
cAMP-Inhibits Cytoplasmic Phospholipase A2 and Protects Neurons against Amyloid-β-Induced Synapse Damage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Primary Cortical Neuronal Cultures
2.2. Isolation of Synaptosomes
2.3. Activated cPLA2 ELISA
2.4. Synaptophysin ELISA
2.5. Western Blotting
2.6. Peptides
2.7. Preparation of Aβ-Containing Medium
2.8. Immunodepletions
2.9. Aβ42 ELISA
2.10. Aβ40 ELISA
2.11. Drugs
2.12. Statistical Methods
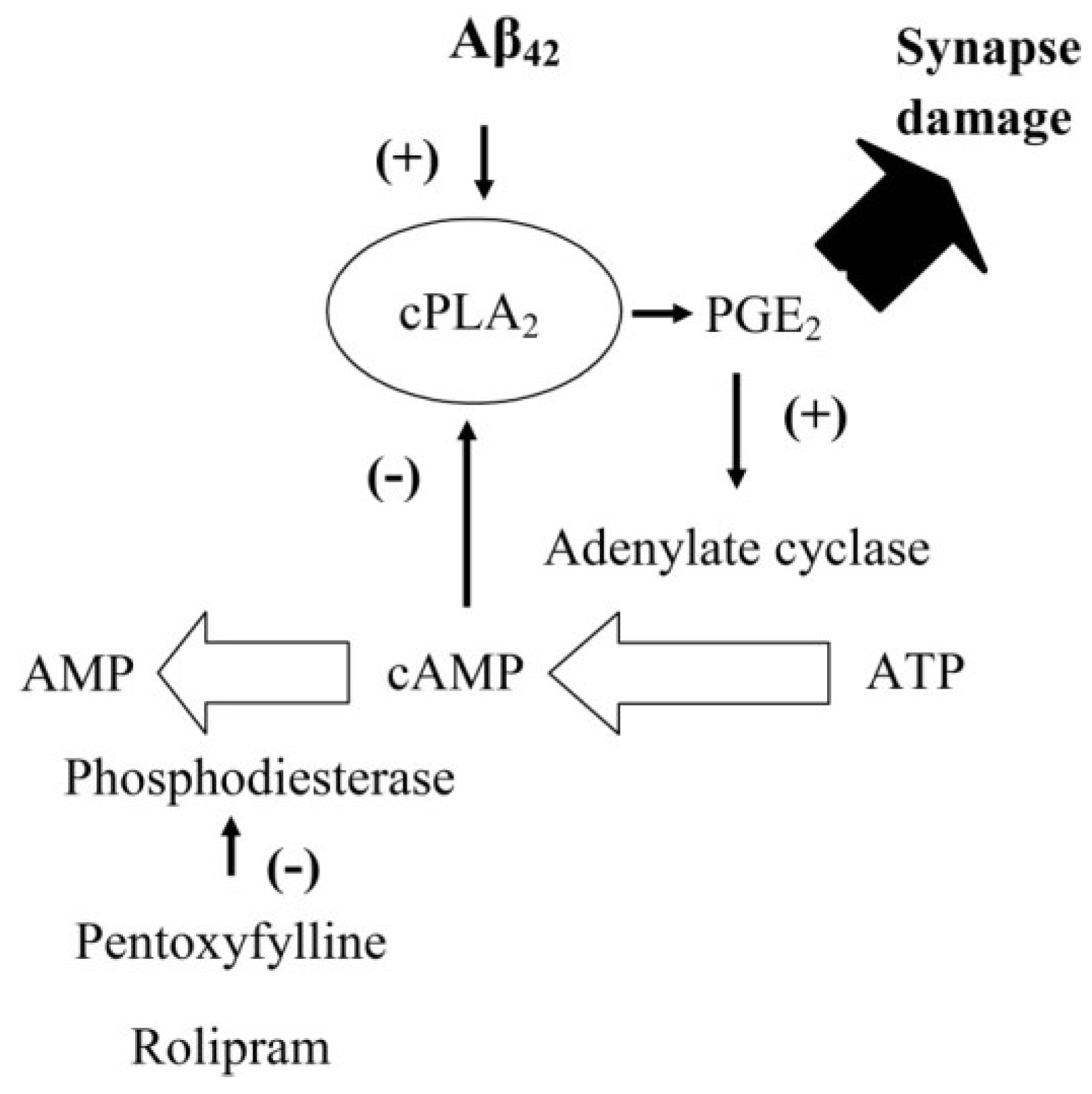
3. Results and Discussion
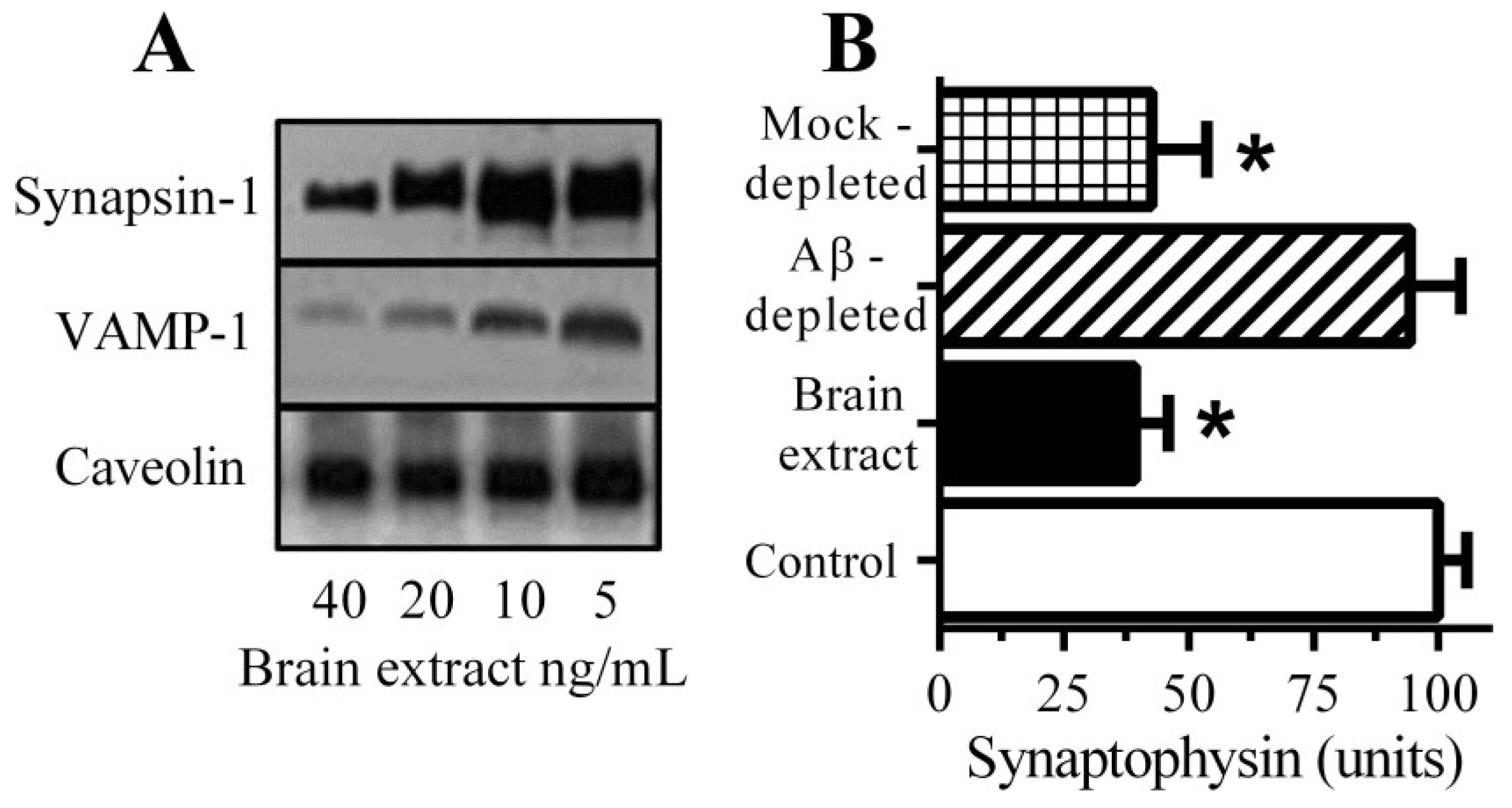
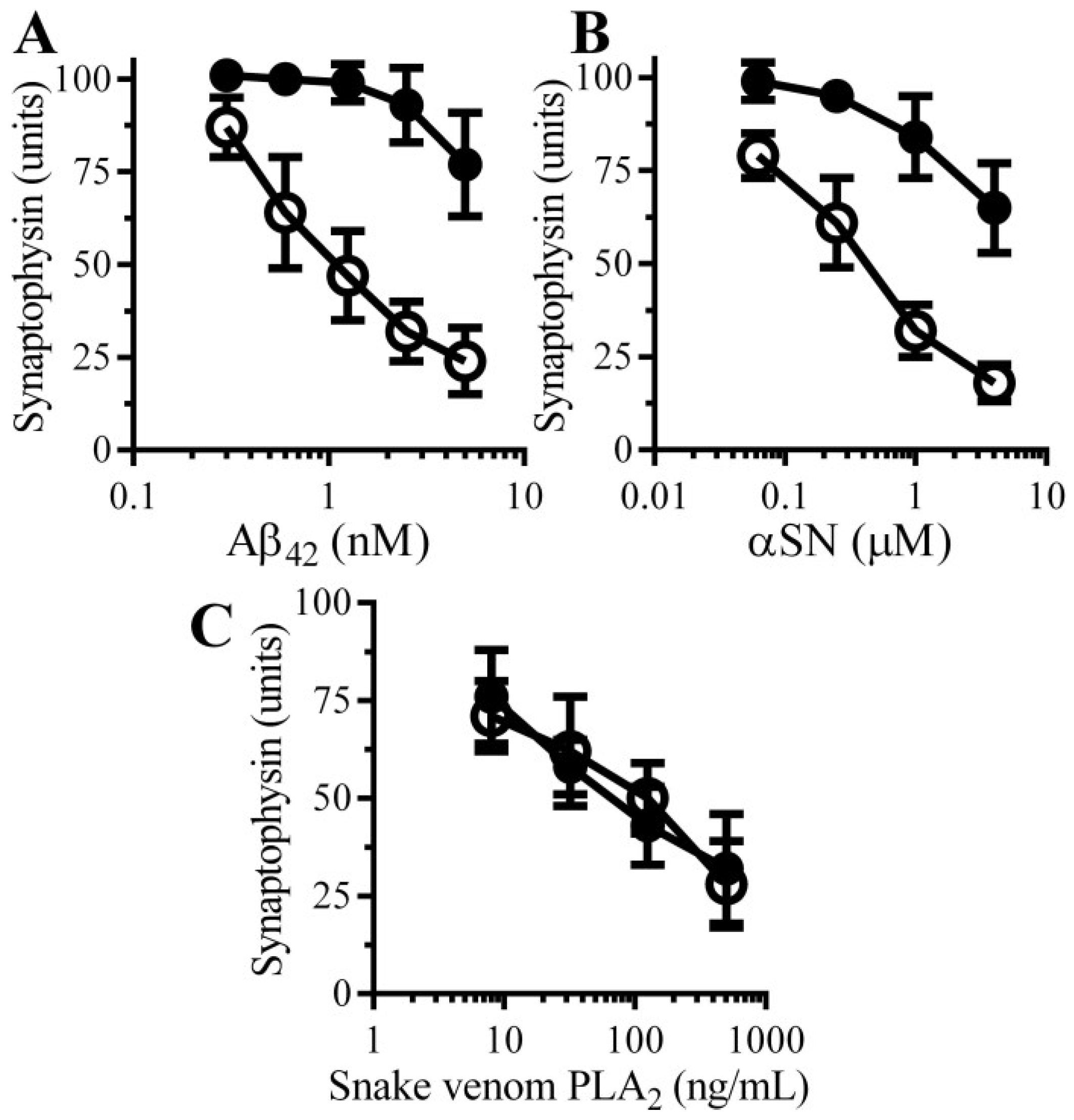
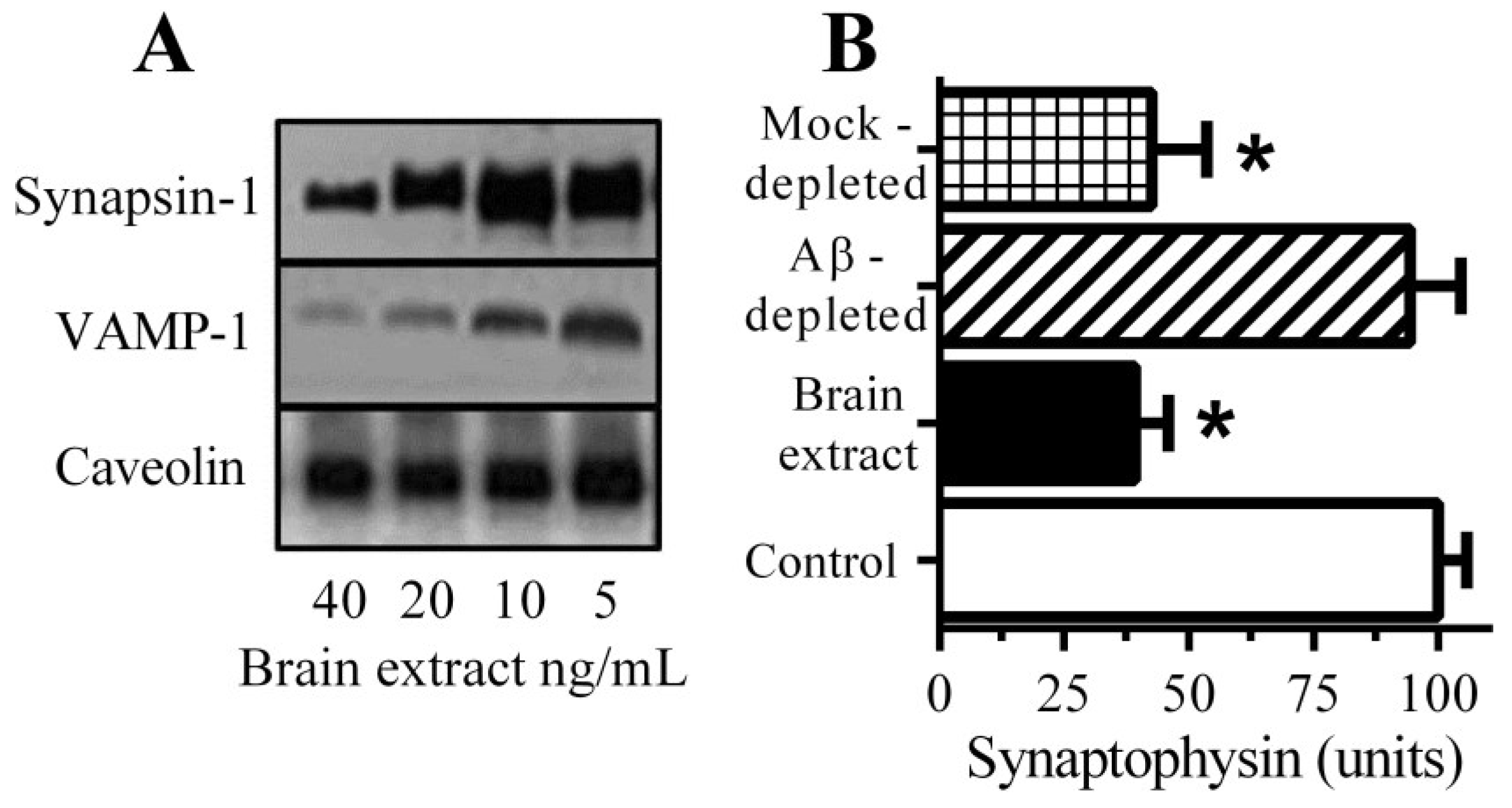
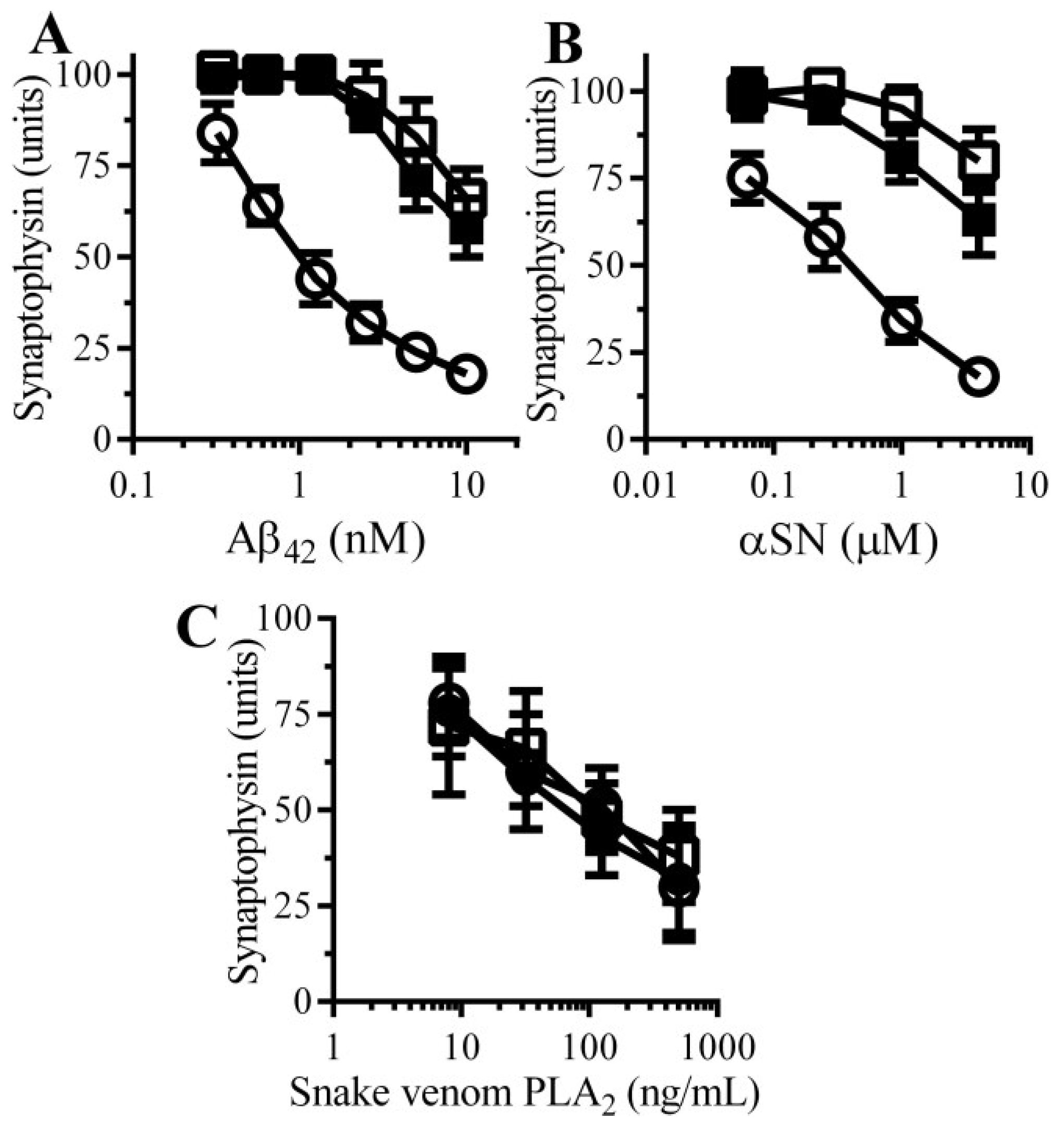
3.1. Aβ Triggered Synapse Damage
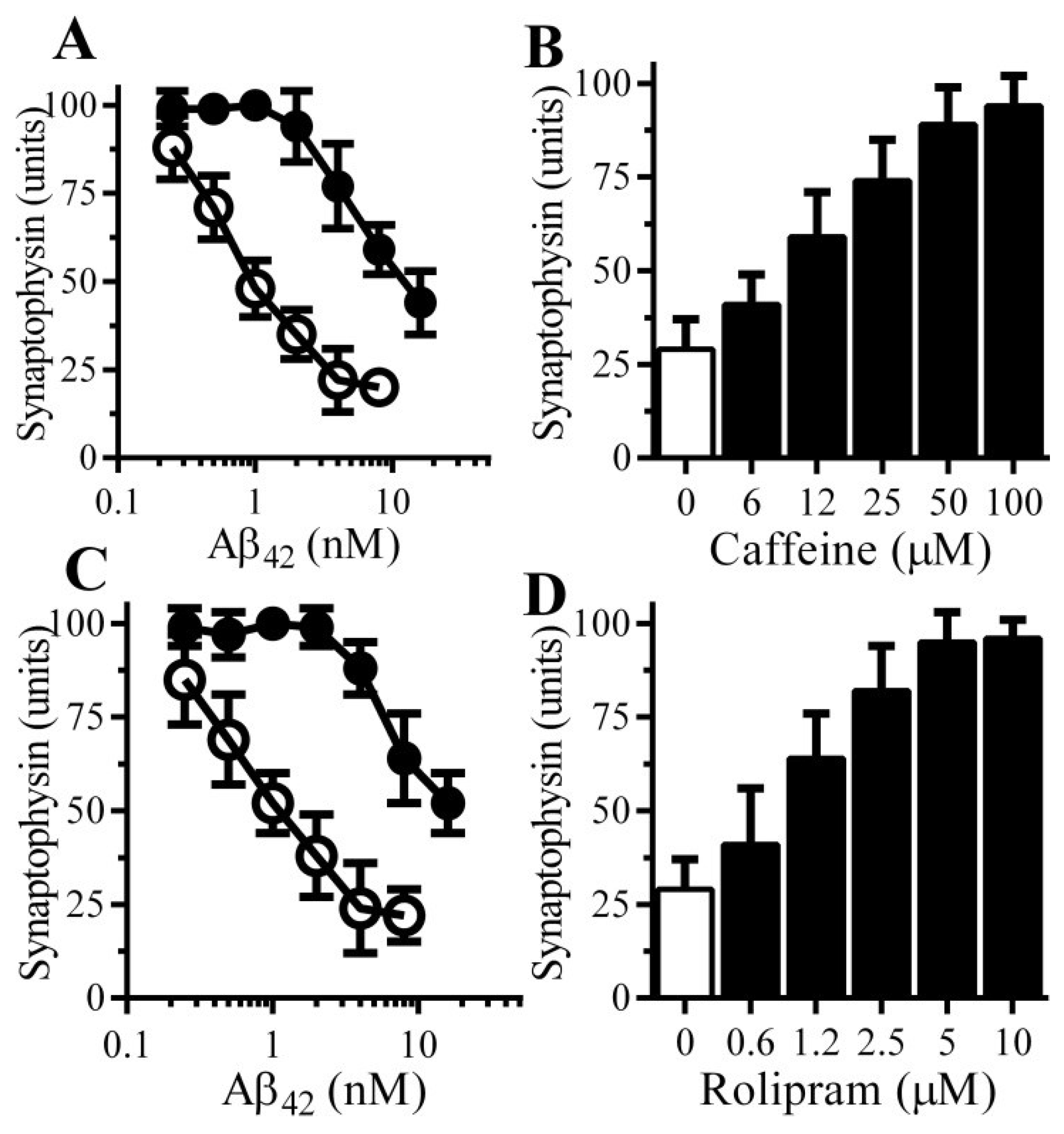
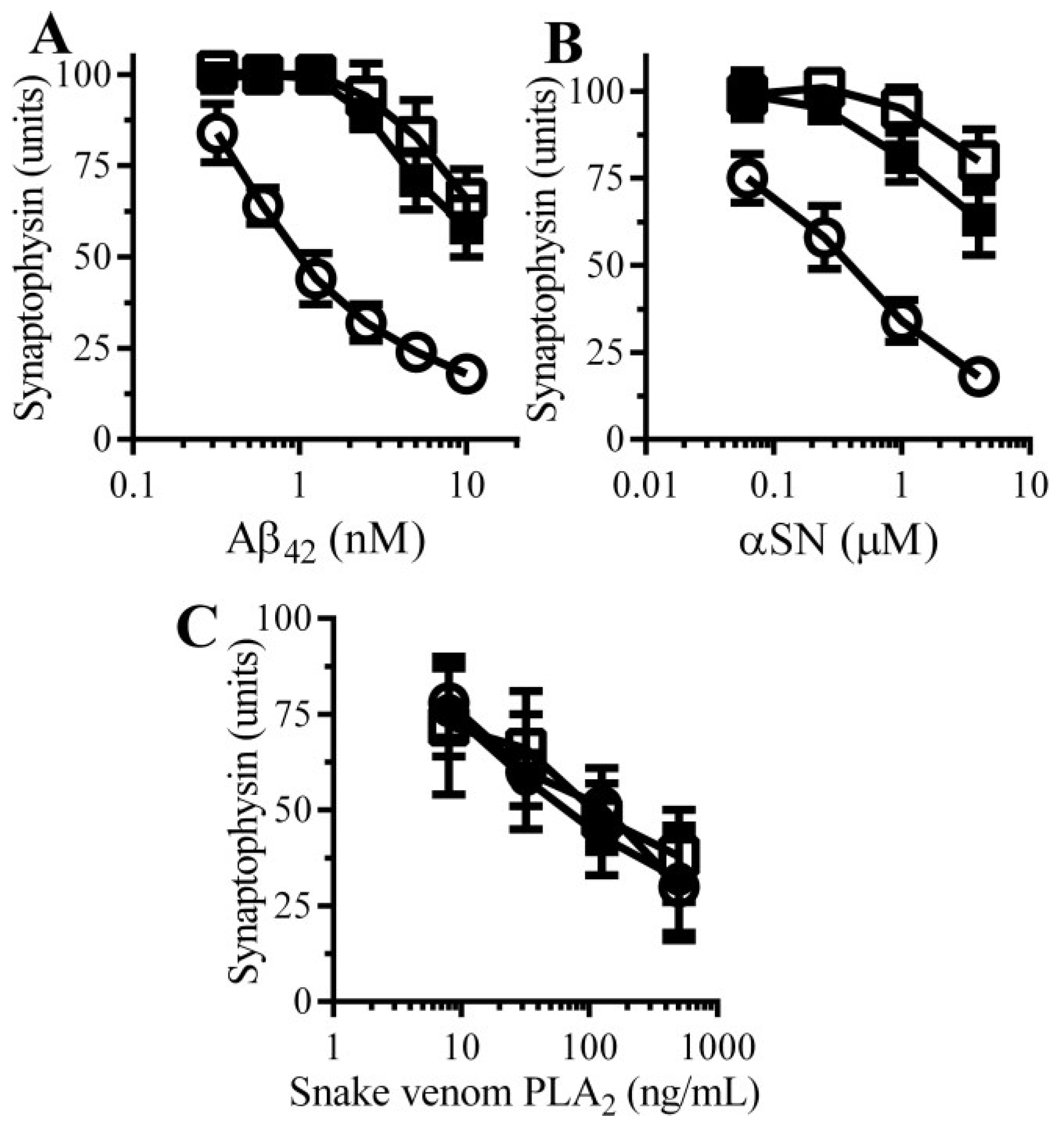
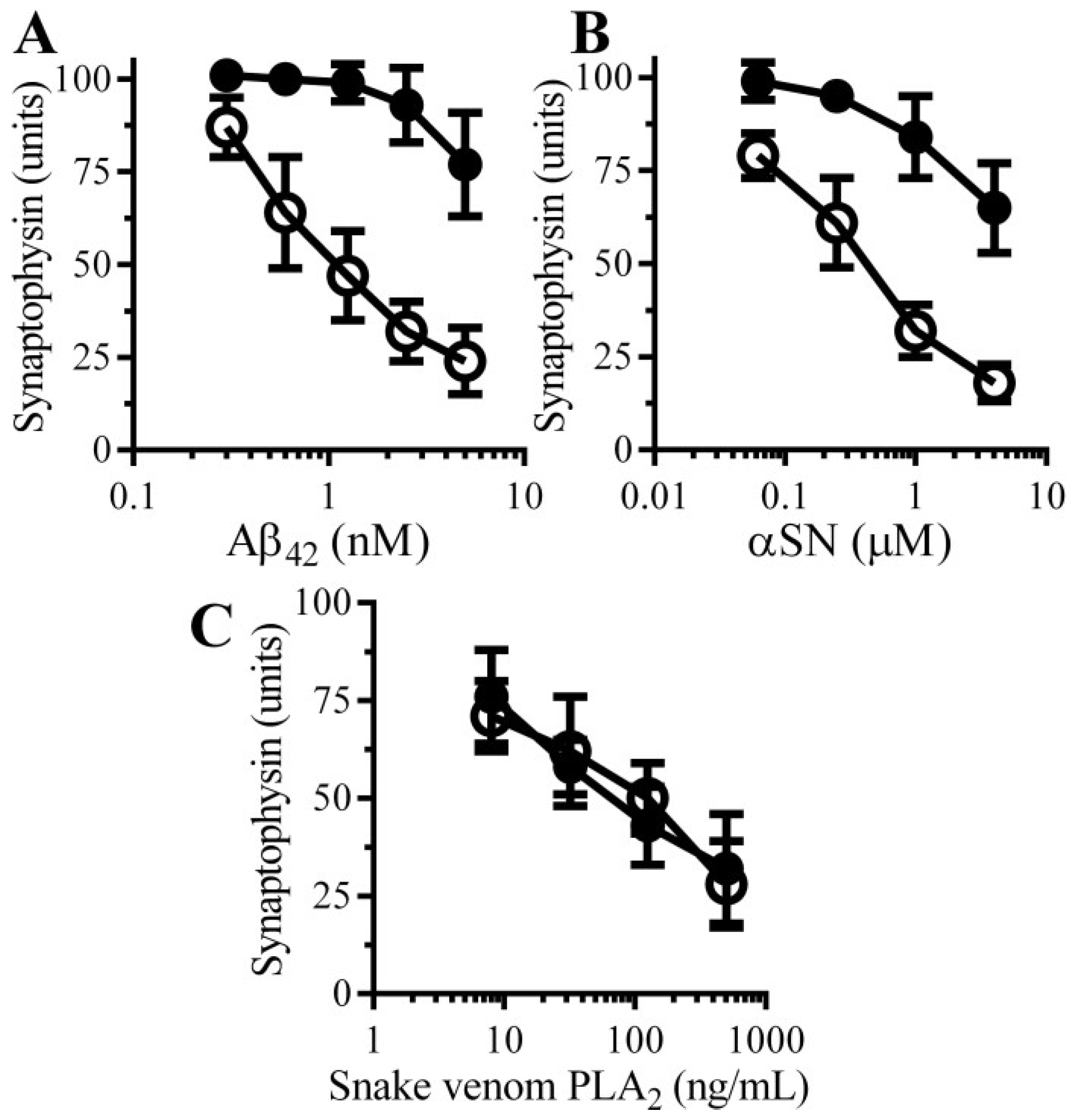
3.2. Pentoxifylline Protects Neurons against Aβ-Induced Synapse Damage
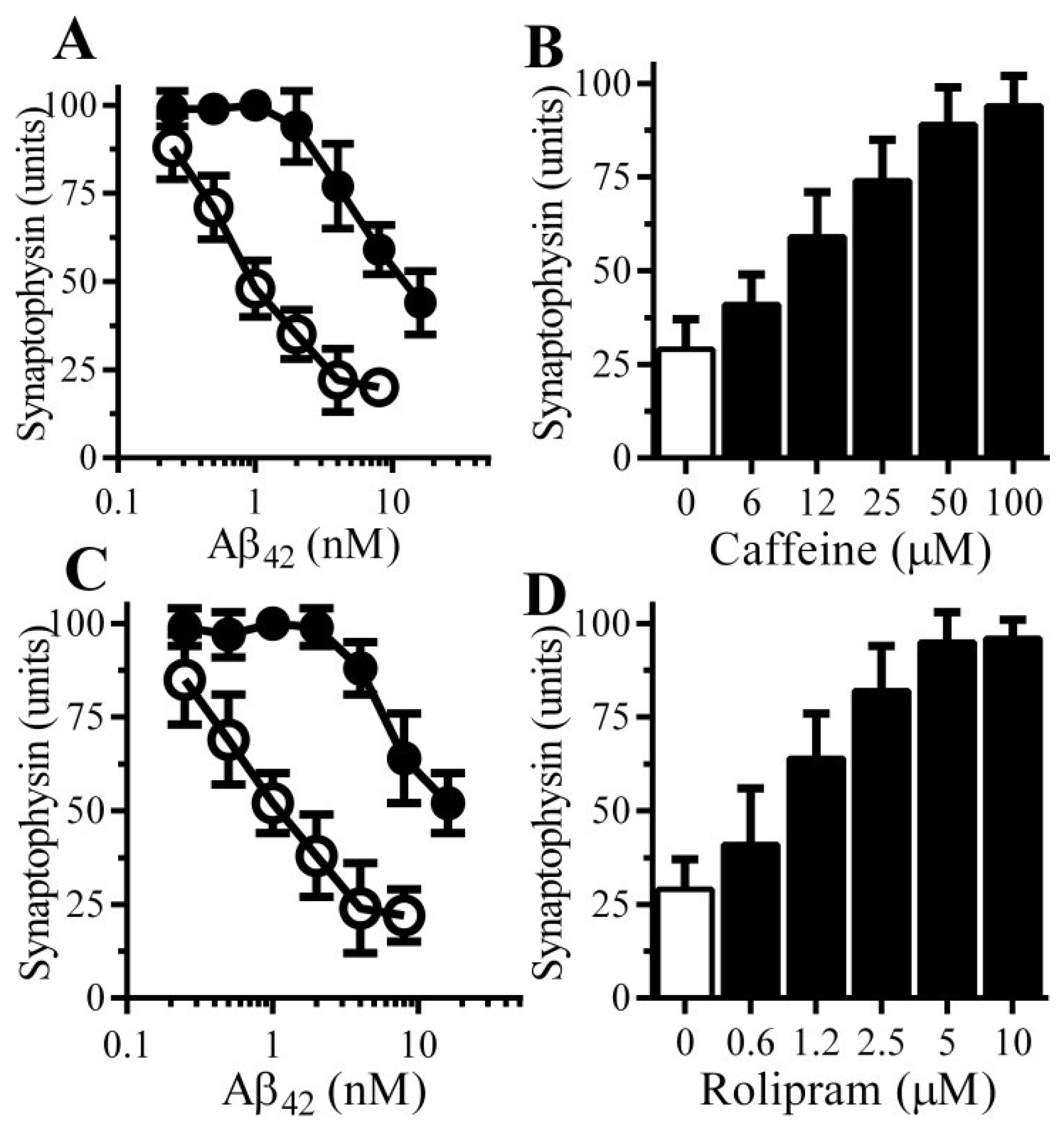
