Next Generation Characterisation of Cereal Genomes for Marker Discovery


Abstract
:1. Introduction
2. DNA Sequencing Technology
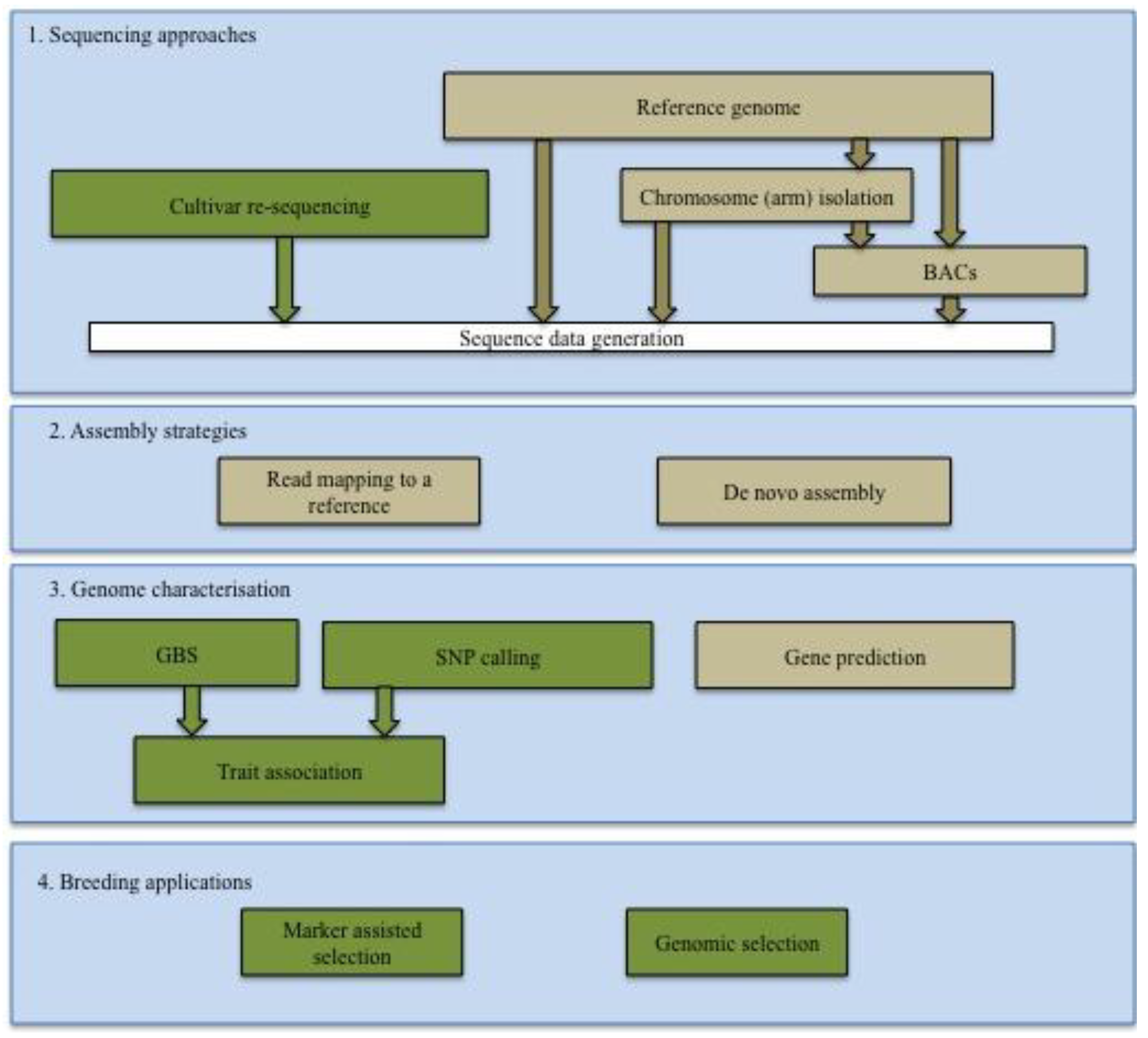
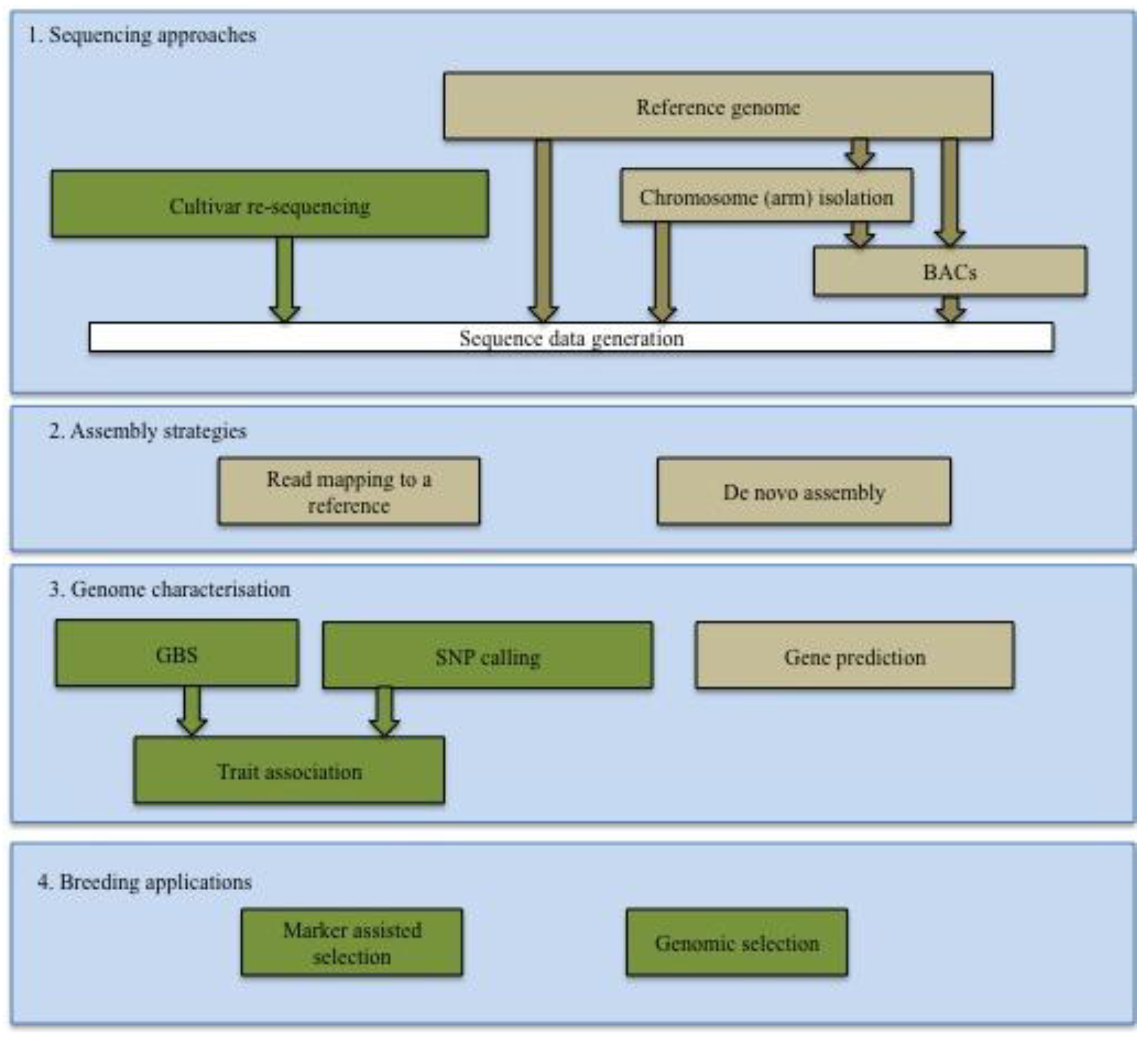
2.1. Sequencing of Cereal Genomes

{kind=link}
| Crop | Assembly/Genome Size (Mb) | Year | Sequencing strategy | Reference |
|---|---|---|---|---|
| Oryza sativa ssp. japonica (Nipponbare) | 370/389 | 2005 | Sanger, BAC-by-BAC | [23] |
| Oryza sativa ssp. japonica (Nipponbare) | 389/420 | 2002 | Sanger, WGS | [38] |
| Oryza sativa ssp. indica | 362/466 | 2002 | Sanger, WGS | [191] |
| Setaria italica (Foxtail Millet) | 423/515 | 2012 | Illumina, WGS | [192] |
| Sorghum bicolor (L.) Moench | 679/730 | 2009 | Sanger, WGS | [28] |
| Zea mays (Palomero Toluqueno) (popcorn) | 177/2100 | 2009 | Sanger, WGS | [193] |
| Zea mays (B73) | 2000/2300 | 2009 | Sanger, BAC-by-BAC | [27] |
| Triticum aestivum (Bread wheat) | */17000 | 2012 | 454, WGS | [25] |
| Hordeum vulgare (Barley) | 4900/5100 | 2012 | 454, BAC-by-BAC | [194] |
| Aegilops tauschii | 4491/4630 | 2013 | Illumina, 454, WGS | [195] |
| Triticum urartu | 3920/4940 | 2013 | Illumina, WGS | [196] |
3. Genome Characterization
3.1. Orthology and Synteny Based Characterisation
3.2. Single Nucleotide Polymorphisms (SNPs)
3.3. Genotyping by Sequencing (GBS)
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Meyers, L.A.; Levin, D.A. On the abundance of polyploids in flowering plants. Evolution 2006, 60, 1198–1206. [Google Scholar]
- Leitch, A.R.; Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lohmueller, K.E.; Albrechtsen, A.; Li, Y.; Korneliussen, T.; Tian, G.; Grarup, N.; Jiang, T.; Andersen, G.; Witte, D.; et al. Estimation of allele frequency and association mapping using next-generation sequencing data. BMC Bioinformatics 2011, 12, 231. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Sanger, F. The Croonian Lecture, 1975. Nucleotide sequences in DNA. Proc. R. Soc. Lond. B Biol. Sci. 1975, 191, 317–333. [Google Scholar] [CrossRef]
- Zimmermann, J.; Voss, H.; Schwager, C.; Stegemann, J.; Ansorge, W. Automated Sanger dideoxy sequencing reaction protocol. FEBS Lett. 1988, 233, 432–436. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; Dewell, S.B.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Edwards, D.; Wilcox, S.; Barrero, R.A.; Fleury, D.; Cavanagh, C.R.; Forrest, K.L.; Hayden, M.J.; Moolhuijzen, P.; Keeble-Gagnère, G.; et al. Bread matters: A national initiative to profile the genetic diversity of Australian wheat. Plant Biotechnol. J. 2012, 10, 703–708. [Google Scholar] [CrossRef]
- Shulaev, V.; Sargent, D.J.; Crowhurst, R.N.; Mockler, T.C.; Folkerts, O.; Delcher, A.L.; Jaiswal, P.; Mockaitis, K.; Liston, A.; Mane, S.P.; et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 2011, 43, 109–116. [Google Scholar] [CrossRef]
- Dong, C.-H.; Li, C.; Yan, X.-H.; Huang, S.-M.; Huang, J.-Y.; Wang, L.-J.; Guo, R.-X.; Lu, G.-Y.; Zhang, X.-K.; Fang, X.-P.; et al. Gene expression profiling of Sinapis alba leaves under drought stress and rewatering growth conditions with Illumina deep sequencing. Mol. Biol. Rep. 2012, 39, 5851–5857. [Google Scholar] [CrossRef]
- Williams-Carrier, R.; Stiffler, N.; Belcher, S.; Kroeger, T.; Stern, D.B.; Monde, R.-A.; Coalter, R.; Barkan, A. Use of Illumina sequencing to identify transposon insertions underlying mutant phenotypes in high-copy mutator lines of maize. Plant J. 2010, 63, 167–177. [Google Scholar]
- Imelfort, M.; Edwards, D. De novo sequencing of plant genomes using second-generation technologies. Brief. Bioinformatics 2009, 10, 609–618. [Google Scholar]
- Edwards, D.; Batley, J.; Snowdon, R.J. Accessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 2013, 126, 1–11. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; Bibillo, A.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Mason, C.E.; Elemento, O. Faster sequencers, larger datasets, new challenges. Genome Biol. 2012, 13, 314. [Google Scholar] [CrossRef]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving PacBio long read accuracy by short read alignment. PLoS ONE 2012, 7, e46679. [Google Scholar]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 13770–13773. [Google Scholar]
- Stoddart, D.; Heron, A.J.; Mikhailova, E.; Maglia, G.; Bayley, H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 7702–7707. [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Flavell, R.B.; Rimpau, J.R.; Smith, D.B. Repeated sequence DNA relationships in four cereal genomes. Chromosoma 1977, 63, 205–222. [Google Scholar] [CrossRef]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.A.; D'Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef]
- Doležel, J.; Kubaláková, M.; Paux, E.; Bartos, J.; Feuillet, C. Chromosome-based genomics in the cereals. Chromosome Res. 2007, 15, 51–66. [Google Scholar] [CrossRef]
- Duran, C.; Edwards, D.; Batley, J. Genetic Maps and the Use of Synteny. In Plant Genomics; Gustafson, J.P., Langridge, P., Somers, D.J., Eds.; Humana Press: New York, NY, USA, 2009; Volume 513, pp. 41–55. [Google Scholar]
- Doležel, J.; Kubaláková, M.; Bartos, J.; Macas, J. Flow cytogenetics and plant genome mapping. Chromosome Res. 2004, 12, 77–91. [Google Scholar] [CrossRef]
