S.C.; Hakel, M. Caffeine and light effects on nighttime melatonin and temperature levels in sleep-deprived humans. Brain Res. 1997, 747, 78–84. [Google Scholar] [CrossRef]
- Elliott, K.J.; Todd Weber, E.; Rea, M.A. Adenosine a1 receptors regulate the response of the hamster circadian clock to light. Eur. J. Pharmacol. 2001, 414, 45–53. [Google Scholar] [CrossRef]
- Sigworth, L.A.; Rea, M.A. Adenosine a1 receptors regulate the response of the mouse circadian clock to light. Brain Res. 2003, 960, 246–251. [Google Scholar] [CrossRef]
- Watanabe, A.; Moriya, T.; Nisikawa, Y.; Araki, T.; Hamada, T.; Shibata, S.; Watanabe, S. Adenosine a1-receptor agonist attenuates the light-induced phase shifts and fos expression in vivo and optic nerve stimulation-evoked field potentials in the suprachiasmatic nucleus in vitro. Brain Res. 1996, 740, 329–336. [Google Scholar] [CrossRef]
- Rogers, N.L.; Dinges, D.F. Interaction of chronic sleep restriction and circadian system in humans. J. Sleep Res. 2008, 17, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.; Lesauter, J. Circadian and homeostatic factors in arousal. Ann. N. Y. Acad. Sci. 2008, 1129, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Deboer, T.; Overeem, S.; Visser, N.A.; Duindam, H.; Frolich, M.; Lammers, G.J.; Meijer, J.H. Convergence of circadian and sleep regulatory mechanisms on hypocretin-1. Neuroscience 2004, 129, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.W.; Gao, X.B. Adenosine inhibits activity of hypocretin/orexin neurons by the a1 receptor in the lateral hypothalamus: A possible sleep-promoting effect. J. Neurophysiol. 2007, 97, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, M.M.; Engemann, S.C.; Walsh, K.M.; Sahota, P.K. Adenosine and the homeostatic control of sleep: Effects of a1 receptor blockade in the perifornical lateral hypothalamus on sleep-wakefulness. Neuroscience 2008, 153, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Inutsuka, A.; Yamanaka, A. The regulation of sleep and wakefulness by the hypothalamic neuropeptide orexin/hypocretin. Nagoya. J. Med. Sci. 2013, 75, 29–36. [Google Scholar] [PubMed]
- Basheer, R.; Bauer, A.; Elmenhorst, D.; Ramesh, V.; McCarley, R.W. Sleep deprivation upregulates a1 adenosine receptors in the rat basal forebrain. Neuroreport 2007, 18, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Elmenhorst, D.; Meyer, P.T.; Winz, O.H.; Matusch, A.; Ermert, J.; Coenen, H.H.; Basheer, R.; Haas, H.L.; Zilles, K.; Bauer, A. Sleep deprivation increases a1 adenosine receptor binding in the human brain: A positron emission tomography study. J. Neurosci. 2007, 27, 2410–2415. [Google Scholar] [CrossRef] [PubMed]
- Florio, C.; Rosati, A.M.; Traversa, U.; Vertua, R. Inhibitory and excitatory effects of adenosine antagonists on spontaneous locomotor activity in mice. Life Sci. 1997, 60, 1477–1486. [Google Scholar] [CrossRef]
- Schmidt, C.; Collette, F.; Cajochen, C.; Peigneux, P. A time to think: Circadian rhythms in human cognition. Cogn. Neuropsychol. 2007, 24, 755–789. [Google Scholar] [CrossRef] [PubMed]
- Harrison, Y.; Horne, J.A. The impact of sleep deprivation on decision making: A review. J. Exp. Psychol. Appl. 2000, 6, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Werth, E.; Achermann, P.; Borbely, A.A. Fronto-occipital eeg power gradients in human sleep. J. Sleep. Res. 1997, 6, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Drummond, S.P.; Brown, G.G.; Gillin, J.C.; Stricker, J.L.; Wong, E.C.; Buxton, R.B. Altered brain response to verbal learning following sleep deprivation. Nature 2000, 403, 655–657. [Google Scholar] [PubMed]
- Maquet, P.; Degueldre, C.; Delfiore, G.; Aerts, J.; Peters, J.M.; Luxen, A.; Franck, G. Functional neuroanatomy of human slow wave sleep. J. Neurosci. 1997, 17, 2807–2812. [Google Scholar] [PubMed]