3.3. Caffeine and Rolipram Protect Neurons against Aβ and αSN-Induced Synapse Damage
3.4. PDE Inhibitors Do Not Alter the Accumulation of Aβ42 in Synapses
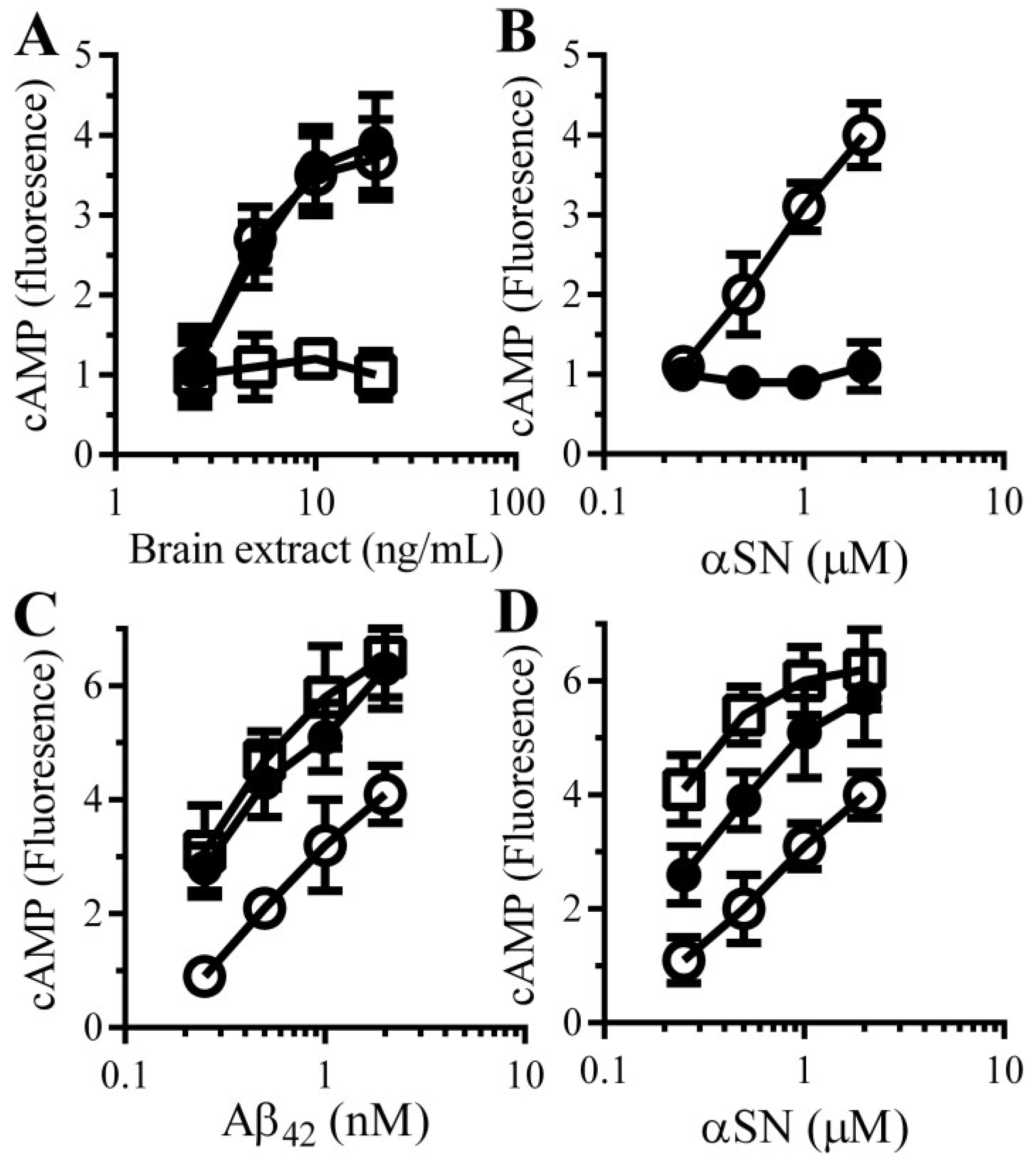
3.5. Aβ and αSN Increase the Concentrations of cAMP in Synaptosomes
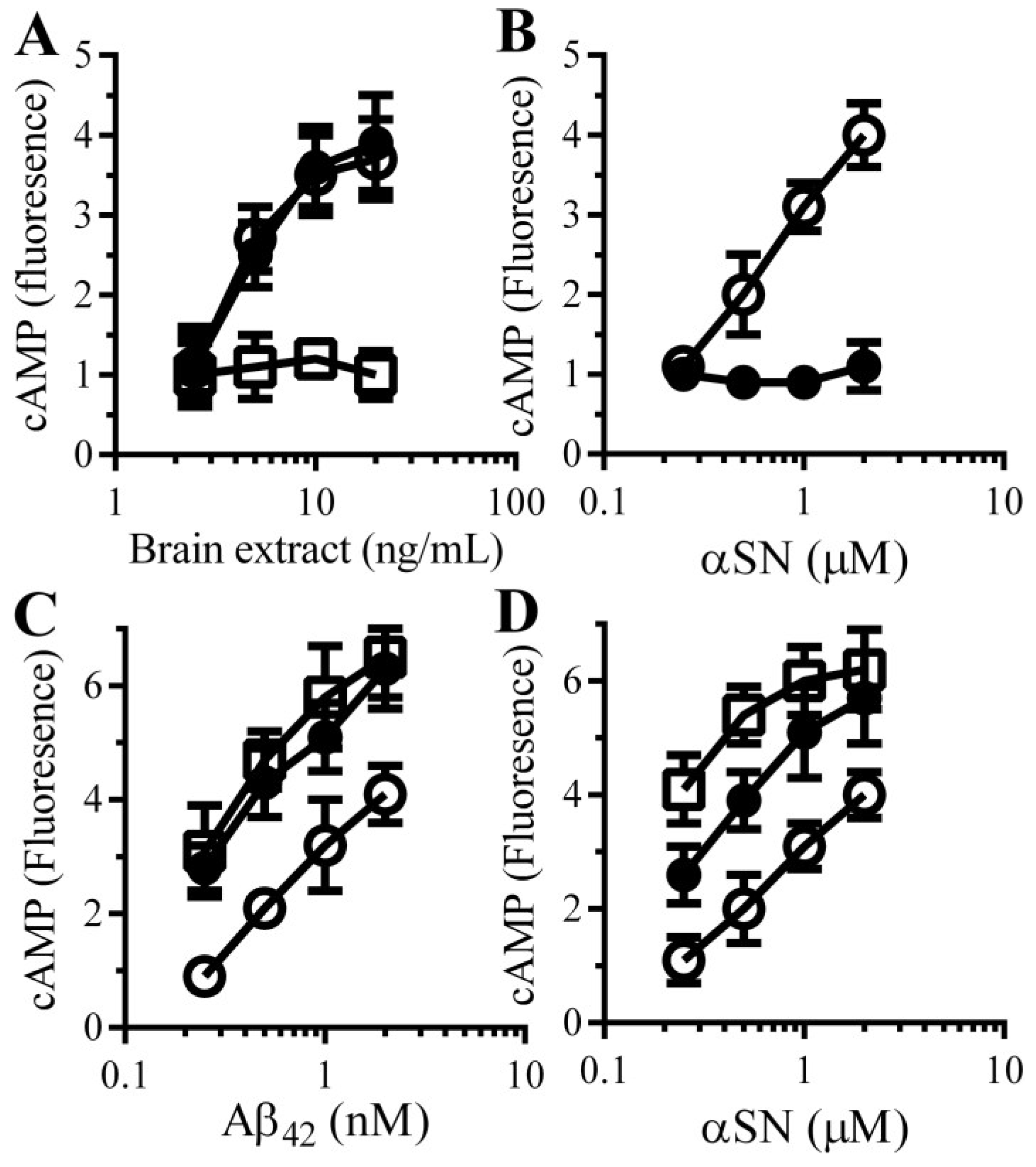
3.6. cAMP Analogues Protect Neurons against Aβ and αSN-Induced Synapse Damage
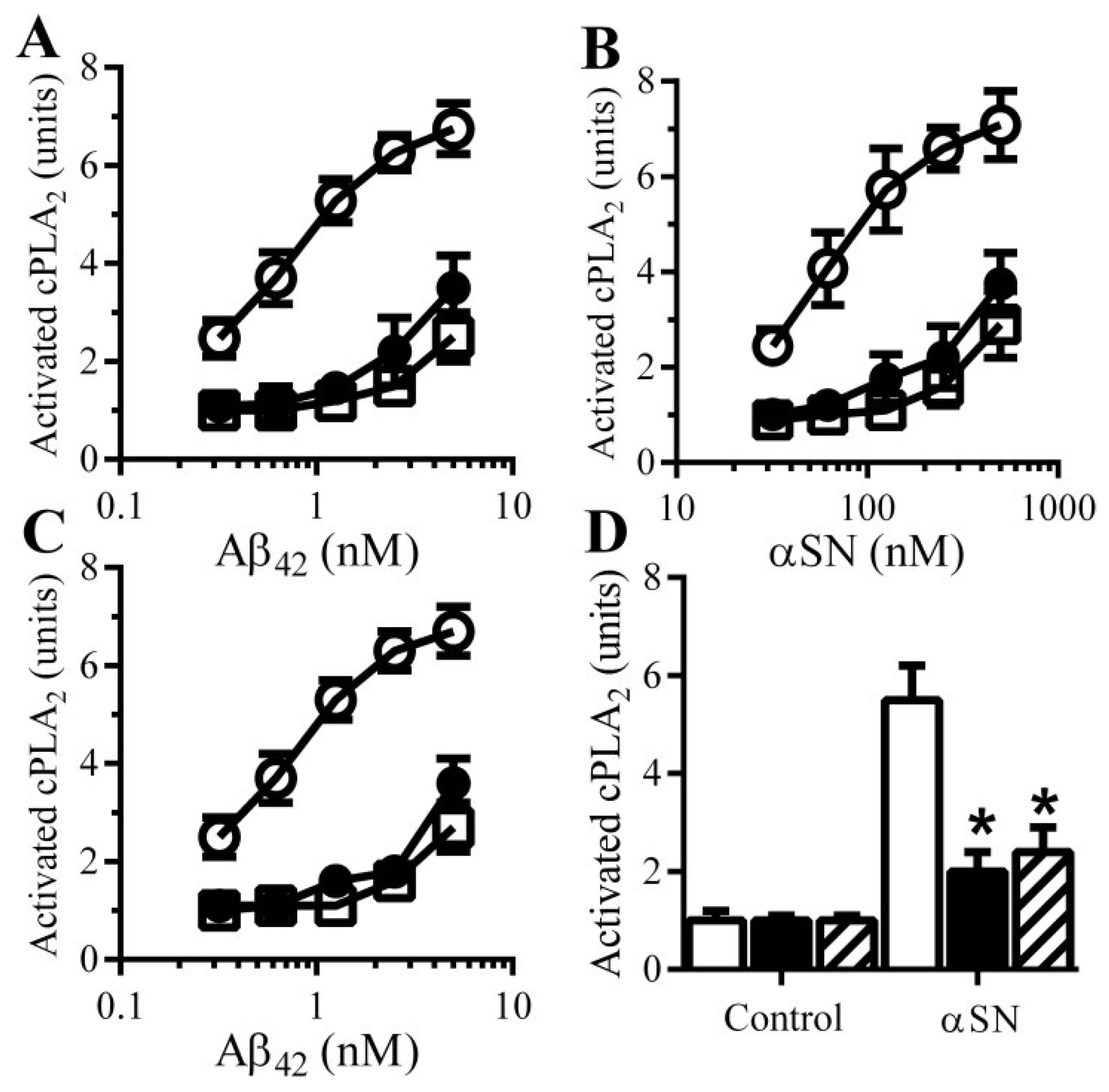
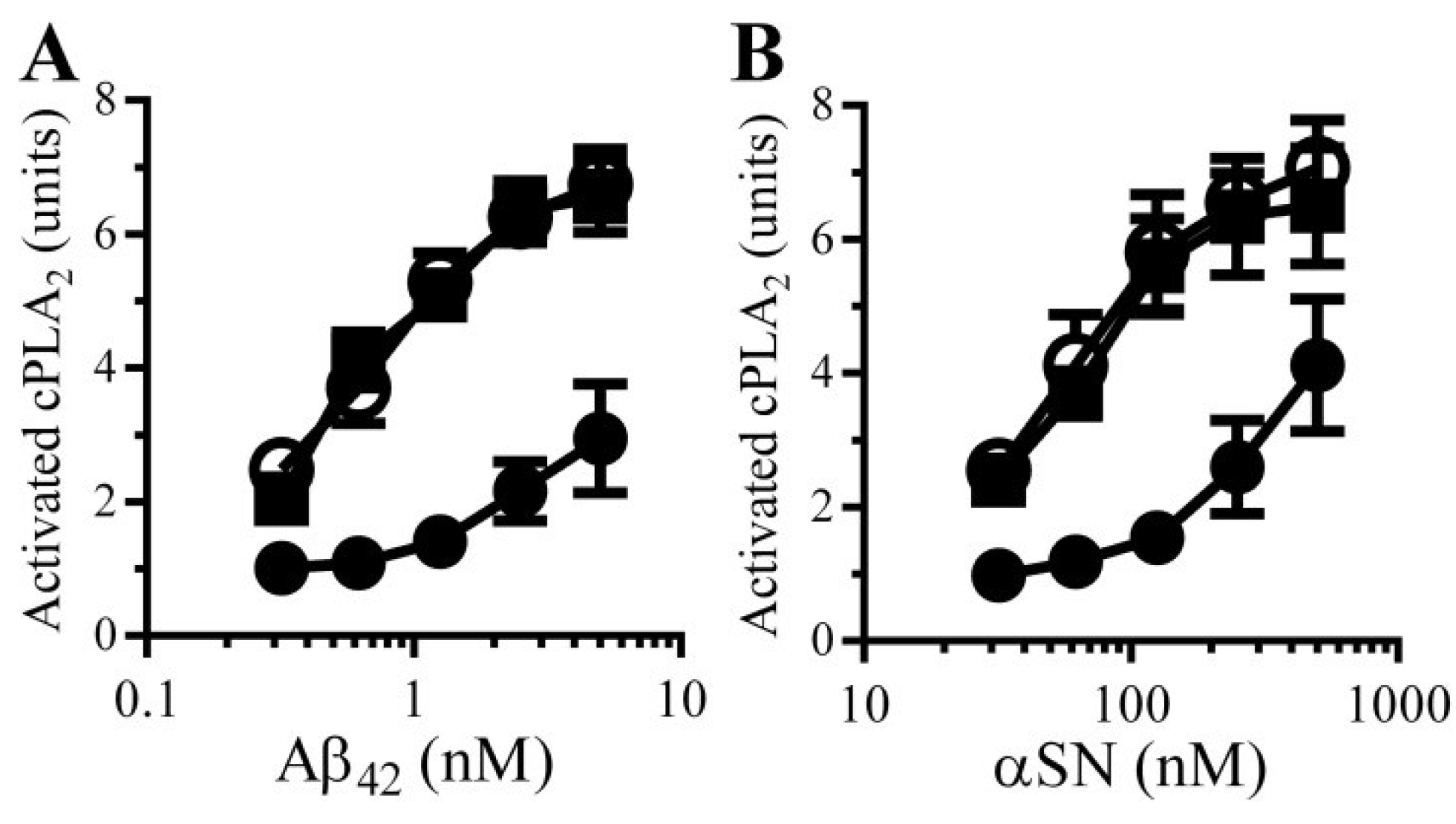
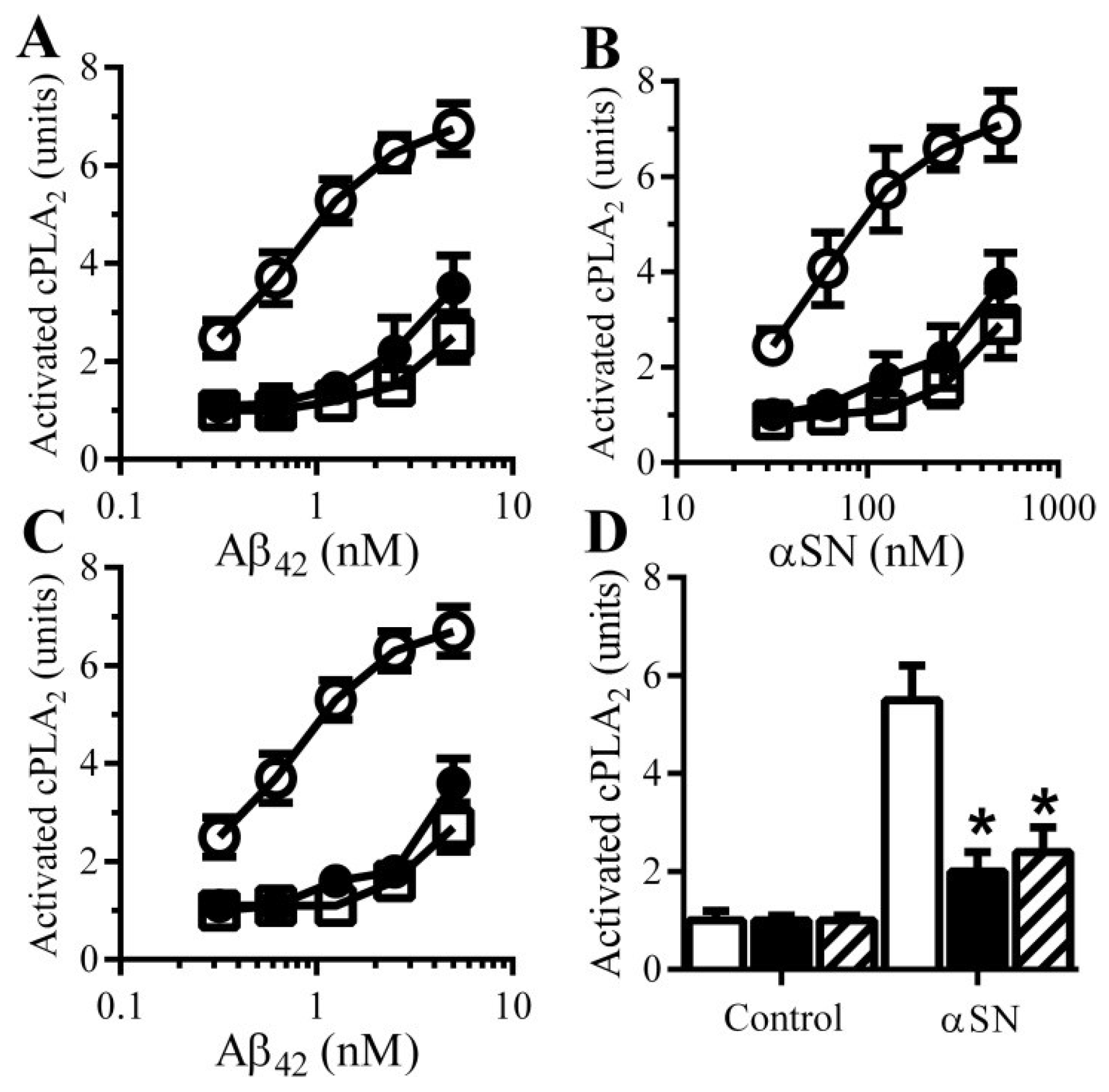
3.7. PDE Inhibitors Reduce the Aβ and αSN-Induced Activation of cPLA2 in Synapses
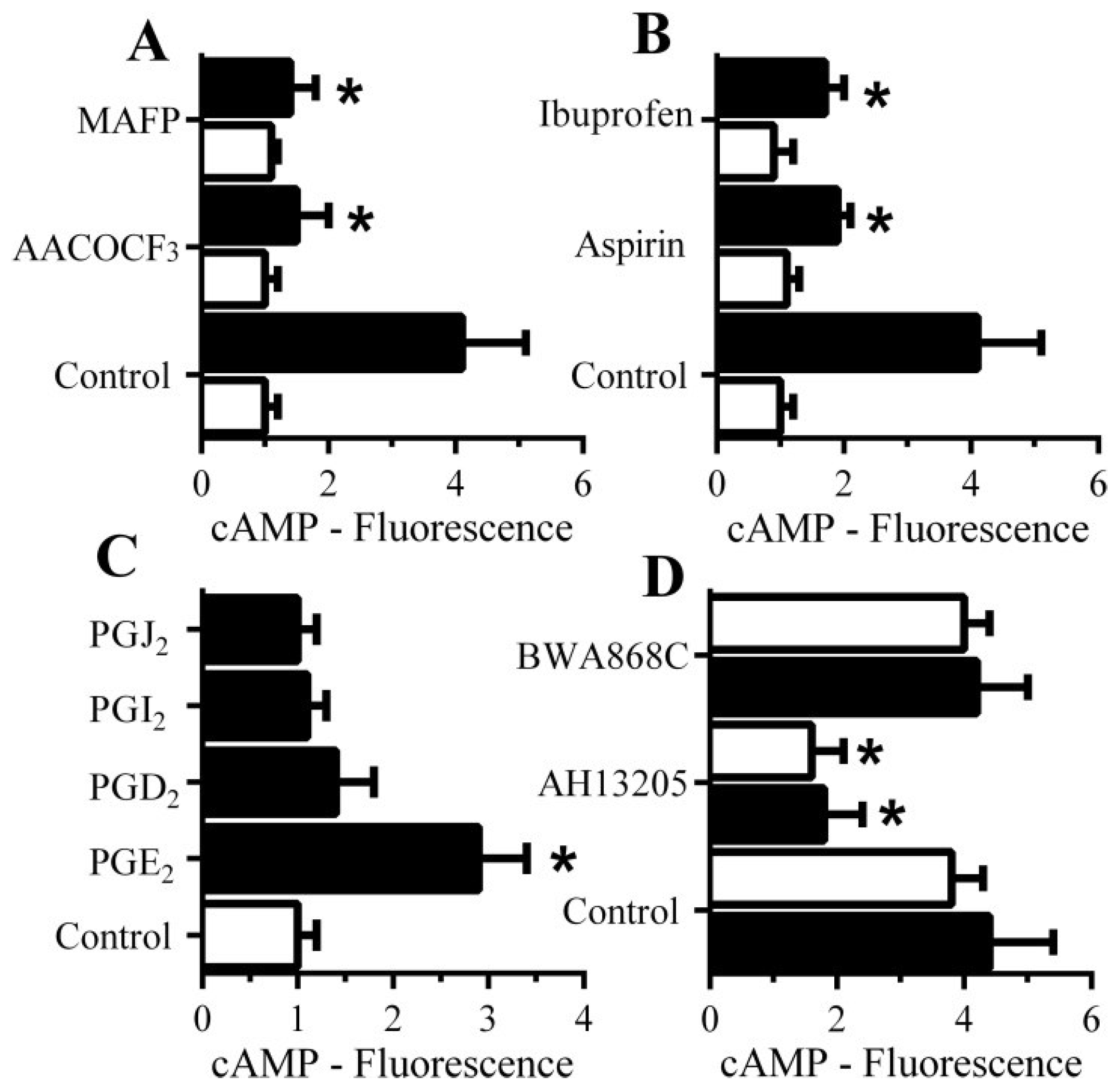
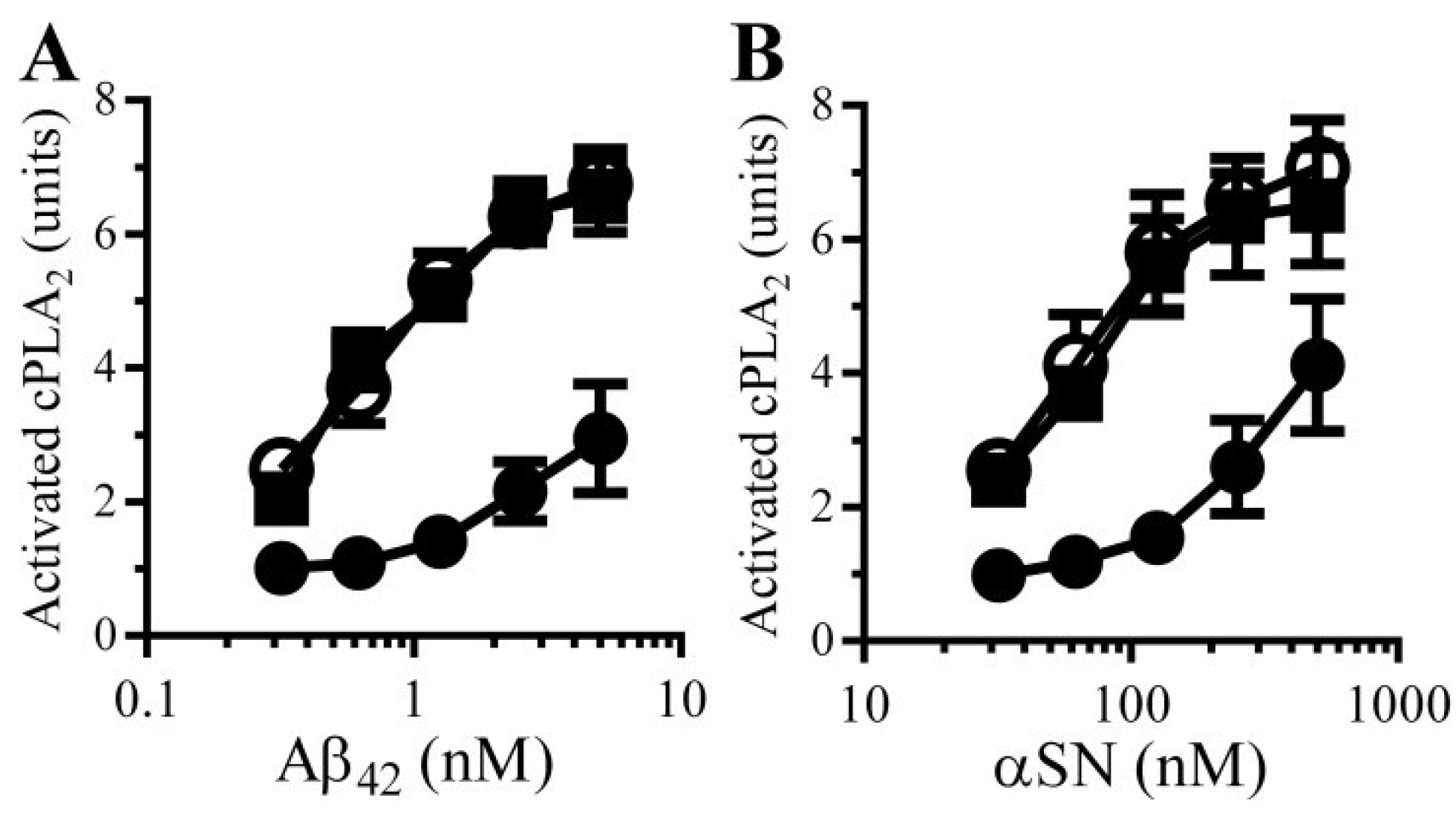
3.8. PGE2-Mediates Aβ-Induced Increase in Camp

3.9. PGE2 Inhibits the Activation of cPLA2 in Synapses
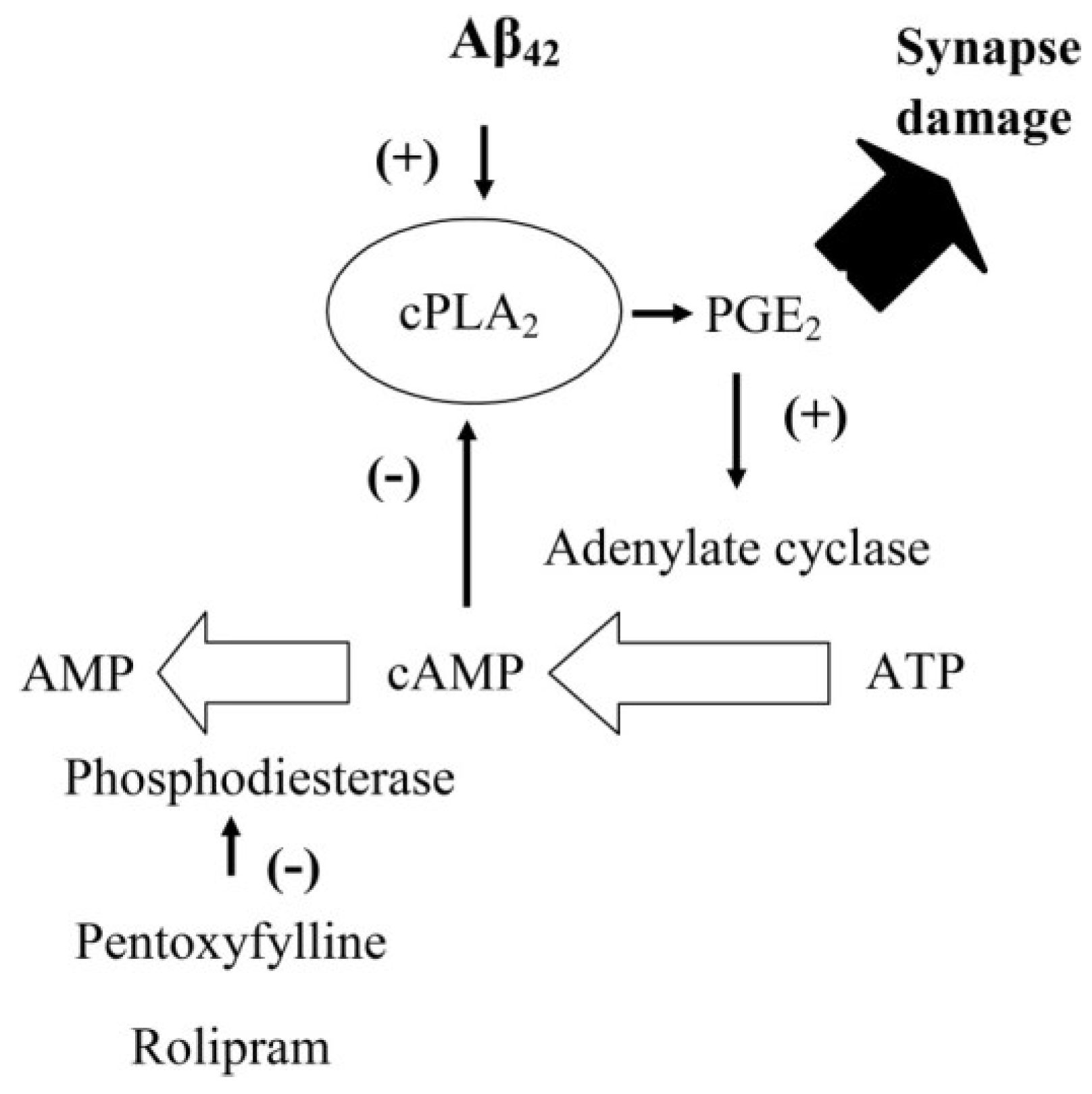
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Hamos, J.E.; DeGennaro, L.J.; Drachman, D.A. Synaptic loss in Alzheimer’s disease and other dementias. Neurology 1989, 39, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Sze, C.I.; Troncoso, J.C.; Kawas, C.; Mouton, P.; Price, D.L.; Martin, L.J. