The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis


Abstract
:1. Introduction: Helicobacter pylori—A Successful Pathogen
2. Helicobacter pylori Adherence-Associated Molecules
2.1. Lipopolysaccharide (LPS)
2.2. Outer Membrane Proteins
2.2.1. BabA
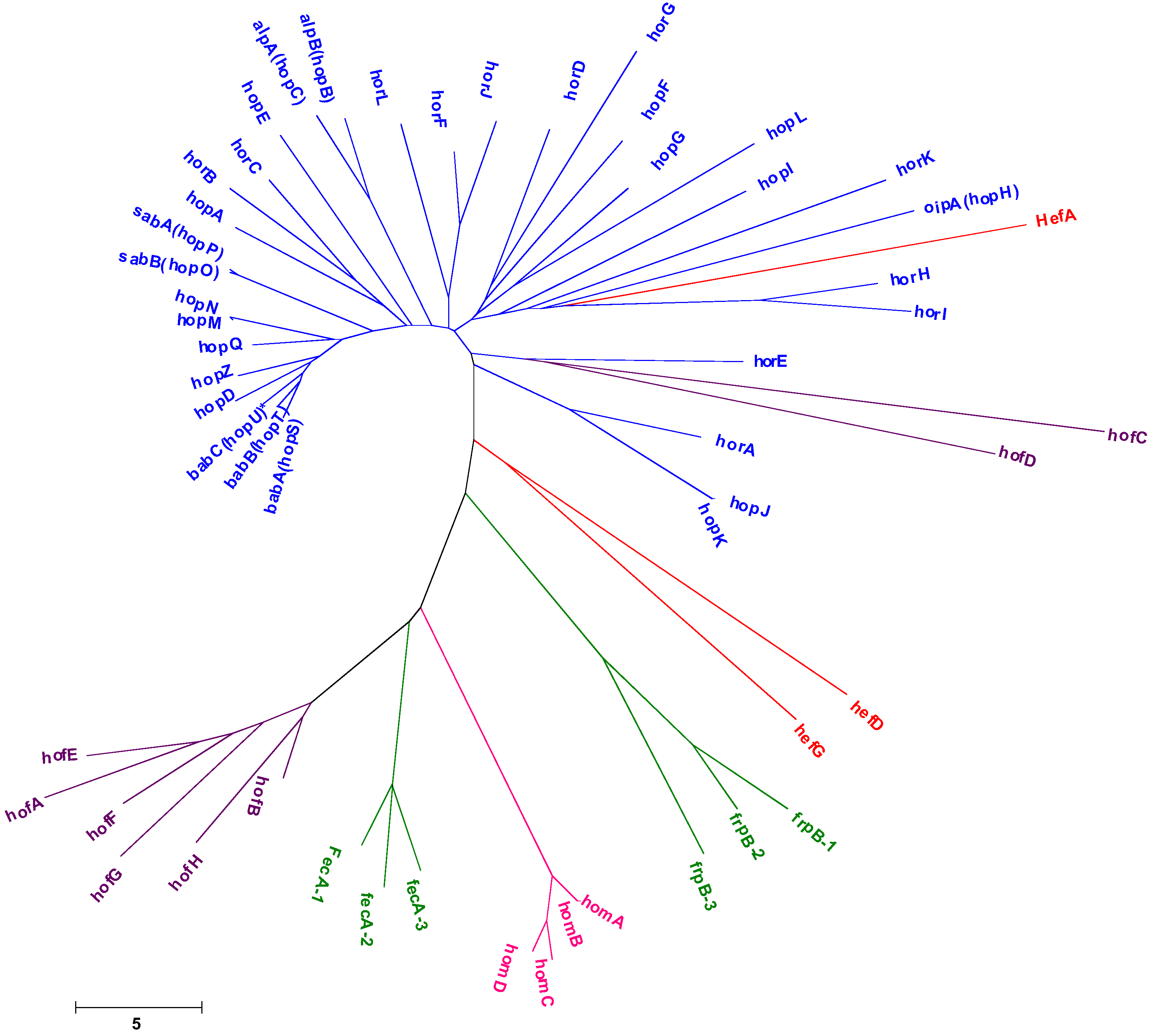

{kind=link}
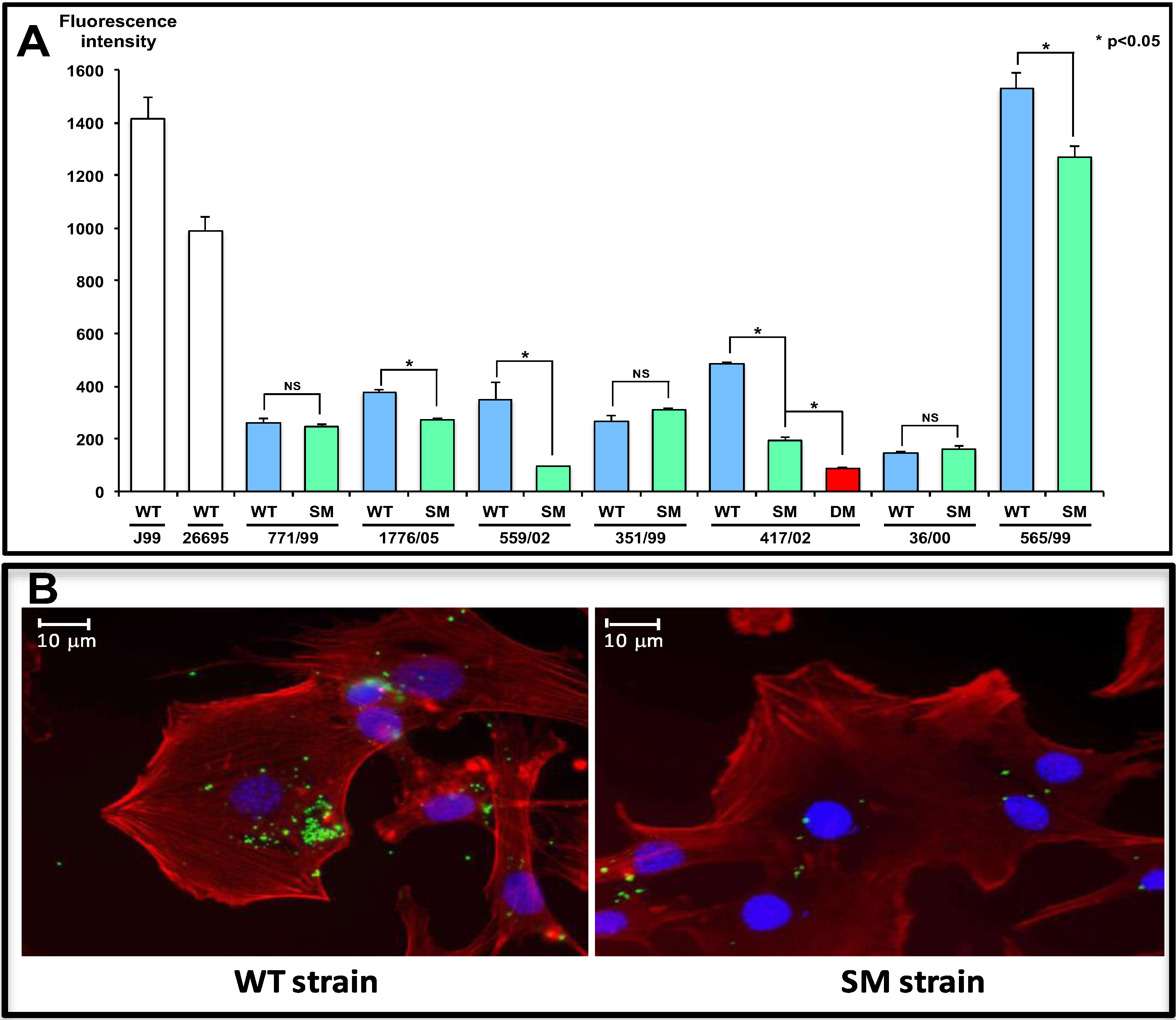{kind=link}
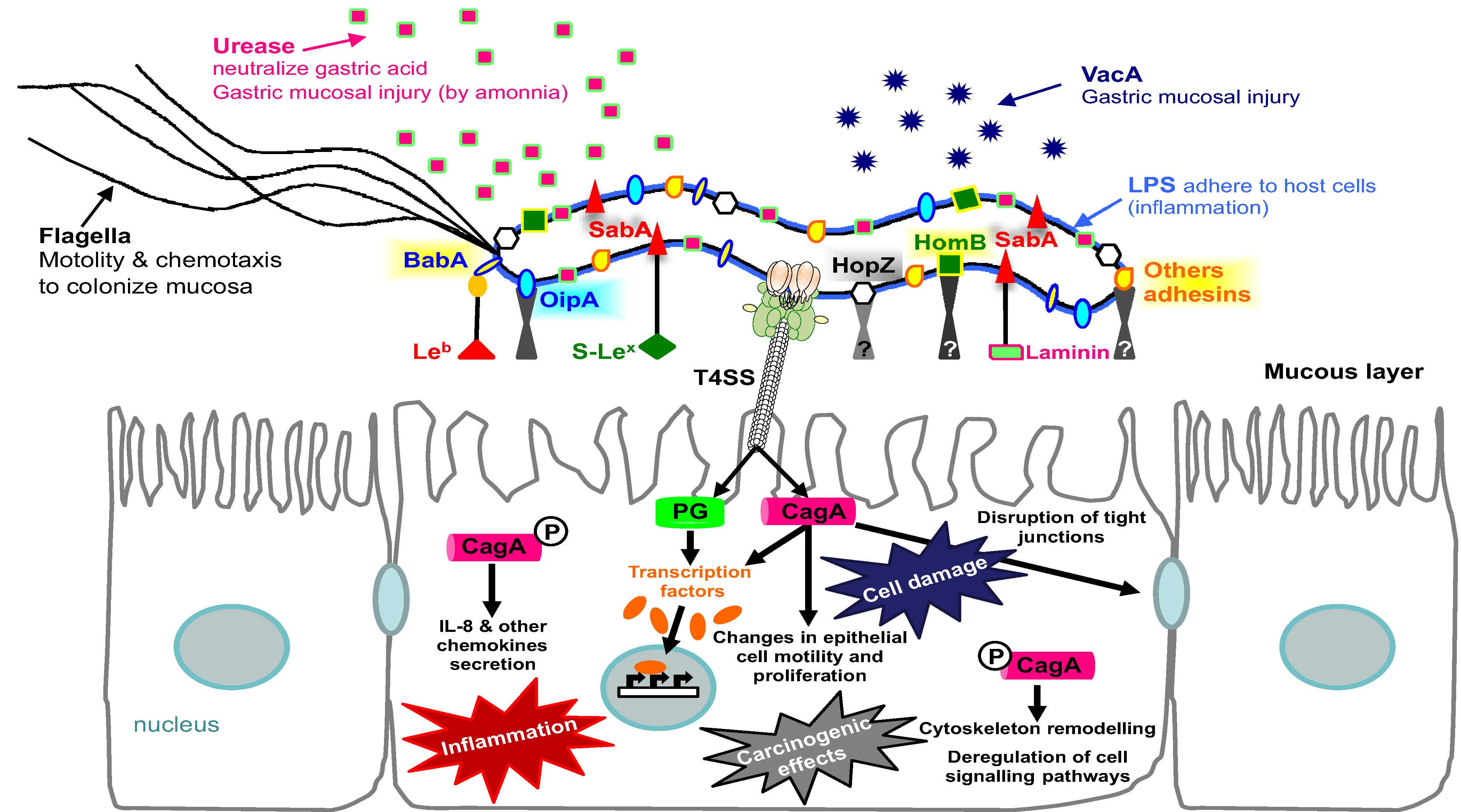{kind=link}
| Adhesins | Receptors identified | References |
|---|---|---|
| BabA | fucosylated Lewis b histo-blood group antigen, Leb and H1 | [27,32] |
| BabA | terminal fucose residues on blood group O (H antigen), A and B antigens | [33] |
| BabA | salivary nonmucin glycoprotein gp-340 | [37] |