- D'Esposito, M.; Postle, B.R. The cognitive neuroscience of working memory. Ann. Rev. Psychol. 2015, 66, 115–142. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, P.A. An overview of the tasks used to test working memory in rodents. Neurosci. Biobehav. Rev. 2004, 28, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Cowan, N. The magical number 4 in short-term memory: A reconsideration of mental storage capacity. Behav. Brain Sci. 2001, 24, 87–185. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.A. The magical number seven plus or minus two: Some limits on our capacity for processing information. Psychol. Rev. 1956, 63, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Backman, L.; Nyberg, L. Dopamine and training-related working-memory improvement. Neurosci. Biobehav. Rev. 2013, 37, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Buschkuehl, M.; Jaeggi, S.M.; Jonides, J. Neuronal effects following working memory training. Dev. Cogn. Neurosci. 2012, 2, S167–S179. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.B.; Chein, J.M. Does working memory training work? The promise and challenges of enhancing cognition by training working memory. Psychon. Bull. Rev. 2011, 18, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Miyake, A.; Friedman, N.P.; Emerson, M.J.; Witzki, A.H.; Howerter, A. The unity and diversity of executive functions and their contributions to complex "frontal lobe" tasks: A latent variable analysis. Cogn. Psychol. 2000, 41, 49–100. [Google Scholar] [CrossRef] [PubMed]
- Collette, F.; van der Linden, M. Brain imaging of the central executive component of working memory. Neurosci. Biobehav. Rev. 2002, 26, 105–125. [Google Scholar] [CrossRef]
- Baddeley, A.D.; Hitch, G.J. Working memory. In The Psychology of Learning and Motivation: Advances in Research and Theory; Bower, G.A., Ed.; Academic: New York, NY, USA, 1974; pp. 47–89. [Google Scholar]
- Baddeley, A. Working memory: Theories, models, and controversies. Annu. Rev. Psychol. 2012, 63, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Conway, A.R.; Kane, M.J.; Bunting, M.F.; Hambrick, D.Z.; Wilhelm, O.; Engle, R.W. Working memory span tasks: A methodological review and user's guide. Psychon. Bull. Rev. 2005, 12, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Tombaugh, T.N. A comprehensive review of the paced auditory serial addition test (pasat). Arch. Clin. Neuropsychol. 2006, 21, 53–76. [Google Scholar] [CrossRef] [PubMed]
- Jaeggi, S.M.; Buschkuehl, M.; Perrig, W.J.; Meier, B. The concurrent validity of the n-back task as a working memory measure. Memory 2010, 18, 394–412. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, D.J.; Pekar, J.J.; Mostofsky, S.H. Meta-analysis of go/no-go tasks demonstrating that fmri activation associated with response inhibition is task-dependent. Neuropsychologia 2008, 46, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.M.; Alexander, G.E. Neuron activity related to short-term memory. Science 1971, 173, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Olton, D.S.; Feustle, W.A. Hippocampal function required for nonspatial working memory. Exp. Brain. Res. 1981, 41, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Owen, A.M.; McMillan, K.M.; Laird, A.R.; Bullmore, E. N-back working memory paradigm: A meta-analysis of normative functional neuroimaging studies. Hum. Brain. Mapp. 2005, 25, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Collette, F.; Hogge, M.; Salmon, E.; van der Linden, M. Exploration of the neural substrates of executive functioning by functional neuroimaging. Neuroscience 2006, 139, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Badre, D.; Wagner, A.D. Left ventrolateral prefrontal cortex and the cognitive control of memory. Neuropsychologia 2007, 45, 2883–2901. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, C.J. The cerebellum and cognition: Evidence from functional imaging studies. Cerebellum 2012, 11, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.C.; Lindenberger, U.; Werkle-Bergner, M. Lifespan age differences in working memory: A two-component framework. Neurosci. Biobehav. Rev. 2012, 36, 2007–2033. [Google Scholar] [CrossRef] [PubMed]