- Berkman, P.J.; Visendi, P.; Lee, H.C.; Stiller, J.; Manoli, S.; Lorenc, M.T.; Lai, K.; Batley, J.; Fleury, D.; Simková, H.; et al. Dispersion and domestication shaped the genome of bread wheat. Plant Biotechnol. J. 2013, 11, 564–571. [Google Scholar] [CrossRef]
- Berkman, P.J.; Skarshewski, A.; Manoli, S.; Lorenc, M.T.; Stiller, J.; Smits, L.; Lai, K.; Campbell, E.; Kubaláková, M.; Simková, H.; et al. Sequencing wheat chromosome arm 7BS delimits the 7BS/4AL translocation and reveals homoeologous gene conservation. Theor. Appl. Genet. 2011, 124, 423–432. [Google Scholar]
- Berkman, P.J.; Skarshewski, A.; Lorenc, M.T.; Lai, K.; Duran, C.; Ling, E.Y.S.; Stiller, J.; Smits, L.; Imelfort, M.; Manoli, S.; et al. Sequencing and assembly of low copy and genic regions of isolated Triticum aestivum chromosome arm 7DS. Plant Biotechnol. J. 2011, 9, 768–775. [Google Scholar] [CrossRef]
- Hernandez, P.; Martis, M.; Dorado, G.; Pfeifer, M.; Gálvez, S.; Schaaf, S.; Jouve, N.; Simková, H.; Valárik, M.; Doležel, J.; et al. Next-generation sequencing and syntenic integration of flow-sorted arms of wheat chromosome 4A exposes the chromosome structure and gene content. Plant J. 2012, 69, 377–386. [Google Scholar] [CrossRef]
- Nie, X.; Li, B.; Wang, L.; Liu, P.; Biradar, S.S.; Li, T.; Doležel, J.; Edwards, D.; Luo, M.; Weining, S. Development of chromosome-arm-specific microsatellite markers in Triticum aestivum (Poaceae) using NGS technology. Am. J. Bot. 2012, 99, e369–e371. [Google Scholar] [CrossRef]
- Barry, G.F. The use of the Monsanto draft rice genome sequence in research. Plant Physiol. 2001, 125, 1164–1165. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.-H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Sci. New Ser. 2002, 296, 92–100. [Google Scholar]
- Mayer, K.F.X.; Waugh, R.; Brown, J.W.S.; Schulman, A.; Langridge, P.; Platzer, M.; Fincher, G.B.; Muehlbauer, G.J.; Sato, K.; Close, T.J.; et al. International Barley Genome Sequencing Consortium, A physical; genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef]
- Salzberg, S.L.; Phillippy, A.M.; Zimin, A.; Puiu, D.; Magoc, T.; Koren, S.; Treangen, T.J.; Schatz, M.C.; Delcher, A.L.; Roberts, M.; et al. A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012, 22, 557–567. [Google Scholar] [CrossRef]
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef]
- Carter, A.H.; Garland-Campbell, K.; Morris, C.F.; Kidwell, K.K. Chromosomes 3B and 4D are associated with several milling and baking quality traits in a soft white spring wheat (Triticum aestivum L.) population. Theor. Appl. Genet. 2012, 124, 1079–1096. [Google Scholar]
- Vitulo, N.; Albiero, A.; Forcato, C.; Campagna, D.; Dal Pero, F.; Bagnaresi, P.; Colaiacovo, M.; Faccioli, P.; Lamontanara, A.; Simková, H.; et al. First survey of the wheat chromosome 5A composition through a next generation sequencing approach. PLoS One 2011, 6, e26421. [Google Scholar] [CrossRef]
- Rustenholz, C.; Choulet, F.; Laugier, C.; Safár, J.; Simková, H.; Doležel, J.; Magni, F.; Scalabrin, S.; Cattonaro, F.; Vautrin, S.; et al. A 3,000-loci transcription map of chromosome 3B unravels the structural and functional features of gene islands in hexaploid wheat. Plant Physiol. 2011, 157, 1596–1608. [Google Scholar] [CrossRef]
- Cseh, A.; Kruppa, K.; Molnár, I.; Rakszegi, M.; Doležel, J.; Molnár-Láng, M. Characterization of a new 4BS.7HL wheat-barley translocation line using GISH, FISH, and SSR markers and its effect on the β-glucan content of wheat. Genome 2011, 54, 795–804. [Google Scholar] [CrossRef]
- Yoshida, T.; Nishida, H.; Zhu, J.; Nitcher, R.; Distelfeld, A.; Akashi, Y.; Kato, K.; Dubcovsky, J. Vrn-D4 is a vernalization gene located on the centromeric region of chromosome 5D in hexaploid wheat. Theor. Appl. Genet. 2010, 120, 543–552. [Google Scholar] [CrossRef]
- Breen, J.; Wicker, T.; Kong, X.; Zhang, J.; Ma, W.; Paux, E.; Feuillet, C.; Appels, R.; Bellgard, M. A highly conserved gene island of three genes on chromosome 3B of hexaploid wheat: diverse gene function and genomic structure maintained in a tightly linked block. BMC Plant Biol. 2010, 10, 98. [Google Scholar] [CrossRef]
- Saintenac, C.; Falque, M.; Martin, O.C.; Paux, E.; Feuillet, C.; Sourdille, P. Detailed recombination studies along chromosome 3B provide new insights on crossover distribution in wheat (Triticum aestivum L.). Genetics 2009, 181, 393–403. [Google Scholar]
- Alfares, W.; Bouguennec, A.; Balfourier, F.; Gay, G.; Bergès, H.; Vautrin, S.; Sourdille, P.; Bernard, M.; Feuillet, C. Fine mapping and marker development for the crossability gene SKr on chromosome 5BS of hexaploid wheat (Triticum aestivum L.). Genetics 2009, 183, 469–481. [Google Scholar] [CrossRef]
- Ren, X.-B.; Lan, X.-J.; Liu, D.-C.; Wang, J.-L.; Zheng, Y.-L. Mapping QTLs for pre-harvest sprouting tolerance on chromosome 2D in a synthetic hexaploid wheat x common wheat cross. J. Appl. Genet. 2008, 49, 333–341. [Google Scholar] [CrossRef]
- Paux, E.; Sourdille, P.; Salse, J.; Saintenac, C.; Choulet, F.; Leroy, P.; Korol, A.; Michalak, M.; Kianian, S.; Spielmeyer, W.; et al. A physical map of the 1-gigabase bread wheat chromosome 3B. Science 2008, 322, 101–104. [Google Scholar] [CrossRef]