| BabA | salivary mucin MUC5B * and proline-rich glycoprotein | [36,37] |
| SabA | sialyl-dimeric-Lex | [28] |
| SabA | salivary mucin MUC7, MUC5B * | [36,37] |
| SabA | salivary glycoproteins (carbonic anhydrase VI, secretory component, heavy chain of secretory IgA1, parotid secretory protein and zinc-α2-glycoprotein) | [37] |
| SabA | sialylated moieties on the extracellular matrix protein laminin | [36] |
| SabA | sialylated structures on the surface of erythrocytes | [40] |
| SabA | sialylated carbohydrates on neutrophils | [41] |
| AlpA | laminin | [42,43] |
| AlpB | laminin | [42,43] |
2.2.2. SabA
2.2.3. AlpA, AlpB
2.2.4. OipA
2.2.5. HopZ
2.2.6. HomB
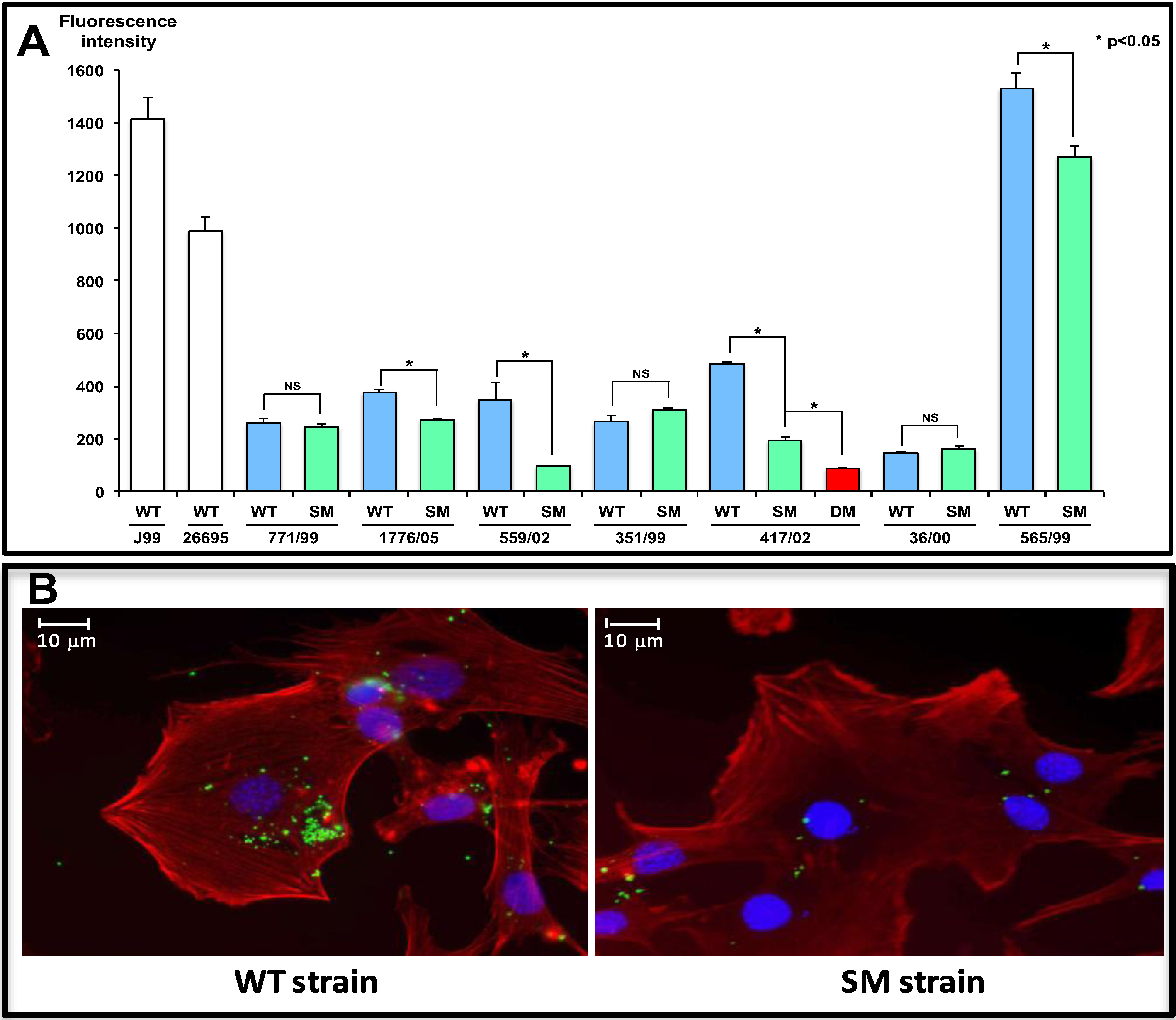
3. Major Gastric Epithelial Receptors of H. pylori
3.1. Lewis Blood Group Antigens
3.2. Sialylated Glycans
4. The Importance of Adherence for Bacterial Colonization and Virulence
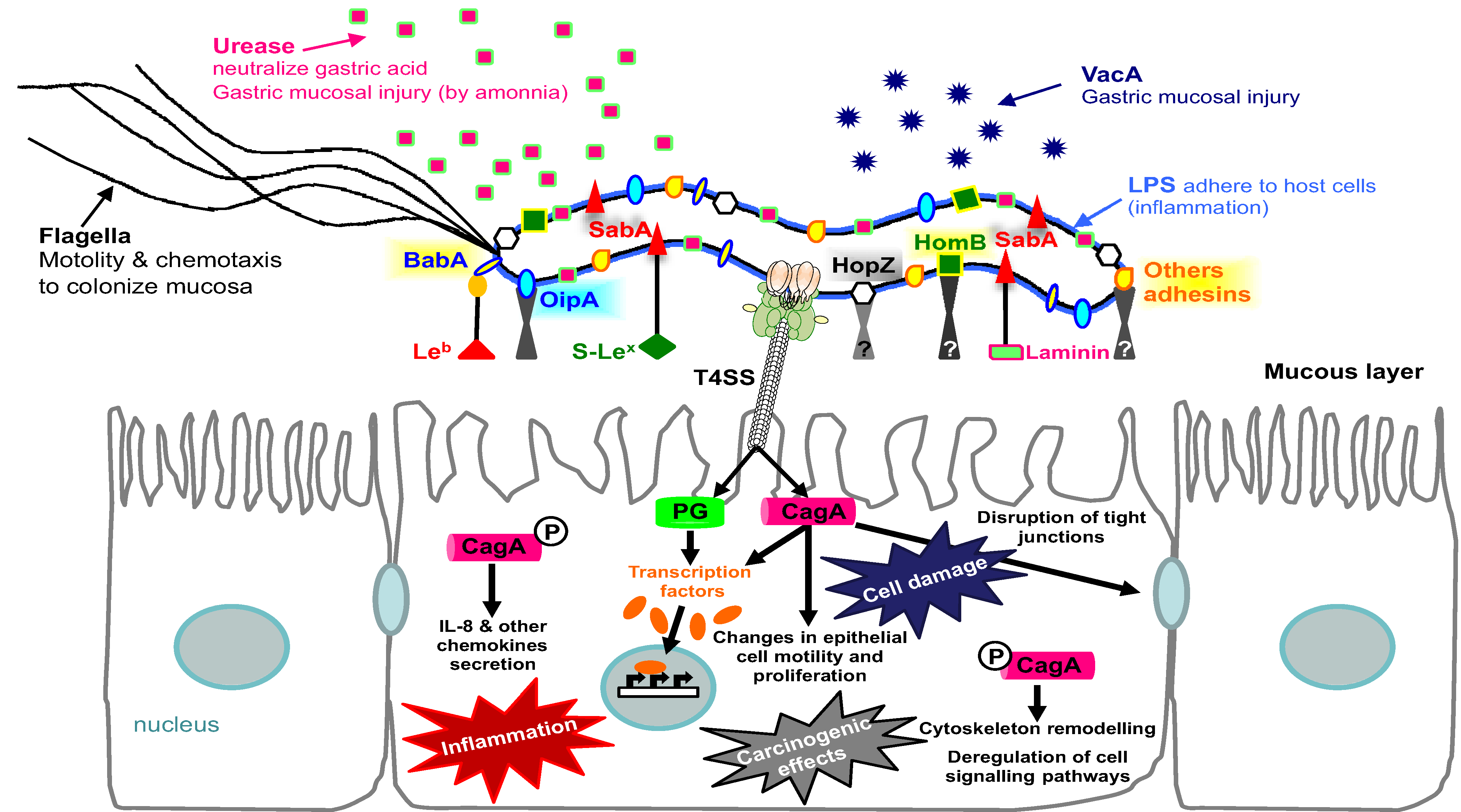
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Traces of human migrations in Helicobacter pylori populations. Science 2003, 299, 1582–1585. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Schistosomes, liver flukes and Helicobacter pylori. In Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization, International Agency for Research on Cancer: Lyon, France, 1994; Volume 61, pp. 1–241.