- Nee, D.E.; Brown, J.W.; Askren, M.K.; Berman, M.G.; Demiralp, E.; Krawitz, A.; Jonides, J. A meta-analysis of executive components of working memory. Cereb Cortex 2013, 23, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Irlbacher, K.; Kraft, A.; Kehrer, S.; Brandt, S.A. Mechanisms and neuronal networks involved in reactive and proactive cognitive control of interference in working memory. Neurosci. Biobehav. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.E. The working memory networks of the human brain. Neuroscientist 2007, 13, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Leszczynski, M. How does hippocampus contribute to working memory processing? Front. Hum. Neurosci. 2011, 5, 168. [Google Scholar] [CrossRef] [PubMed]
- Yonelinas, A.P. The hippocampus supports high-resolution binding in the service of perception, working memory and long-term memory. Behav. Brain Res. 2013, 254, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Robbins, T.W.; Roberts, A.C. Differential regulation of fronto-executive function by the monoamines and acetylcholine. Cereb Cortex 2007, 17, i151–i160. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Moore, A.N.; Kobori, N.; Runyan, J.D. Molecular activity underlying working memory. Learn Mem. 2007, 14, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.U.; Muly, E.C. Molecular mechanisms of working memory. Behav. Brain Res. 2011, 219, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Noudoost, B. The role of prefrontal catecholamines in attention and working memory. Front. Neural. Circuits 2014, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Barch, D.M.; Ceaser, A. Cognition in schizophrenia: Core psychological and neural mechanisms. Trends Cogn. Sci. 2012, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Chen, J.F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.M. Adenosine and brain function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [PubMed]
- Ohno, M.; Watanabe, S. Working memory failure by stimulation of hippocampal adenosine a1 receptors in rats. Neuroreport 1996, 7, 3013–3016. [Google Scholar] [CrossRef] [PubMed]
- Von Lubitz, D.K.; Paul, I.A.; Bartus, R.T.; Jacobson, K.A. Effects of chronic administration of adenosine a1 receptor agonist and antagonist on spatial learning and memory. Eur. J. Pharmacol. 1993, 249, 271–280. [Google Scholar] [CrossRef]
- Shen, H.Y.; Singer, P.; Lytle, N.; Wei, C.J.; Lan, J.Q.; Williams-Karnesky, R.L.; Chen, J.F.; Yee, B.K.; Boison, D. Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J. Clin. Invest. 2012, 122, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Yee, B.K.; Singer, P.; Chen, J.F.; Feldon, J.; Boison, D. Transgenic overexpression of adenosine kinase in brain leads to multiple learning impairments and altered sensitivity to psychomimetic drugs. Eur. J. Neurosci. 2007, 26, 3237–3252. [Google Scholar] [CrossRef] [PubMed]
- Augusto, E.; Matos, M.; Sevigny, J.; El-Tayeb, A.; Bynoe, M.S.; Muller, C.E.; Cunha, R.A.; Chen, J.F. Ecto-5'-nucleotidase (cd73)-mediated formation of adenosine is critical for the striatal adenosine a2a receptor functions. J. Neurosci. 2013, 33, 11390–11399. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Llort, L.; Schiffmann, S.N.; Shmidt, T.; Canela, L.; Camon, L.; Wassholm, M.; Canals, M.; Terasmaa, A.; Fernandez-Teruel, A.; Tobena, A.; et al. Working memory deficits in transgenic rats overexpressing human adenosine a2a receptors in the brain. Neurobiol. Learn Mem. 2007, 87, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Zlomuzica, A.; Burghoff, S.; Schrader, J.; Dere, E. Superior working memory and behavioural habituation but diminished psychomotor coordination in mice lacking the ecto-5'-nucleotidase (cd73) gene. Purinergic. Signal. 