- Maccaferri, M.; Mantovani, P.; Tuberosa, R.; Deambrogio, E.; Giuliani, S.; Demontis, A.; Massi, A.; Sanguineti, M.C. A major QTL for durable leaf rust resistance widely exploited in durum wheat breeding programs maps on the distal region of chromosome arm 7BL. Theor. Appl. Genet. 2008, 117, 1225–1240. [Google Scholar] [CrossRef]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef]
- Wicker, T.; Mayer, K.F.X.; Gundlach, H.; Martis, M.; Steuernagel, B.; Scholz, U.; Simková, H.; Kubaláková, M.; Choulet, F.; Taudien, S.; et al. Frequent gene movement and pseudogene evolution is common to the large and complex genomes of wheat, barley, and their relatives. Plant Cell 2011, 23, 1706–1718. [Google Scholar] [CrossRef]
- Bossolini, E.; Wicker, T.; Knobel, P.A.; Keller, B. Comparison of orthologous loci from small grass genomes Brachypodium and rice: Implications for wheat genomics and grass genome annotation. Plant J. 2007, 49, 704–717. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Asakura, N.; Mori, N.; Nakamura, C.; Ohtsuka, I. Genotyping of the Q locus in wheat by a simple PCR-RFLP method. Genes Genet. Syst. 2009, 84, 233–237. [Google Scholar] [CrossRef]
- Han, F.P.; Fedak, G.; Benabdelmouna, A.; Armstrong, K.; Ouellet, T. Characterization of six wheat x Thinopyrum intermedium derivatives by GISH, RFLP, and multicolor GISH. Genome 2003, 46, 490–495. [Google Scholar] [CrossRef]
- Ma, X.F.; Ross, K.; Gustafson, J.P. Physical mapping of restriction fragment length polymorphism (RFLP) markers in homoeologous groups 1 and 3 chromosomes of wheat by in situ hybridization. Genome 2001, 44, 401–412. [Google Scholar] [CrossRef]
- Sim, S.; Chang, T.; Curley, J.; Warnke, S.E.; Barker, R.E.; Jung, G. Chromosomal rearrangements differentiating the ryegrass genome from the Triticeae, oat, and rice genomes using common heterologous RFLP probes. Theor. Appl. Genet. 2005, 110, 1011–1019. [Google Scholar] [CrossRef]
- Singh, R.K.; Mishra, R.P.N.; Jaiswal, H.K.; Kumar, V.; Pandey, S.P.; Rao, S.B.; Annapurna, K. Isolation and identification of natural endophytic rhizobia from rice (Oryza sativa L.) through rDNA PCR-RFLP and sequence analysis. Curr. Microbiol. 52, 2006, 345–349. [Google Scholar]
- Huang, W.; Wang, L.; Yi, P.; Tan, X.-L.; Zhang, X.-M.; Zhang, Z.-J.; Li, Y.-S.; Zhu, Y.-G. RFLP analysis for mitochondrial genome of CMS-rice. Yi Chuan Xue Bao 2006, 33, 330–338. [Google Scholar]
- Xu, X.F.; Mei, H.W.; Luo, L.J.; Cheng, X.N.; Li, Z.K. RFLP-facilitated investigation of the quantitative resistance of rice to brown planthopper ( Nilaparvata lugens). Theor. Appl. Genet. 2002, 104, 248–253. [Google Scholar] [CrossRef]
- Lu, B.-R.; Zheng, K.L.; Qian, H.R.; Zhuang, J.Y. Genetic differentiation of wild relatives of rice as assessed by RFLP analysis. Theor. Appl. Genet. 2002, 106, 101–106. [Google Scholar]
- Maestri, E.; Malcevschi, A.; Massari, A.; Marmiroli, N. Genomic analysis of cultivated barley (Hordeum vulgare) using sequence-tagged molecular markers. Estimates of divergence based on RFLP and PCR markers derived from stress-responsive genes, and simple-sequence repeats (SSRs). Mol. Genet. Genomics 2002, 267, 186–201. [Google Scholar] [CrossRef]
- Künzel, G.; Waugh, R. Integration of microsatellite markers into the translocation-based physical RFLP map of barley chromosome 3H. Theor. Appl. Genet. 2002, 105, 660–665. [Google Scholar] [CrossRef]
- Saeki, K.; Miyazaki, C.; Hirota, N.; Saito, A.; Ito, K.; Konishi, T. RFLP mapping of BaYMV resistance gene rym3 in barley (Hordeum vulgare). Theor. Appl. Genet. 1999, 99, 727–732. [Google Scholar] [CrossRef]
- Michalek, W.; Künzel, G.; Graner, A. Sequence analysis and gene identification in a set of mapped RFLP markers in barley (Hordeum vulgare). Genome 1999, 42, 849–853. [Google Scholar]
- Jordan, D.R.; Tao, Y.; Godwin, I.D.; Henzell, R.G.; Cooper, M.; McIntyre, C.L. Prediction of hybrid performance in grain sorghum using RFLP markers. Theor. Appl. Genet. 2003, 106, 559–567. [Google Scholar]
- Schloss, J.; Mitchell, E.; White, M.; Kukatla, R.; Bowers, E.; Paterson, H.; Kresovich, S. Characterization of RFLP probe sequences for gene discovery and SSR development in Sorghum bicolor (L.) Moench. Theor. Appl. Genet. 2002, 105, 912–920. [Google Scholar] [CrossRef]
- Haussmann, G.; Hess, E.; Seetharama, N.; Welz, G.; Geiger, H. Construction of a combined sorghum linkage map from two recombinant inbred populations using AFLP, SSR, RFLP, and RAPD markers, and comparison with other sorghum maps. Theor. Appl. Genet. 2002, 105, 629–637. [Google Scholar] [CrossRef]
- Subudhi, P.K.; Nguyen, H.T. Linkage group alignment of sorghum RFLP maps using a RIL mapping population. Genome 2000, 43, 240–249. [Google Scholar] [CrossRef]
- Gauthier, P.; Gouesnard, B.; Dallard, J.; Redaelli, R.; Rebourg, C.; Charcosset, A.; Boyat, A. RFLP diversity and relationships among traditional European maize populations. Theor. Appl. Genet. 2002, 105, 91–99. [Google Scholar] [CrossRef]
- Dubreuil, P.; Charcosset, A. Relationships among maize inbred lines and populations from European and North-American origins as estimated using RFLP markers. Theor. Appl. Genet. 1999, 99, 473–480. [Google Scholar] [CrossRef]