- González, C.; Megraud, F.; Buissonniere, A.; Lujan Barroso, L.; Agudo, A.; Duell, E.J.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Palli, D.; Krogh, V.; et al. Helicobacter pylori infection assessed by ELISA and by immunoblot and noncardia gastric cancer risk in a prospective study: The Eurgast-EPIC project. Ann. Oncol. 2012, 23, 1320–1324. [Google Scholar] [CrossRef]
- Banic, M.; Franceschi, F.; Babic, Z.; Gasbarrini, A. Extragastric manifestations of Helicobacter pylori infection. Helicobacter 2012, 17, 49–55. [Google Scholar]
- Fukase, K.; Kato, M.; Kikuchi, S.; Inoue, K.; Uemura, N.; Okamoto, S.; Terao, S.; Amagai, K.; Hayashi, S.; Asaka, M.; The Japan Gast Study Group. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet 2008, 372, 392–397. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.; Bazzoli, F.; El-Omar, E.; Graham, D.; Hunt, R.; Rokkas, T.; Vakil, N.; Kuipers, E.J.; et al. Current concepts in the management of Helicobacter pylori infection: The Maastricht III Consensus Report. Gut 2007, 56, 772–781. [Google Scholar] [CrossRef]
- Backert, S.; Clyne, M.; Tegtmeyer, N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun. Signal 2011, 9, 28. [Google Scholar] [CrossRef]
- Suerbaum, S.; Achtman, M. Evolution of Helicobacter pylori: The role of recombination. Trends Microbiol. 1999, 7, 182–184. [Google Scholar] [CrossRef]
- Falush, D.; Kraft, C.; Taylor, N.S.; Correa, P.; Fox, J.G.; Achtman, M.; Suerbaum, S. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: Estimates of clock rates, recombination size, and minimal age. Proc. Natl. Acad. Sci. USA 2001, 98, 15056–15061. [Google Scholar]
- Odenbreit, S.; Swoboda, K.; Barwig, I.; Ruhl, S.; Borén, T.; Koletzko, S.; Haas, R. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infect. Immun. 2009, 77, 3782–3790. [Google Scholar] [CrossRef]
- Suerbaum, S.; Josenhans, C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007, 5, 441–452. [Google Scholar] [CrossRef]
- Morelli, G.; Didelot, X.; Kusecek, B.; Schwarz, S.; Bahlawane, C.; Falush, D.; Suerbaum, S.; Achtman, M. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010, 6, e1001036. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Israel, D.A.; Kusters, J.G.; Gerrits, M.M.; Weel, J.; van der Ende, A.; van der Hulst, R.; Wirth, H.P.; Höök-Nikanne, J.; Thompson, S.A.; et al. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart from the same host. J. Infect. Dis. 2000, 181, 273–282. [Google Scholar] [CrossRef]
- Muotiala, A.; Helander, I.M.; Pyhala, L.; Kosunen, T.U.; Moran, A.P. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect. Immun. 1992, 60, 1714–1716. [Google Scholar]
- Monteiro, M.A.; Appelmelk, B.J.; Rasko, D.A.; Moran, A.P.; Hynes, S.O.; MacLean, L.L.; Chan, K.H.; Michael, F.S.; Logan, S.M.; O’Rourke, J.; et al. Lipopolysaccharide structures of Helicobacter pylori genomic strains 26695 and J99, mouse model H-pylori Sydney strain, H-pylori P466 carrying sialyl Lewis X, and H-pylori UA915 expressing Lewis B—Classification of H-pylori lipopolysaccharides into glycotype families. Eur. J. Biochem. 2000, 267, 305–320. [Google Scholar] [CrossRef]
- Appelmelk, B.J.; Shiberu, B.; Trinks, C.; Tapsi, N.; Zheng, P.Y.; Verboom, T.; Maaskant, J.; Hokke, C.H.; Schiphorst, W.E.; Blanchard, D.; et al. Phase variation in Helicobacter pylori lipopolysaccharide. Infect. Immun. 1998, 66, 70–76. [Google Scholar]
- Edwards, N.J.; Monteiro, M.A.; Faller, G.; Walsh, E.J.; Moran, A.P.; Roberts, I.S.; High, N.J. Lewis X structures in the O antigen side-chain promote adhesion of Helicobacter pylori to the gastric epithelium. Mol. Microbiol. 2000, 35, 1530–1539. [Google Scholar]
- Monteiro, M.A.; Chan, K.H.N.; Rasko, D.A.; Taylor, D.E.; Zheng, P.Y.; Appelmelk, B.J.; Wirth, H.P.; Yang, M.; Blaser, M.J.; Hynes, S.O.; et al. Simultaneous expression of type 1 and type 2 Lewis blood group antigens by Helicobacter pylori lipopolysaccharides. J. Biol. Chem. 1998, 273, 11533–11543. [Google Scholar] [CrossRef]
- Osaki, T.; Yamaguchi, H.; Taguchi, H.; Fukuda, M.; Kawakami, H.; Hirano, H.; Watanabe, S.; Takagi, A.; Kamiya, S. Establishment and characterisation of a monoclonal antibody to inhibit adhesion of Helicobacter pylori to gastric epithelial cells. J. Med. Microbiol. 1998, 47, 505–512. [Google Scholar]
- Fowler, M.; Thomas, R.J.; Atherton, J.; Roberts, I.S.; High, N.J. Galectin-3 binds to Helicobacter pylori O-antigen: It is upregulated and rapidly secreted by gastric epithelial cells in response to H. pylori adhesion. Cell. Microbiol. 2006, 8, 44–54. [Google Scholar] [CrossRef]
- Odenbreit, S.; Faller, G.; Haas, R. Role of the AlpAB proteins and lipopolysaccharide in adhesion of Helicobacter pylori to human gastric tissue. Int. Med. Microbiol. 2002, 292, 247–256. [Google Scholar] [CrossRef]
- Mahdavi, J.; Boren, T.; Vandenbroucke Grauls, C.; Appelmelk, B.J. Limited role of lipopolysaccharide lewis antigens in adherence of Helicobacter pylori to the human gastric epithelium. Infect. Immun. 2003, 71, 2876–2880. [Google Scholar] [CrossRef]