2013, 9, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.J.; Zhu, M.E.; Shu, D.; Du, X.P.; Song, X.H.; Wang, X.T.; Zheng, R.Y.; Cai, X.H.; Chen, J.F.; He, J.C. Preferential enhancement of working memory in mice lacking adenosine a(2a) receptors. Brain Res. 2009, 1303, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.J.; Singer, P.; Coelho, J.; Boison, D.; Feldon, J.; Yee, B.K.; Chen, J.F. Selective inactivation of adenosine a(2a) receptors in striatal neurons enhances working memory and reversal learning. Learn Mem. 2011, 18, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.; McGarrity, S.; Shen, H.Y.; Boison, D.; Yee, B.K. Working memory and the homeostatic control of brain adenosine by adenosine kinase. Neuroscience 2012, 213, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F. Adenosine receptor control of cognition in normal and disease. Int. Rev. Neurobiol. 2014, 119, 257–307. [Google Scholar] [PubMed]
- Haller, S.; Rodriguez, C.; Moser, D.; Toma, S.; Hofmeister, J.; Sinanaj, I.; van de Ville, D.; Giannakopoulos, P.; Lovblad, K.O. Acute caffeine administration impact on working memory-related brain activation and functional connectivity in the elderly: A bold and perfusion mri study. Neuroscience 2013, 250, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, E.B.; de Groot, R.H.; Evers, E.A.; Snel, J.; Veerman, E.C.; Ligtenberg, A.J.; Jolles, J.; Veltman, D.J. The effect of caffeine on working memory load-related brain activation in middle-aged males. Neuropharmacolog 2013, 64, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Koppelstaetter, F.; Poeppel, T.D.; Siedentopf, C.M.; Ischebeck, A.; Verius, M.; Haala, I.; Mottaghy, F.M.; Rhomberg, P.; Golaszewski, S.; Gotwald, T.; et al. Does caffeine modulate verbal working memory processes? An fmri study. Neuroimage 2008, 39, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Dinges, D.F. A meta-analysis of the impact of short-term sleep deprivation on cognitive variables. Psychol. Bull. 2010, 136, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Killgore, W.D. Effects of sleep deprivation on cognition. Prog. Brain Res. 2010, 185, 105–129. [Google Scholar] [PubMed]
- Tucker, A.M.; Whitney, P.; Belenky, G.; Hinson, J.M.; van Dongen, H.P. Effects of sleep deprivation on dissociated components of executive functioning. Sleep 2010, 33, 47–57. [Google Scholar] [PubMed]
- Drummond, S.P.; Paulus, M.P.; Tapert, S.F. Effects of two nights sleep deprivation and two nights recovery sleep on response inhibition. J. Sleep Res. 2006, 15, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Couyoumdjian, A.; Sdoia, S.; Tempesta, D.; Curcio, G.; Rastellini, E.; de Gennaro, L.; Ferrara, M. The effects of sleep and sleep deprivation on task-switching performance. J. Sleep Res. 2010, 19, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Chee, M.W.; Chuah, L.Y. Functional neuroimaging insights into how sleep and sleep deprivation affect memory and cognition. Curr. Opin. Neurol. 2008, 21, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Chee, M.W.; Choo, W.C. Functional imaging of working memory after 24 hr of total sleep deprivation. J. Neurosci. 2004, 24, 4560–4567. [Google Scholar] [CrossRef] [PubMed]
- Chee, M.W.; Chuah, L.Y.; Venkatraman, V.; Chan, W.Y.; Philip, P.; Dinges, D.F. Functional imaging of working memory following normal sleep and after 24 and 35 h of sleep deprivation: Correlations of fronto-parietal activation with performance. Neuroimage 2006, 31, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Choo, W.C.; Lee, W.W.; Venkatraman, V.; Sheu, F.S.; Chee, M.W. Dissociation of cortical regions modulated by both working memory load and sleep deprivation and by sleep deprivation alone. Neuroimage 2005, 25, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Chuah, Y.M.; Venkatraman, V.; Dinges, D.F.; Chee, M.W. The neural basis of interindividual variability in inhibitory efficiency after sleep deprivation. J. Neurosci. 2006, 26, 7156–7162. [Google Scholar] [CrossRef] [PubMed]