- Lin, B.Y.; Peng, S.F.; Chen, Y.J.; Chen, H.S.; Kao, C.F. Physical mapping of RFLP markers on four chromosome arms in maize using terminal deficiencies. Mol. Gen. Genet. 1997, 256, 509–516. [Google Scholar] [CrossRef]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van de Lee, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef]
- Tautz, D.; Schlötterer, C. Simple sequences. Curr. Opin. Genet. Dev. 1994.
- Batley, J.; Jewell, E.; Edwards, D. Automated Discovery of Single Nucleotide Polymorphism and Simple Sequence Repeat Molecular Genetic Markers. In Methods in Molecular Biology; Edwards, D., Ed.; Humana Press: New York, NY, USA, 2007; Volume 406, pp. 473–494. [Google Scholar]
- Batley, J.; Edwards, D. Mining for Single Nucleotide Polymorphism (SNP) and Simple Sequence Repeat (SSR) Molecular Genetic Markers. In Bioinformatics for DNA Sequence Analysis; Posada, D., Ed.; Humana Press: New York, NY, USA, 2009; pp. 303–322. [Google Scholar]
- Duran, C.; Edwards, D.; Batley, J. Molecular marker discovery and genetic map visualisation. Bioinformatics 2009, 4, 165–189. [Google Scholar] [CrossRef]
- Hartings, H.; Berardo, N.; Mazzinelli, G.F.; Valoti, P.; Verderio, A.; Motto, M. Assessment of genetic diversity and relationships among maize (Zea mays L.) Italian landraces by morphological traits and AFLP profiling. Theor. Appl. Genet. 2008, 117, 831–842. [Google Scholar] [CrossRef]
- Zhang, Z.F.; Wang, Y.; Zheng, Y.L. AFLP and PCR-based markers linked to Rf3, a fertility restorer gene for S cytoplasmic male sterility in maize. Mol. Genet. Genomics 2006, 276, 162–169. [Google Scholar] [CrossRef]
- Zhang, F.; Wan, X.-Q.; Pan, G.-T. QTL mapping of Fusarium moniliforme ear rot resistance in maize. 1. Map construction with microsatellite and AFLP markers. J. Appl. Genet. 2006, 47, 9–15. [Google Scholar] [CrossRef]
- Schrag, T.A.; Melchinger, A.E.; Sørensen, A.P.; Frisch, M. Prediction of single-cross hybrid performance for grain yield and grain dry matter content in maize using AFLP markers associated with QTL. Theor. Appl. Genet. 2006, 113, 1037–1047. [Google Scholar] [CrossRef]
- Peng, S.-F.; Lin, Y.-P.; Lin, B.-Y. Characterization of AFLP sequences from regions of maize B chromosome defined by 12 B-10L translocations. Genetics 2005, 169, 375–388. [Google Scholar] [CrossRef]
- Miranda Oliveira, K.; Rios Laborda, P.; Augusto F Garcia, A.; Zagatto Paterniani, M.E. A.G.; de Souza, A.P. Evaluating genetic relationships between tropical maize inbred lines by means of AFLP profiling. Hereditas 2004, 140, 24–33. [Google Scholar] [CrossRef]
- Cai, H.-W.; Gao, Z.-S.; Yuyama, N.; Ogawa, N. Identification of AFLP markers closely linked to the rhm gene for resistance to southern corn leaf blight in maize by using bulked segregant analysis. Mol. Genet. Genomics 2003, 269, 299–303. [Google Scholar]
- Agrama, H.A.; Houssin, S.F.; Tarek, M.A. Cloning of AFLP markers linked to resistance to Peronosclerospora sorghi in maize. Mol. Genet. Genomics 2002, 267, 814–819. [Google Scholar] [CrossRef]
- Legesse, B.W.; Myburg, A.A.; Pixley, K.V.; Botha, A.M. Genetic diversity of African maize inbred lines revealed by SSR markers. Hereditas 2007, 144, 10–17. [Google Scholar] [CrossRef]
- Wen, L.; Tang, H.V.; Chen, W.; Chang, R.; Pring, D.R.; Klein, P.E.; Childs, K.L.; Klein, R.R. Development and mapping of AFLP markers linked to the sorghum fertility restorer gene rf4. Theor. Appl. Genet. 2002, 104, 577–585. [Google Scholar] [CrossRef]
- Klein, P.E.; Klein, R.R.; Cartinhour, S.W.; Ulanch, P.E.; Dong, J.; Obert, J.A.; Morishige, D.T.; Schlueter, S.D.; Childs, K.L.; Ale, M.; et al. A high-throughput AFLP-based method for constructing integrated genetic and physical maps: progress toward a sorghum genome map. Genome Res. 2000, 10, 789–807. [Google Scholar] [CrossRef]
- Zhang, D.; Ding, Y. Genetic diversity of wild close relatives of barley in Tibet of China revealed by AFLP. Yi Chuan 2007, 29, 725–730. [Google Scholar] [CrossRef]
- Takahashi, H.; Akagi, H.; Mori, K.; Sato, K.; Takeda, K. Genomic distribution of MITEs in barley determined by MITE-AFLP mapping. Genome 2006, 49, 1616–1620. [Google Scholar] [CrossRef]
- Komatsuda, T.; Maxim, P.; Senthil, N.; Mano, Y. High-density AFLP map of nonbrittle rachis 1 (btr1) and 2 (btr2) genes in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2004, 109, 986–995. [Google Scholar] [CrossRef]
- He, C.; Sayed-Tabatabaei, B.E.; Komatsuda, T. AFLP targeting of the 1-cM region conferring the vrs1 gene for six-rowed spike in barley, Hordeum vulgare L. Genome 2004, 47, 1122–1129. [Google Scholar] [CrossRef]
- Turpeinen, T.; Vanhala, T.; Nevo, E.; Nissilä, E. AFLP genetic polymorphism in wild barley (Hordeum spontaneum) populations in Israel. Theor. Appl. Genet. 2003, 106, 1333–1339. [Google Scholar]
- Wang, Y.; Zhu, J.; Zhao, H.M.; Lei, D.H.; Wang, Z.Y.; Peng, Y.K.; Xie, C.J.; Sun, Q.X.; Liu, Z.Y.; Yang, Z.M. Screening and identification of the AFLP markers linked to a new powdery mildew resistance gene in wheat cultivar Brock. Fen Zi Xi Bao Sheng Wu Xue Bao 2008, 41, 294–300. [Google Scholar]