- Alm, R.A.; Bina, J.; Andrews, B.M.; Doig, P.; Hancock, R.E.W.; Trust, T.J. Comparative genomics of Helicobacter pylori: Analysis of the outer membrane protein families. Infect. Immun. 2000, 68, 4155–4168. [Google Scholar] [CrossRef]
- Doig, P.; Exner, M.M.; Hancock, R.E.W.; Trust, T.J. Isolation and characterization of a conserved porin protein from Helicobacter pylori. J. Bacteriol. 1995, 177, 5447–5452. [Google Scholar]
- Exner, M.M.; Doig, P.; Trust, T.J.; Hancock, R.E. Isolation and characterization of a family of porin proteins from Helicobacter pylori. Infect. Immun. 1995, 63, 1567–1572. [Google Scholar]
- Ilver, D.; Arnqvist, A.; Ögren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Borén, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef]
- Mahdavi, J.; Sonden, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Ăngström, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef]
- Oleastro, M.; Cordeiro, R.; Ferrand, J.; Nunes, B.; Lehours, P.; Carvalho-Oliveira, I.; Mendes, A.L.; Monteiro, L.; Mégraud, F.; Ménard, A. Evaluation of the clinical significance of homB, a novel candidate marker of Helicobacter pylori strains associated with peptic ulcer disease. J. Infect. Dis. 2008, 198, 1379–1387. [Google Scholar] [CrossRef]
- Yamaoka, Y. Roles of Helicobacter pylori BabA in gastroduodenal pathogenesis. World J. Gastroenterol. 2008, 14, 4265–4272. [Google Scholar] [CrossRef]
- Moore, M.; Borén, T.; Solnick, J. Life at the margins: Modulation of attachment proteins in Helicobacter pylori. Gut Microbes 2011, 2, 42–46. [Google Scholar] [CrossRef]
- Borén, T.; Falk, P.; Roth, K.A.; Larson, G.; Normark, S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993, 262, 1892–1895. [Google Scholar]
- Aspholm-Hurtig, M.; Dailide, G.; Lahmann, M.; Kalia, A.; Ilver, D.; Roche, N.; Vikström, S.; Lindén, S.; Bäckström, A.; Lundberg, C.; et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 2004, 305, 519–522. [Google Scholar] [CrossRef]
- Sakamoto, S.; Watanabe, T.; Tokumaru, T.; Takagi, H.; Nakazato, H.; Lloyd, K.O. Expression of Lewisa, Lewisb, Lewisx, Lewisy, siayl-Lewisa, and sialyl-Lewisx blood group antigens in human gastric carcinoma and in normal gastric tissue. Cancer Res. 1989, 49, 745–752. [Google Scholar]
- Benktander, J.; Ångström, J.; Breimer, M.; Teneberg, S. Redefinition of the carbohydrate binding specificity of Helicobacter pylori BabA adhesin. J. Biol. Chem. 2012, 287, 31712–31724. [Google Scholar]
- Walz, A.; Odenbreit, S.; Mahdavi, J.; Boren, T.; Ruhl, S. Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology 2005, 15, 700–708. [Google Scholar] [CrossRef]
- Walz, A.; Odenbreit, S.; Stühler, K.; Wattenberg, A.; Meyer, H.E.; Mahdavi, J.; Borén, T.; Ruhl, S. Identification of glycoprotein receptors within the human salivary proteome for the lectin-like BabA and SabA adhesins of Helicobacter pylori by fluorescence-based 2-D bacterial overlay. Proteomics 2009, 9, 1582–1592. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Aspholm, M.; Olfat, F.O.; Nordén, J.; Sondén, B.; Lundberg, C.; Sjöström, R.; Altraja, S.; Odenbreit, S.; Haas, R.; Wadström, T.; et al. SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog 2006, 2, e110. [Google Scholar] [CrossRef]
- Unemo, M.; Aspholm-Hurtig, M.; Ilver, D.; Bergström, J.; Borén, T.; Danielsson, D.; Teneberg, S. The sialic acid binding SabA adhesin of Helicobacter pylori is essential for nonopsonic activation of human neutrophils. J. Biol. Chem. 2005, 280, 15390–15397. [Google Scholar]
- Odenbreit, S.; Till, M.; Hofreuter, D.; Faller, G.; Haas, R. Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol. Microbiol. 1999, 31, 1537–1548. [Google Scholar] [CrossRef]
- Senkovich, O.A.; Yin, J.; Ekshyyan, V.; Conant, C.; Traylor, J.; Adegboyega, P.; McGee, D.J.; Rhoads, R.E.; Slepenkov, S.; Testerman, T.L. Helicobacter pylori AlpA and AlpB bind host laminin and influence gastric inflammation in gerbils. Infect. Immun. 2011, 79, 3106–3116. [Google Scholar] [CrossRef]
- Pride, D.T.; Blaser, M.J. Concerted evolution between duplicated genetic elements in Helicobacter pylori. J. Mol. Biol. 2002, 316, 629–642. [Google Scholar] [CrossRef]
- Pride, D.T.; Meinersmann, R.J.; Blaser, M.J. Allelic variation within Helicobacter pylori babA and babB. Infect. Immun. 2001, 69, 1160–1171. [Google Scholar] [CrossRef]
- Colbeck, J.C.; Hansen, L.M.; Fong, J.M.; Solnick, J.V. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori. Infect. Immun. 2006, 74, 4375–4378. [Google Scholar] [CrossRef]
- Backstrom, A.; Lundberg, C.; Kersulyte, D.; Berg, D.E.; Boren, T.; Arnqvist, A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc. Natl. Acad. Sci. USA 2004, 101, 16923–16928. [Google Scholar]
- Solnick, J.V.; Hansen, L.M.; Salama, N.R.; Boonjakuakul, J.K.; Syvanen, M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of Rhesus macaques. Proc. Natl. Acad. Sci. USA 2004, 101, 2106–2111. [Google Scholar]