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Klyubin, I.; Betts, V.; Welzel, A.T.; Blennow, K.; Zetterberg, H.; Wallin, A.; Lemere, C.A.; Cullen, W.K.; Peng, Y.; Wisniewski, T.; et al. Amyloid-β Protein Dimer-Containing Human CSF Disrupts Synaptic Plasticity: Prevention by Systemic Passive Immunization. J. Neurosci. 2008, 28, 4231–4237. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.M.; Cullum, C.M.; Satumtira, S.; Sontag, E.; Hynan, L.S.; White, C.L., 3rd; Bigio, E.H. Contribution of asymmetric synapse loss to lateralizing clinical deficits in frontotemporal dementias. Arch. Neurol. 2001, 58, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic α-Synuclein Aggregates, Not Lewy Bodies, Cause Neurodegeneration in Dementia with Lewy Bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Parkinson’s Disease Linked to Pathological α-Synuclein: New Targets for Drug Discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Gentleman, S.; Williams, A. α-synuclein induced synapse damage is enhanced by amyloid-β1–42. Mol. Neurodegener. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Tayebi, M.; Williams, A. Phospholipase A2 inhibitors protect against prion and Aβ mediated synapse degeneration. Mol. Neurodegener. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mix, E.; Winblad, B. The antidepressant and antiinflammatory effects of rolipram in the central nervous system. CNS Drug Rev. 2001, 7, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, J.Y.; Holopainen, J.M.; Kinnunen, P.K. Activation of phospholipase A2 by amyloid β-peptides in vitro. Biochemistry 1996, 35, 9407–9414. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.-Q.; Zhou, Y.; Halabisky, B.; Cisse, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; Huh, S.E.; Elsen, F.P.; Carroll, M.S.; Hodge, R.D.; Bedogni, F.; Turner, M.S.; Hevner, R.F.; Ramirez, J.M. Prostaglandin