- Ozbek, O.; Millet, E.; Anikster, Y.; Arslan, O.; Feldman, M. Spatio-temporal genetic variation in populations of wild emmer wheat, Triticum turgidum ssp. dicoccoides, as revealed by AFLP analysis. Theor. Appl. Genet. 2007, 115, 19–26. [Google Scholar] [CrossRef]
- Xu, D.H.; Ban, T. Conversion of AFLP markers associated with FHB resistance in wheat into STS markers with an extension-AFLP method. Genome 2004, 47, 660–665. [Google Scholar] [CrossRef]
- Tyrka, M. Fingerprinting of common wheat cultivars with an Alw44I-based AFLP method. J. Appl. Genet. 2004, 45, 405–410. [Google Scholar]
- Zhou, W.; Kolb, F.L.; Bai, G.; Shaner, G.; Domier, L.L. Genetic analysis of scab resistance QTL in wheat with microsatellite and AFLP markers. Genome 2002, 45, 719–727. [Google Scholar] [CrossRef]
- Ng'uni, D.; Geleta, M.; Bryngelsson, T. Genetic diversity in sorghum (Sorghum bicolor (L.) Moench) accessions of Zambia as revealed by simple sequence repeats (SSR). Hereditas 2011, 148, 52–62. [Google Scholar] [CrossRef]
- Balfourier, F.; Roussel, V.; Strelchenko, P.; Exbrayat-Vinson, F.; Sourdille, P.; Boutet, G.; Koenig, J.; Ravel, C.; Mitrofanova, O.; Beckert, M.; Charmet, G. A worldwide bread wheat core collection arrayed in a 384-well plate. Theor. Appl. Genet. 2007, 114, 1265–1275. [Google Scholar] [CrossRef]
- Ashfaq, M.; Khan, A.S. Genetic diversity in basmati rice (Oryza sativa L.) germplasm as revealed by microsatellite (SSR) markers. Genetika 2012, 48, 62–71. [Google Scholar]
- Zhang, P.; Li, J.; Li, X.; Liu, X.; Zhao, X.; Lu, Y. Population structure and genetic diversity in a rice core collection (Oryza sativa L.) investigated with SSR markers. PLoS One 2011, 6, e27565. [Google Scholar]
- Hao, C.; Wang, L.; Ge, H.; Dong, Y.; Zhang, X. Genetic diversity and linkage disequilibrium in Chinese bread wheat (Triticum aestivum L.) revealed by SSR markers. PLoS One 2011, 6, e17279. [Google Scholar]
- Achtar, S.; Moualla, M.Y.; Kalhout, A.; Röder, M.S.; MirAli, N. Assessment of genetic diversity among Syrian durum (Triticum turgidum ssp. durum) and bread wheat (Triticum aestivum L.) using SSR markers. Genetika 2010, 46, 1500–1506. [Google Scholar]
- Roussel, V.; Leisova, L.; Exbrayat, F.; Stehno, Z.; Balfourier, F. SSR allelic diversity changes in 480 European bread wheat varieties released from 1840 to 2000. Theor. Appl. Genet. 2005, 111, 162–170. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Chen, P.; Liu, D. Assessment of genetic diversity of Yunnan, Tibetan, and Xinjiang wheat using SSR markers. J. Genet. Genomics 2007, 34, 623–633. [Google Scholar] [CrossRef]
- Yao, Q.; Yang, K.; Pan, G.; Rong, T. Genetic diversity of maize (Zea mays L.) landraces from southwest China based on SSR data. J. Genet. Genomics 2007, 34, 851–859. [Google Scholar] [CrossRef]
- Singh, R.K.; Bhatia, V.S.; Bhat, K.V.; Mohapatra, T.; Singh, N.K.; Bansal, K.C.; Koundal, K.R. SSR and AFLP based genetic diversity of soybean germplasm differing in photoperiod sensitivity. Genet. Mol. Biol. 2010, 33, 319–324. [Google Scholar]
- Hu, X.; Wang, J.; Lu, P.; Zhang, H. Assessment of genetic diversity in broomcorn millet (Panicum miliaceum L.) using SSR markers. J. Genet. Genomics 2009, 36, 491–500. [Google Scholar] [CrossRef]
- Zeid, M.; Belay, G.; Mulkey, S.; Poland, J.; Sorrells, M.E. QTL mapping for yield and lodging resistance in an enhanced SSR-based map for tef. Theor. Appl. Genet. 2011, 122, 77–93. [Google Scholar] [CrossRef]
- Apotikar, D.B.; Venkateswarlu, D.; Ghorade, R.B.; Wadaskar, R.M.; Patil, J.V.; Kulwal, P.L. Mapping of shoot fly tolerance loci in sorghum using SSR markers. J. Genet. 2011, 90, 59–66. [Google Scholar] [CrossRef]
- Fu, S.; Zhan, Y.; Zhi, H.; Gai, J.; Yu, D. Mapping of SMV resistance gene Rsc-7 by SSR markers in soybean. Genetica 2006, 128, 63–69. [Google Scholar] [CrossRef]
- Liu, J.-C.; Chu, Q.; Cai, H.-G.; Mi, G.-H.; Chen, F.-J. SSR linkage map construction and QTL mapping for leaf area in maize. Yi Chuan 2010, 32, 625–631. [Google Scholar] [CrossRef]
- Ha, B.-K.; Robbins, R.T.; Han, F.; Hussey, R.S.; Soper, J.F.; Boerma, H.R. SSR mapping and confirmation of soybean QTL from PI 437654 conditioning resistance to reniform nematode. Crop Sci. 2007, 47, 1336. [Google Scholar]
- Su, C.-C.; Zhai, H.-Q.; Wang, C.-M.; Sun, L.-H.; Wan, J.-M. SSR mapping of brown planthopper resistance gene Bph9 in kaharamana, an indica rice (Oryza sativa L.). Yi Chuan Xue Bao 2006, 33, 262–268. [Google Scholar]
- Maccaferri, M.; Sanguineti, M.C.; Demontis, A.; El-Ahmed, A.; Garcia del Moral, L.; Maalouf, F.; Nachit, M.; Nserallah, N.; Ouabbou, H.; Rhouma, S.; et al. Association mapping in durum wheat grown across a broad range of water regimes. J. Exp. Bot. 2011, 62, 409–438. [Google Scholar] [CrossRef]
- Gupta, K.; Balyan, S.; Edwards, J.; Isaac, P.; Korzun, V.; Röder, M.; Gautier, M.F.; Joudrier, P.; Schlatter, R.; Dubcovsky, J.; et al. Genetic mapping of 66 new microsatellite (SSR) loci in bread wheat. Theor. Appl. Genet. 2002, 105, 413–422. [Google Scholar] [CrossRef]
- Röder, M.S.; Korzun, V.; Wendehake, K.; Plaschke, J.; Tixier, M.H.; Leroy, P.; Ganal, M.W. A microsatellite map of wheat. Genetics 1998, 149, 2007–2023. [Google Scholar]
- Somers, D.J.; Isaac, P.; Edwards, K. A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2004, 109, 1105–1114. [Google Scholar] [CrossRef]