- Habeck, C.; Rakitin, B.C.; Moeller, J.; Scarmeas, N.; Zarahn, E.; Brown, T.; Stern, Y. An event-related fmri study of the neurobehavioral impact of sleep deprivation on performance of a delayed-match-to-sample task. Cogn. Brain Res. 2004, 18, 306–321. [Google Scholar] [CrossRef]
- Mu, Q.; Nahas, Z.; Johnson, K.A.; Yamanaka, K.; Mishory, A.; Koola, J.; Hill, S.; Horner, M.D.; Bohning, D.E.; George, M.S. Decreased cortical response to verbal working memory following sleep deprivation. Sleep 2005, 28, 55–67. [Google Scholar] [PubMed]
- Mu, Q.; Mishory, A.; Johnson, K.A.; Nahas, Z.; Kozel, F.A.; Yamanaka, K.; Bohning, D.E.; George, M.S. Decreased brain activation during a working memory task at rested baseline is associated with vulnerability to sleep deprivation. Sleep 2005, 28, 433–446. [Google Scholar] [PubMed]
- Chee, M.W.; van Dongen, H.P. Functional imaging of inter-individual differences in response to sleep deprivation. In Neuroimaging of Sleep and Sleep Disorders; Nofzinger, E., Maquet, P., Thorpy, M.J., Eds.; Cambridge University Press: New York, NY, USA, 2013; pp. 154–162. [Google Scholar]
- Wesensten, N.J.; Killgore, W.D.; Balkin, T.J. Performance and alertness effects of caffeine, dextroamphetamine, and modafinil during sleep deprivation. J. Sleep Res. 2005, 14, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Bjorness, T.E.; Kelly, C.L.; Gao, T.; Poffenberger, V.; Greene, R.W. Control and function of the homeostatic sleep response by adenosine a1 receptors. J. Neurosci. 2009, 29, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Monk, T.H. Circadian rhythms in human performance and mood under constant conditions. J. Sleep Res. 1997, 6, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.F.; Maire, M.; Gabel, V.; Viola, A.U.; Götz, T.; Scheffler, K.; Klarhöfer, M.; Strobel, W.; Cajochen, C.; Schmidt, S. Centre for Chronobiology, Psychiatric Hospital of the University of Basel, Basel, Switzerland. Unpublished work. 2015. [Google Scholar]
- Cirelli, C.; Tononi, G. Is sleep essential? PLoS Biol 2008, 6, e216. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.H.; Arand, D.L. Caffeine use as a model of acute and chronic insomnia. Sleep 1992, 15, 526–536. [Google Scholar] [PubMed]
- Retey, J.V.; Adam, M.; Khatami, R.; Luhmann, U.F.; Jung, H.H.; Berger, W.; Landolt, H.P. A genetic variation in the adenosine a2a receptor gene (adora2a) contributes to individual sensitivity to caffeine effects on sleep. Clin. Pharmacol. Ther. 2007, 81, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Koppelstaetter, F.; Poeppel, T.D.; Siedentopf, C.M.; Ischebeck, A.; Kolbitsch, C.; Mottaghy, F.M.; Felber, S.R.; Jaschke, W.R.; Krause, B.J. Caffeine and cognition in functional magnetic resonance imaging. J. Alzheimer Dis. 2010, 20, S71–S84. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reichert, C.F.; Maire, M.; Schmidt, C.; Cajochen, C. Sleep-Wake Regulation and Its Impact on Working Memory Performance: The Role of Adenosine. Biology 2016, 5, 11. https://doi.org/10.3390/biology5010011
Reichert CF, Maire M, Schmidt C, Cajochen C. Sleep-Wake Regulation and Its Impact on Working Memory Performance: The Role of Adenosine. Biology. 2016; 5(1):11. https://doi.org/10.3390/biology5010011
Chicago/Turabian StyleReichert, Carolin Franziska, Micheline Maire, Christina Schmidt, and Christian Cajochen. 2016. "Sleep-Wake Regulation and Its Impact on Working Memory Performance: The Role of Adenosine" Biology 5, no. 1: 11. https://doi.org/10.3390/biology5010011
APA StyleReichert, C. F., Maire, M., Schmidt, C., & Cajochen, C. (2016). Sleep-Wake Regulation and Its Impact on Working Memory Performance: The Role of Adenosine. Biology, 5(1), 11. https://doi.org/10.3390/biology5010011