- Matteo, M.; Armitano, R.; Romeo, M.; Wonaga, A.; Olmos, M.; Catalano, M. Helicobacter pylori bab genes during chronic colonization. Int. J. Mol. Epidemiol. Genet. 2011, 30, 286–291. [Google Scholar]
- Styer, C.M.; Hansen, L.M.; Cooke, C.L.; Gundersen, A.M.; Choi, S.S.; Berg, D.E.; Benghezal, M.; Peek, R.M.; Borén, T.; Solnick, J.V.; et al. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect. Immun. 2010, 78, 1593–1600. [Google Scholar] [CrossRef]
- Ohno, T.; Vallström, A.; Rugge, M.; Ota, H.; Graham, D.Y.; Arnqvist, A.; Yamaoka, Y. Effects of blood group antigen-binding adhesin expression during Helicobacter pylori infection of Mongolian gerbils. J. Infect. Dis. 2011, 203, 726–735. [Google Scholar] [CrossRef]
- Hennig, E.; Allen, J.; Cover, T. Multiple chromosomal loci for the babA gene in Helicobacter pylori. Infect. Immun. 2006, 74, 3046–3051. [Google Scholar] [CrossRef]
- Armitano, R.; Matteo, M.; Goldman, C.; Wonaga, A.; Viola, L.A.; de Palma, G.Z.; Catalano, M. Helicobacter pylori heterogeneity in patients with gastritis and peptic ulcer disease. Infect. Genet. Evol. 2013, 16, 377–385. [Google Scholar] [CrossRef]
- Ishijima, N.; Suzuki, M.; Ashida, H.; Ichikawa, Y.; Kanegae, Y.; Saito, I.; Borén, T.; Haas, R.; Sasakawa, C.; Mimuro, H. BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J. Biol. Chem. 2011, 286, 25256–25264. [Google Scholar] [CrossRef]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.O.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef]
- Fujimoto, S.; Olaniyi, O.O.; Arnqvist, A.; Wu, J.Y.; Odenbreit, S.; Haas, R.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori BabA expression, gastric mucosal injury, and clinical outcome. Clin. Gastroenterol. Hepatol. 2007, 5, 49–58. [Google Scholar] [CrossRef]
- Azevedo, M.; Eriksson, S.; Mendes, N.; Serpa, J.; Figueiredo, C.; Resende, L.P.; Ruvoën-Clouet, N.; Haas, R.; Borën, T.; Le Pedun, J.; et al. Infection by Helicobacter pylori expressing the BabA adhesin is influenced by the secretor phenotype. J. Pathol. 2008, 215, 308–316. [Google Scholar] [CrossRef]
- Gerhard, M.; Lehn, N.; Neumayer, N.; Borén, T.; Rad, R.; Schepp, W.; Miehlke, S.; Classen, M.; Prinz, C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc. Natl. Acad. Sci. USA 1999, 96, 12778–12783. [Google Scholar] [CrossRef]
- Yamaoka, Y. Increasing evidence of the role of Helicobacter pylori SabA in the pathogenesis of gastroduodenal disease. J. Infect. Dev. Ctries. 2008, 2, 174–181. [Google Scholar] [CrossRef]
- Talarico, S.; Whitefield, S.E.; Fero, J.; Haas, R.; Salama, N.R. Regulation of Helicobacter pylori adherence by gene conversion. Mol. Microbiol. 2012, 84, 1050–1061. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Imamura, S.; Ohno, T.; Sawai, N.; Ishimaru, A.; Imanishi, J.; Graham, D.Y. Helicobacter pylori infection in mice: Role of outer membrane proteins in colonization and inflammation. Gastroenterology 2002, 123, 1992–2004. [Google Scholar] [CrossRef]
- Loh, J.T.; Torres, V.J.; Algood, H.M.; McClain, M.S.; Cover, T.L. Helicobacter pylori HopQ outer membrane protein attenuates bacterial adherence to gastric epithelial cells. FEMS Microbiol. Lett. 2008, 289, 53–58. [Google Scholar] [CrossRef]
- Merrell, D.S.; Goodrich, M.L.; Otto, G.; Tompkins, L.S.; Falkow, S. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 2003, 71, 3529–3539. [Google Scholar]
- Yamaoka, Y.; Ojo, O.; Fujimoto, S.; Odenbreit, S.; Haas, R.; Gutierrez, O.; El-Zimaity, H.M.T.; Reddy, R.; Arnqvist, A.; Graham, D.Y. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut 2006, 55, 775–781. [Google Scholar] [CrossRef]
- Saunders, N.J.; Boonmee, P.; Peden, J.F.; Jarvis, S.A. Inter-species horizontal transfer resulting in core-genome and niche-adaptive variation within Helicobacter pylori. BMC Genomics 2005, 6, 9. [Google Scholar]
- Kao, C.Y.; Sheu, B.S.; Sheu, S.M.; Yang, H.B.; Chang, W.L.; Cheng, H.C.; Wu, J.J. Higher motility enhances bacterial density and inflammatory response in dyspeptic patients infected with Helicobacter pylori. Helicobacter 2012, 17, 411–416. [Google Scholar] [CrossRef]
- Sheu, B.; Odenbreit, S.; Hung, K.H.; Liu, C.P.; Sheu, S.M.; Yang, H.B.; Wu, J.J. Interaction between host gastric Sialyl-Lewis X and H. pylori SabA enhances H. pylori density in patients lacking gastric Lewis B antigen. Am. J. Gastroenterol. 2006, 101, 36–44. [Google Scholar] [CrossRef]
- Marcos, N.T.; Magalhães, A.; Ferreira, B.; Oliveira, M.J.; Carvalho, A.S.; Mendes, N.; Gilmartin, T.; Head, S.R.; Figueiredo, C.; David, L.; et al. Helicobacter pylori induces beta3GnT5 in human gastric cell lines, modulating expression of the SabA ligand sialyl-Lewis x. J. Clin. Invest. 2008, 118, 2325–2336. [Google Scholar]
- Bernarde, C.; Lehours, P.; Lasserre, J.P.; Castroviejo, M.; Mégraud, F.; Ménard, A. Complexomics study of two Helicobacter pylori strains of two pathological origins: Potential targets for vaccine development and new insight in bacteria metabolism. Mol. Cell Proteomics 2010, 9, 2796–2826. [Google Scholar] [CrossRef]