E2-induced synaptic plasticity in neocortical networks of organotypic slice cultures. J. Neurosci. 2010, 30, 11678–11687. [Google Scholar] [CrossRef] [PubMed]
- Rosso, A.; Mossey, J.; Lippa, C.F. Caffeine: Neuroprotective functions in cognition and Alzheimer’s disease. Am. J. Alzheimers Dis. Other Dement. 2008, 23, 417–422. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Vitolo, O.V.; Trinchese, F.; Liu, S.; Shelanski, M.; Arancio, O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Investig. 2004, 114, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Pozueta, J.; Gong, B.; Arancio, O.; Shelanski, M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. USA 2009, 106, 16877–16882. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.R.; van den Hove, D.L.A.; Pfau, F.; Philippens, M.; Bruno, O.; Fedele, E.; Ricciarelli, R.; Steinbusch, H.W.M.; Vanmierlo, T.; Prickaerts, J. Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 2014, 77, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef] [PubMed]
- Rigoni, M.; Caccin, P.; Gschmeissner, S.; Koster, G.; Postle, A.D.; Rossetto, O.; Schiavo, G.; Montecucco, C.; Montecucco, C.; Rossetto, O. Equivalent effects of snake PLA2 neurotoxins and lysophospholipid-fatty acid mixtures. How do presynaptic PLA2 neurotoxins block nerve terminals? Science 2005, 310, 1678–1680. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic Targeting by Alzheimer’s-Related Amyloid β Oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- Menegon, A.; Bonanomi, D.; Albertinazzi, C.; Lotti, F.; Ferrari, G.; Kao, H.-T.; Benfenati, F.; Baldelli, P.; Valtorta, F. Protein Kinase A-Mediated Synapsin I Phosphorylation Is a Central Modulator of Ca2+-Dependent Synaptic Activity. J. Neurosci. 2006, 26, 11670–11681. [Google Scholar] [CrossRef] [PubMed]