- Guyomarc'h, H.; Sourdille, P.; Charmet, G.; Edwards, J.; Bernard, M. Characterisation of polymorphic microsatellite markers from Aegilops tauschii and transferability to the D-genome of bread wheat. Theor. Appl. Genet. 2002, 104, 1164–1172. [Google Scholar] [CrossRef]
- Song, Q.J.; Fickus, E.W.; Cregan, P.B. Characterization of trinucleotide SSR motifs in wheat. Theor. Appl. Genet. 2002, 104, 286–293. [Google Scholar] [CrossRef]
- Song, Q.J.; Shi, J.R.; Singh, S.; Fickus, E.W.; Costa, J.M.; Lewis, J.; Gill, B.S.; Ward, R.; Cregan, P.B. Development and mapping of microsatellite (SSR) markers in wheat. Theor. Appl. Genet. 2005, 110, 550–560. [Google Scholar] [CrossRef]
- Stephenson, P.; Bryan, G.; Kirby, J.; Collins, A.; Devos, K.; Busso, C.; Gale, M. Fifty new microsatellite loci for the wheat genetic map. Theor. Appl. Genet. 1998, 97, 946–949. [Google Scholar] [CrossRef]
- Yu, J.-K.; La Rota, M.; Kantety, R.V.; Sorrells, M.E. EST derived SSR markers for comparative mapping in wheat and rice. Mol. Genet. Genomics 2004, 271, 742–751. [Google Scholar]
- Jia, X.; Zhang, Z.; Liu, Y.; Zhang, C.; Shi, Y.; Song, Y.; Wang, T.; Li, Y. Development and genetic mapping of SSR markers in foxtail millet (Setaria italica (L.) P. Beauv.). Theor. Appl. Genet. 2009, 118, 821–829. [Google Scholar] [CrossRef]
- Lin, H.-S.; Chiang, C.-Y.; Chang, S.-B.; Kuoh, C.-S. Development of simple sequence repeats (SSR) markers in Setaria italica (Poaceae) and cross-amplification in related species. Int. J. Mol. Sci. 2011, 12, 7835–7845. [Google Scholar] [CrossRef]
- Maccaferri, M.; Sanguineti, M.C.; Corneti, S.; Ortega, J.L.A.; Salem, M.B.; Bort, J.; DeAmbrogio, E.; del Moral, L.F.G.; Demontis, A.; El-Ahmed, A.; Maalouf, F.; et al. Quantitative trait loci for grain yield and adaptation of durum wheat (Triticum durum Desf.) across a wide range of water availability. Genetics 2008, 178, 489–511. [Google Scholar] [CrossRef]
- Breseghello, F.; Sorrells, M.E. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 2006, 172, 1165–1177. [Google Scholar] [CrossRef]
- Li, S.; Jia, J.; Wei, X.; Zhang, X.; Li, L.; Chen, H.; Fan, Y.; Sun, H.; Zhao, X.; Lei, T.; et al. A intervarietal genetic map and QTL analysis for yield traits in wheat. Mol Breeding 2007, 20, 167–178. [Google Scholar] [CrossRef]
- Emebiri, L.C. EST-SSR markers derived from an elite barley cultivar (Hordeum vulgare L. “Morex”): Polymorphism and genetic marker potential. Genome 2013, 52, 665–676. [Google Scholar]
- Dong, P.; Wei, Y.-M.; Chen, G.-Y.; Li, W.; Wang, J.-R.; Nevo, E.; Zheng, Y.-L. EST-SSR diversity correlated with ecological and genetic factors of wild emmer wheat in Israel. Hereditas 2009, 146, 1–10. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Wei, Y.-M.; Yan, Z.-H.; Zheng, Y.-L. EST-SSR DNA polymorphism in durum wheat (Triticum durum L.) collections. J. Appl. Genet. 2007, 48, 35–42. [Google Scholar]
- Mullan, D.J.; Platteter, A.; Teakle, N.L.; Appels, R.; Colmer, T.D.; Anderson, J.M.; Francki, M.G. EST-derived SSR markers from defined regions of the wheat genome to identify Lophopyrum elongatum specific loci. Genome 2005, 48, 811–822. [Google Scholar]
- Duran, C.; Singhania, R.; Raman, H.; Batley, J.; Edwards, D. Predicting polymorphic EST-SSRs in silico. Mol. Ecol. Resour. 2013, 13, 538–545. [Google Scholar] [CrossRef]
- Sim, S.-C.; Yu, J.-K.; Jo, Y.-K.; Sorrells, M.E.; Jung, G. Transferability of cereal EST-SSR markers to ryegrass. Genome 2009, 52, 431–437. [Google Scholar] [CrossRef]
- Appleby, N.; Edwards, D.; Batley, J. New Technologies for Ultra-high Throughput Genotyping in Plants. In Plant Genomics; Gustafson, J.P., Langridge, P., Somers, D.J., Eds.; Humana press: New York, NY, USA, 2009; Volume 513, pp. 19–39. [Google Scholar]
- Edwards, D.; Forster, J.W.; Cogan, N.O.I.; Batley, J.; Chagné, D. Single Nucleotide Polymorphism Discovery. In Association Mapping in Plants; Oraguzie, N.C., Rikkerink, E.H.A., Gardiner, S.E., De Silva, D.H.N., Eds.; Springer: New York, NY, USA, 2007; pp. 53–76. [Google Scholar]
- Batley, J.; Edwards, D. SNP Applications in Plants. In Association Mapping in Plants; Oraguzie, D.N.C., Rikkerink, D.E.H.A., Gardiner, D.S.E., De Silva, D.H.N., Eds.; Springer: New York, NY, USA, 2007; pp. 95–102. [Google Scholar]
- Edwards, D.; Forster, J.W.; Chagné, D.; Batley, J. What are SNPs? In Association Mapping in Plants; Oraguzie, D.N.C., Rikkerink, D.E.H.A., Gardiner, D.S.E., De Silva, D.H.N., Eds.; Springer: New York, NY, USA, 2007; pp. 41–52. [Google Scholar]
- Hao, Z.; Li, X.; Xie, C.; Weng, J.; Li, M.; Zhang, D.; Liang, X.; Liu, L.; Liu, S.; Zhang, S. Identification of functional genetic variations underlying drought tolerance in maize using SNP markers. J. Integr. Plant. Biol. 2011, 53, 641–652. [Google Scholar] [CrossRef]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef]
- Simillion, C.; Vandepoele, K.; Van Montagu, M.C.E.; Zabeau, M.; Van de Peer, Y. The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13627–13632. [Google Scholar]
- Vandepoele, K.; Simillion, C.; Van de Peer, Y. Evidence that rice and other cereals are ancient aneuploids. Plant Cell 2003, 15, 2192–2202. [Google Scholar] [CrossRef]