- Alm, R.A.; Ling, L.S.L.; Moir, D.T.; King, B.L.; Brown, E.D.; Doig, P.C.; Smith, D.R.; Noonan, B.; Guild, B.C.; deJonge, B.L.; et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 1999, 397, 176–180. [Google Scholar] [CrossRef]
- Baik, S.C.; Kim, K.M.; Song, S.M.; Kim, D.S.; Jun, J.S.; Lee, S.G.; Song, J.Y.; Park, J.U.; Kang, H.L.; Lee, W.K.; et al. Proteomic analysis of the sarcosine-insoluble outer membrane fraction of Helicobacter pylori strain 26695. J. Bacteriol. 2004, 186, 949–955. [Google Scholar] [CrossRef]
- Odenbreit, S.; Till, M.; Hofreuter, D.; Haas, R. Outer membrane proteins AlpA and AlpB are involved in H. pylori binding to epithelial cells. Gut 1997, 41, A107. [Google Scholar] [CrossRef]
- De Jonge, R.; Durrani, Z.; Rijpkema, S.G.; Kuipers, E.J.; van Vliet, A.H.; Kusters, J.G. Role of the Helicobacter pylori outer-membrane proteins AlpA and AlpB in colonization of the guinea pig stomach. J. Med. Microbiol. 2004, 53, 375–379. [Google Scholar] [CrossRef]
- Sugimoto, M.; Ohno, T.; Graham, D.; Yamaoka, Y. Helicobacter pylori outer membrane proteins on gastric mucosal interleukin 6 and 11 expression in Mongolian gerbils. J. Gastroenterol. Hepatol. 2011, 26, 1677–1684. [Google Scholar] [CrossRef]
- Lu, H.; Wu, J.Y.; Beswick, E.J.; Ohno, T.; Odenbreit, S.; Haas, R.; Reyes, V.E.; Kita, M.; Graham, D.Y.; Yamaoka, Y. Functional and intracellular signaling differences associated with the Helicobacter pylori AlpAB adhesin from Western and East Asian strains. J. Biol. Chem. 2007, 282, 6242–6254. [Google Scholar]
- Huang, C.H.; Chiou, S.H. Proteomic analysis of upregulated proteins in Helicobacter pylori under oxidative stress induced by hydrogen peroxide. Kaohsiung J. Med. Sci. 2011, 27, 544–553. [Google Scholar]
- Xue, J.; Bai, Y.; Chen, Y.; Wang, J.D.; Zhang, Z.S.; Zhang, Y.L.; Zhou, D.Y. Expression of Helicobacter pylori AlpA protein and its immunogenicity. World J. Gastroenterol. 2005, 11, 2260–2263. [Google Scholar]
- Bai, Y.; Zhang, Y.; Wang, J.; Lin, H.; Zhang, Z.; Zhou, D. Conservative region of the genes encoding four adhesins of Helicobacter pylori: Cloning, sequence analysis and biological information analysis. Di Yi Jun Yi Da Xue Xue Bao 2002, 22, 869–871. [Google Scholar]
- Sun, Z.L.; Bi, Y.W.; Bai, C.M.; Gao, D.D.; Li, Z.H.; Dai, Z.X.; Li, J.F.; Xu, W.M. Expression of Helicobacter pylori alpA gene in Lactococcus lactis and its immunogenicity analysis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2010, 26, 203–206. [Google Scholar]
- Yamaoka, Y.; Kwon, D.H.; Graham, D.Y. A M(r) 34,000 proinflammatory outer membrane protein (OipA) of Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2000, 97, 111–133. [Google Scholar]
- Yamaoka, Y.; Kikuchi, S.; El-Zimaity, H.M.T.; Gutierrez, O.; Osato, M.S.; Graham, D.Y. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 2002, 123, 414–424. [Google Scholar] [CrossRef]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori-associated regulation of forkhead transcription factors FoxO1/3a in human gastric cells. Helicobacter 2012, 17, 193–202. [Google Scholar] [CrossRef]
- Dossumbekova, A.; Prinz, C.; Mages, J.; Lang, R.; Kusters, J.G.; van Vliet, A.H.; Reindl, W.; Backert, S.; Saur, D.; Schmid, R.M.; et al. Helicobacter pylori HopH (OipA) and bacterial pathogenicity: Genetic and functional genomic analysis of hopH gene polymorphisms. J. Infect. Dis. 2006, 194, 1346–1355. [Google Scholar] [CrossRef]
- Franco, A.T.; Johnston, E.; Krishna, U.; Yamaoka, Y.; Israel, D.A.; Nagy, T.A.; Wroblewski, L.E.; Piazuelo, M.B.; Correa, P.; Peek, R.M., Jr. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008, 68, 379–387. [Google Scholar] [CrossRef]
- Teymournejad, O.; Mobarez, A.M.; Hassan, Z.M.; Moazzeni, S.M.; Yakhchali, B.; Eskandari, V. In silico prediction of exposure amino acid sequences of outer inflammatory protein A of Helicobacter pylori for surface display on Escherichia coli. Indian J. Hum. Genet. 2012, 18, 83–86. [Google Scholar] [CrossRef]
- Saunders, N.J.; Peden, J.F.; Hood, D.W.; Moxon, E.R. Simple sequence repeats in the Helicobacter pylori genome. Mol. Microbiol. 1998, 27, 1091–1098. [Google Scholar] [CrossRef]
- Markovska, R.; Boyanova, L.; Yordanov, D.; Gergova, G.; Mitov, I. Helicobacter pylori oipA genetic diversity and its associations with both disease and cagA, vacA s,m, and i alleles among Bulgarian patients. Diagn. Microbiol. Infect. Dis. 2011, 71, 335–340. [Google Scholar] [CrossRef]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. OipA plays a role in Helicobacter pylori-induced focal adhesion kinase activation and cytoskeletal re-organization. Cell. Microbiol. 2008, 10, 1008–1020. [Google Scholar] [CrossRef]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. Paxillin is a novel cellular target for converging Helicobacter pylori-induced cellular signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G601–G611. [Google Scholar] [CrossRef]
- Chen, J.; Lin, M.; Li, N.; Lin, L.; She, F. Therapeutic vaccination with Salmonella-delivered codon-optimized outer inflammatory protein DNA vaccine enhances protection in Helicobacter pylori infected mice. Vaccine 2012, 30, 5310–5315. [Google Scholar] [CrossRef]