- Munno, D.W.; Prince, D.J.; Syed, N.I. Synapse Number and Synaptic Efficacy Are Regulated by Presynaptic cAMP and Protein Kinase A. J. Neurosci. 2003, 23, 4146–4155. [Google Scholar] [PubMed]
- Desbene, C.; Malaplate-Armand, C.; Youssef, I.; Garcia, P.; Stenger, C.; Sauvee, M.; Fischer, N.; Rimet, D.; Koziel, V.; Escanye, M.C.; et al. Critical role of cPLA(2) in Abeta oligomer-induced neurodegeneration and memory deficit. Neurobiol. Aging 2012, 33, 1123.e17–1123.e29. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S.; Makhlouf, G.M. Differential Regulation of Phospholipase A2(PLA2)-dependent Ca2+ Signaling in Smooth Muscle by cAMP- and cGMP-dependent Protein Kinases. J. Biol. Chem. 1998, 273, 34519–34526. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.Y.; Xu, J.; Jensen, M.D.; Simonyi, A. Phospholipase A2 in the central nervous system: Implications for neurodegenerative diseases. J. Lipid Res. 2004, 45, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Bazan, N.G. Endogenous PGE2 Regulates Membrane Excitability and Synaptic Transmission in Hippocampal CA1 Pyramidal Neurons. J. Neurphysiol. 2005, 93, 929–941. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bate, C.; Williams, A. cAMP-Inhibits Cytoplasmic Phospholipase A2 and Protects Neurons against Amyloid-β-Induced Synapse Damage. Biology 2015, 4, 591-606. https://doi.org/10.3390/biology4030591
Bate C, Williams A. cAMP-Inhibits Cytoplasmic Phospholipase A2 and Protects Neurons against Amyloid-β-Induced Synapse Damage. Biology. 2015; 4(3):591-606. https://doi.org/10.3390/biology4030591
Chicago/Turabian StyleBate, Clive, and Alun Williams. 2015. "cAMP-Inhibits Cytoplasmic Phospholipase A2 and Protects Neurons against Amyloid-β-Induced Synapse Damage" Biology 4, no. 3: 591-606. https://doi.org/10.3390/biology4030591
APA StyleBate, C., & Williams, A. (2015). cAMP-Inhibits Cytoplasmic Phospholipase A2 and Protects Neurons against Amyloid-β-Induced Synapse Damage. Biology, 4(3), 591-606. https://doi.org/10.3390/biology4030591