- Gore, M.A.; Chia, J.-M.; Elshire, R.J.; Sun, Q.; Ersoz, E.S.; Hurwitz, B.L.; Peiffer, J.A.; McMullen, M.D.; Grills, G.S.; Ross-Ibarra, J.; et al. A first-generation haplotype map of maize. Science 2009, 326, 1115–1117. [Google Scholar] [CrossRef]
- Barker, G.; Batley, J.; O' Sullivan, H.; Edwards, K.J.; Edwards, D. Redundancy based detection of sequence polymorphisms in expressed sequence tag data using autoSNP. Bioinformatics 2003, 19, 421–422. [Google Scholar] [CrossRef]
- Coulondre, C.; Miller, J.H.; Farabaugh, P.J.; Gilbert, W. Molecular basis of base substitution hotspots in Escherichia coli. Nature 1978, 274, 775–780. [Google Scholar]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar]
- Berkman, P.J.; Lai, K.; Lorenc, M.T.; Edwards, D. Next-generation sequencing applications for wheat crop improvement. Am. J. Bot. 2012, 99, 365–371. [Google Scholar] [CrossRef]
- Lai, K.; Lorenc, M.T.; Edwards, D. Genomic databases for crop improvement. Agronomy 2012, 2, 62–73. [Google Scholar] [CrossRef]
- Lorenc, M.T.; Hayashi, S.; Stiller, J.; Lee, H.; Manoli, S.; Ruperao, P.; Visendi, P.; Berkman, P.J.; Lai, K.; Batley, J.; et al. Discovery of single nucleotide polymorphisms in complex genomes using SGSautoSNP. Biology 2012, 1, 370–382. [Google Scholar] [CrossRef]
- Lai, K.; Berkman, P.J.; Lorenc, M.T.; Duran, C.; Smits, L.; Manoli, S.; Stiller, J.; Edwards, D. WheatGenome.info: An integrated database and portal for wheat genome information. Plant Cell Physiol. 2012, 53, e2. [Google Scholar] [CrossRef]
- Allen, A.M.; Barker, G.L.A.; Berry, S.T.; Coghill, J.A.; Gwilliam, R.; Kirby, S.; Robinson, P.; Brenchley, R.C.; D'Amore, R.; McKenzie, N.; Waite, D.; et al. Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotechnol. J. 2011, 9, 1086–1099. [Google Scholar]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010, 42, 1027–1030. [Google Scholar] [CrossRef]
- Lee, H.C.; Lai, K.; Lorenc, M.T.; Imelfort, M.; Duran, C.; Edwards, D. Bioinformatics tools and databases for analysis of next-generation sequence data. Brief. Funct. Genomics 2012, 11, 12–24. [Google Scholar] [CrossRef]
- Duran, C.; Eales, D.; Marshall, D.; Imelfort, M.; Stiller, J.; Berkman, P.J.; Clark, T.; McKenzie, M.; Appleby, N.; Batley, J.; et al. Future tools for association mapping in crop plants. Genome 2010, 53, 1017–1023. [Google Scholar] [CrossRef]
- Marshall, D.J.; Hayward, A.; Eales, D.; Imelfort, M.; Stiller, J.; Berkman, P.J.; Clark, T.; McKenzie, M.; Lai, K.; Duran, C.; et al. Targeted identification of genomic regions using TAGdb. Plant Methods 2010, 6, 19. [Google Scholar] [CrossRef]
- Imelfort, M.; Duran, C.; Batley, J.; Edwards, D. Discovering genetic polymorphisms in next-generation sequencing data. Plant Biotechnol. J. 2009, 7, 312–317. [Google Scholar] [CrossRef]
- Duran, C.; Appleby, N.; Edwards, D.; Batley, J. Molecular genetic markers: discovery, applications, data storage and visualisation. Current Bioinformatics 2009, 4, 16–27. [Google Scholar] [CrossRef]
- Lai, K.; Duran, C.; Berkman, P.J.; Lorenc, M.T.; Stiller, J.; Manoli, S.; Hayden, M.J.; Forrest, K.L.; Fleury, D.; Baumann, U.; et al. Single nucleotide polymorphism discovery from wheat next-generation sequence data. Plant Biotechnol. J. 2012, 10, 743–749. [Google Scholar] [CrossRef]
- Duran, C.; Appleby, N.; Clark, T.; Wood, D.; Imelfort, M.; Batley, J.; Edwards, D. AutoSNPdb: an annotated single nucleotide polymorphism database for crop plants. Nucleic Acids Res. 2009, 37, D951–D953. [Google Scholar] [CrossRef]
- Duran, C.; Appleby, N.; Vardy, M.; Imelfort, M.; Edwards, D.; Batley, J. Single nucleotide polymorphism discovery in barley using autoSNPdb. Plant Biotechnol. J. 2009, 7, 326–333. [Google Scholar] [CrossRef]
- Batley, J.; Barker, G.; O'Sullivan, H.; Edwards, K.J.; Edwards, D. Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data. Plant Physiol. 2003, 132, 84–91. [Google Scholar] [CrossRef]
- Bundock, P.C.; Eliott, F.G.; Ablett, G.; Benson, A.D.; Casu, R.E.; Aitken, K.S.; Henry, R.J. Targeted single nucleotide polymorphism (SNP) discovery in a highly polyploid plant species using 454 sequencing. Plant Biotechnol. J. 2009, 7, 347–354. [Google Scholar] [CrossRef]
- You, F.M.; Huo, N.; Deal, K.R.; Gu, Y.Q.; Luo, M.-C.; McGuire, P.E.; Dvorak, J.; Anderson, O.D. Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence. BMC Genomics 2011, 12, 59. [Google Scholar]
- Close, T.J.; Bhat, P.R.; Lonardi, S.; Wu, Y.; Rostoks, N.; Ramsay, L.; Druka, A.; Stein, N.; Svensson, J.T.; Wanamaker, S.; et al. Development and implementation of high-throughput SNP genotyping in barley. BMC Genomics 2009, 10, 582. [Google Scholar] [CrossRef]
- Cavanagh, C.R.; Chao, S.; Wang, S.; Huang, B.E.; Stephen, S.; Kiani, S.; Forrest, K.; Saintenac, C.; Brown-Guedira, G.L.; Akhunova, A.; et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8057–8062. [Google Scholar] [CrossRef]
- Ganal, M.W.; Durstewitz, G.; Polley, A.; Bérard, A.; Buckler, E.S.; Charcosset, A.; Clarke, J.D.; Graner, E.-M.; Hansen, M.; Joets, J.; et al. A large maize (Zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS One 2011, 6, e28334. [Google Scholar] [CrossRef]
- Seeb, J.E.; Carvalho, G.; Hauser, L.; Naish, K.; Roberts, S.; Seeb, L.W. Single-nucleotide polymorphism (SNP) discovery and applications of SNP genotyping in nonmodel organisms. Mol Ecol Resour 2011, 11, 1–8. [Google Scholar]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol 2012, 30, 105–111. [Google Scholar]
- Jin, J.; Huang, W.; Gao, J.-P.; Yang, J.; Shi, M.; Zhu, M.-Z.; Luo, D.; Lin, H.-X. Genetic control of rice plant architecture under domestication. Nat. Genet. 2008, 40, 1365–1369. [Google Scholar] [CrossRef]
- Tan, L.; Li, X.; Liu, F.; Sun, X.; Li, C.; Zhu, Z.; Fu, Y.; Cai, H.; Wang, X.; Xie, D.; et al. Control of a key transition from prostrate to erect growth in rice domestication. Nat. Genet. 2008, 40, 1360–1364. [Google Scholar] [CrossRef]
- Li, C.; Zhou, A.; Sang, T. Rice domestication by reducing shattering. Science 2006, 311, 1936–1939. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Yu, H.; Xie, W.; Wang, J.; Xing, Y.; Xu, C.; Li, X.; Xiao, J. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 2011, 6, e17595. [Google Scholar]
- Xie, W.; Feng, Q.; Yu, H.; Huang, X.; Zhao, Q.; Xing, Y.; Yu, S.; Han, B.; Zhang, Q. Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10578–10583. [Google Scholar]
- van Poecke, R.M.P.; Maccaferri, M.; Tang, J.; Truong, H.T.; Janssen, A.; van Orsouw, N.J.; Salvi, S.; Sanguineti, M.C.; Tuberosa, R.; van der Vossen, E.A.G. Sequence-based SNP genotyping in durum wheat. Plant Biotechnol. J. 2013, 11, 809–817. [Google Scholar] [CrossRef]
- Hollister, J.D.; Arnold, B.J.; Svedin, E.; Xue, K.S.; Dilkes, B.P.; Bomblies, K. Genetic adaptation associated with genome-doubling in autotetraploid Arabidopsis arenosa. PLoS Genet. 2012, 8, e1003093. [Google Scholar] [CrossRef]
- Mammadov, J.A.; Chen, W.; Ren, R.; Pai, R.; Marchione, W.; Yalçin, F.; Witsenboer, H.; Greene, T.W.; Thompson, S.A.; Kumpatla, S.P. Development of highly polymorphic SNP markers from the complexity reduced portion of maize (Zea mays L.) genome for use in marker-assisted breeding. Theor. Appl. Genet. 2010, 121, 577–588. [Google Scholar] [CrossRef]
- van Orsouw, N.J.; Hogers, R.C.J.; Janssen, A.; Yalçin, F.; Snoeijers, S.; Verstege, E.; Schneiders, H.; van der Poel, H.; van Oeveren, J.; Verstegen, H.; et al. Complexity reduction of polymorphic sequences (CRoPS): A novel approach for large-scale polymorphism discovery in complex genomes. PLoS One 2007, 2, e1172. [Google Scholar] [CrossRef]
- Trebbi, D.; Maccaferri, M.; de Heer, P.; Sørensen, A.; Giuliani, S.; Salvi, S.; Sanguineti, M.C.; Massi, A.; van der Vossen, E.A.G.; Tuberosa, R. High-throughput SNP discovery and genotyping in durum wheat (Triticum durum Desf.). Theor. Appl. Genet. 2011, 123, 555–569. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011, 6, e19379. [Google Scholar]
- Chutimanitsakun, Y.; Nipper, R.W.; Cuesta-Marcos, A.; Cistué, L.; Corey, A.; Filichkina, T.; Johnson, E.A.; Hayes, P.M. Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley. BMC Genomics 2011, 12, 4. [Google Scholar]
- Batley, J.; Edwards, D. Genome sequence data: management, storage, and visualization. BioTechniques 2009, 46, 333–336. [Google Scholar] [CrossRef]
- Edwards, D.; Batley, J. Plant bioinformatics: from genome to phenome. Trends in Biotechniques 2004, 22, 232–237. [Google Scholar] [CrossRef]
- Tetz, V.V. The pangenome concept: a unifying view of genetic information. Med. Sci. Monit. 2005, 11, HY24–HY29. [Google Scholar]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef]
- Vielle-Calzada, J.-P.; Martínez de la Vega, O.; Hernández-Guzmán, G.; Ibarra-Laclette, E.; Alvarez-Mejía, C.; Vega-Arreguín, J.C.; Jiménez-Moraila, B.; Fernández-Cortés, A.; Corona-Armenta, G.; Herrera-Estrella, L.; et al. The Palomero genome suggests metal effects on domestication. Science 2009, 326, 1078–1078. [Google Scholar] [CrossRef]
- International Barley Genome Sequencing Consortium; Mayer, K.F.X.; Waugh, R.; Brown, J.W.S.; Schulman, A.; Langridge, P.; Platzer, M.; Fincher, G.B.; Muehlbauer, G.J.; Sato, K.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar]
- Jia, J.; Zhao, S.; Kong, X.; Li, Y.; Zhao, G.; He, W.; Appels, R.; Pfeifer, M.; Tao, Y.; Zhang, X.; et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 2013, 496, 91–95. [Google Scholar] [CrossRef]
- Ling, H.-Q.; Zhao, S.; Liu, D.; Wang, J.; Sun, H.; Zhang, C.; Fan, H.; Li, D.; Dong, L.; Tao, Y.; et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 2013, 496, 87–90. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Visendi, P.; Batley, J.; Edwards, D. Next Generation Characterisation of Cereal Genomes for Marker Discovery. Biology 2013, 2, 1357-1377. https://doi.org/10.3390/biology2041357
Visendi P, Batley J, Edwards D. Next Generation Characterisation of Cereal Genomes for Marker Discovery. Biology. 2013; 2(4):1357-1377. https://doi.org/10.3390/biology2041357
Chicago/Turabian StyleVisendi, Paul, Jacqueline Batley, and David Edwards. 2013. "Next Generation Characterisation of Cereal Genomes for Marker Discovery" Biology 2, no. 4: 1357-1377. https://doi.org/10.3390/biology2041357
APA StyleVisendi, P., Batley, J., & Edwards, D. (2013). Next Generation Characterisation of Cereal Genomes for Marker Discovery. Biology, 2(4), 1357-1377. https://doi.org/10.3390/biology2041357