- Chen, J.; Lin, L.; Li, N.; She, F. Enhancement of Helicobacter pylori outer inflammatory protein DNA vaccine efficacy by co-delivery of interleukin-2 and B subunit heat-labile toxin gene encoded plasmids. Microbiol. Immunol. 2012, 56, 85–92. [Google Scholar] [CrossRef]
- Peck, B.; Ortkamp, M.; Diehl, K.D.; Hundt, E.; Knapp, B. Conservation, localization and expression of HopZ, a protein involved in adhesion of Helicobacter pylori. Nucl. Acids Res. 1999, 27, 3325–3333. [Google Scholar] [CrossRef]
- Giannakis, M.; Bäckhed, H.K.; Chen, S.L.; Faith, J.J.; Wu, M.; Guruge, J.L.; Engstrand, L.; Gordon, J.I. Response of gastric epithelial progenitors to Helicobacter pylori isolates obtained from Swedish patients with chronic atrophic gastritis. J. Biol. Chem. 2009, 284, 30383–30394. [Google Scholar] [CrossRef]
- Kennemann, L.; Didelot, X.; Aebischer, T.; Kuhn, S.; Drescher, B.; Droege, M.; Reinhardt, R.; Correa, P.; Meyer, T.F.; Josenhans, C.; et al. Helicobacter pylori genome evolution during human infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5033–5038. [Google Scholar] [CrossRef]
- Kennemann, L.; Brenneke, B.; Andres, S.; Engstrand, L.; Meyer, T.F.; Aebischer, T.; Josenhans, C.; Suerbaum, S. In vivo sequence variation in HopZ, a phase-variable outer membrane protein of Helicobacter pylori. Infect. Immun. 2012, 80, 4364–4373. [Google Scholar] [CrossRef]
- De Jonge, R.; Pot, R.G.J.; Loffeld, R.J.L.F.; van Vliet, A.H.M.; Kuipers, E.J.; Kusters, J.G. The functional status of the Helicobacter pylori sabB adhesin gene as a putative marker for disease outcome. Helicobacter 2004, 9, 158–164. [Google Scholar] [CrossRef]
- Chiarini, A.; Calà, C.; Bonura, C.; Gullo, A.; Giuliana, G.; Peralta, S.; D’Arpa, F.; Giammanco, A. Prevalence of virulence-associated genotypes of Helicobacter pylori and correlation with severity of gastric pathology in patients from western Sicily, Italy. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 437–446. [Google Scholar] [CrossRef]
- Lehours, P.; Ménard, A.; Dupouy, S.; Bergey, B.; Richy, F.; Zerbib, F.; Ruskoné-Fourmestraux, A.; Delchier, J.C.; Mégraud, F. Evaluation of the association of nine Helicobacter pylori virulence factors with strains involved in low-grade gastric mucosa-associated lymphoid tissue lymphoma. Infect. Immun. 2004, 72, 880–888. [Google Scholar] [CrossRef]
- Oleastro, M.; Cordeiro, R.; Ménard, A.; Yamaoka, Y.; Queiroz, D.; Mégraud, F.; Monteiro, L. Allelic diversity and phylogeny of homB, a novel co-virulence marker of Helicobacter pylori. BMC Microbiol. 2009, 9, 248. [Google Scholar] [CrossRef]
- Oleastro, M.; Cordeiro, R.; Ménard, A.; Gomes, J.P. Allelic diversity among Helicobacter pylori outer membrane protein genes homB and homA generated by recombination. J. Bacteriol. 2010, 192, 3961–3968. [Google Scholar] [CrossRef]
- Oleastro, M.; Monteiro, L.; Lehours, P.; Mégraud, F.; Ménard, A. Identification of markers for Helicobacter pylori strains isolated from children with peptic ulcer disease by suppressive subtractive hybridization. Infect. Immun. 2006, 74, 4064–4074. [Google Scholar] [CrossRef]
- Oleastro, M.; Cordeiro, R.; Yamaoka, Y.; Queiroz, D.; Mégraud, F.; Monteiro, L.; Ménard, A. Disease association with two Helicobacter pylori duplicate outer membrane protein genes, homB and homA. Gut Pathog 2009. [Google Scholar] [CrossRef]
- Jung, S.W.; Sugimoto, M.; Graham, D.Y.; Yamaoka, Y. homB status of Helicobacter pylori as a novel marker to distinguish gastric cancer from duodenal ulcer. J. Clin. Microbiol. 2009, 47, 3241–3245. [Google Scholar] [CrossRef]
- Abadi, A.T.B.; Rafiei, A.; Ajami, A.; Hosseini, V.; Taghvaei, T.; Jones, K.R.; Merrell, D.S. Helicobacter pylori homB, but not cagA, is associated with gastric cancer in Iran. J. Clin. Microbiol. 2011, 49, 3191–3197. [Google Scholar] [CrossRef]
- Hussein, N.R. A study of Helicobacter pylori outer-membrane proteins (hom) A and B in Iraq and Turkey. J. Infect. Public Health 2011, 4, 135–139. [Google Scholar] [CrossRef]
- Kang, J.; Jones, K.R.; Jang, S.; Olsen, C.H.; Yoo, Y.J.; Merrell, D.S.; Cha, J.H. The geographic origin of Helicobacter pylori influences the association of the homB gene with gastric cancer. J. Clin. Microbiol. 2012, 50, 1082–1085. [Google Scholar] [CrossRef]
- Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef]
- Morris, A.; Nicholson, G.; Zwi, J.; Vanderwee, M. Campylobacter pylori infection in Meckel’s diverticula containing gastric mucosa. Gut 1989, 30, 1233–1235. [Google Scholar] [CrossRef]
- Kestemberg, A.; Mariño, G.; de Lima, E.; Garcia, F.; Carrascal, E.; Arredondo, J.L. Gastric heterotopic mucosa in the rectum with Helicobacter pylori-like organisms: A rare cause of rectal bleeding. Int. J. Colorectal Dis. 1993, 8, 9–12. [Google Scholar]
- Genta, R.M.; Gurer, I.E.; Graham, D.Y.; Krishnan, B.; Segura, A.M.; Gutierrez, O.; Kim, J.G.; Burchette, J.L., Jr. Adherence of Helicobacter pylori to areas of incomplete intestinal metaplasia in the gastric mucosa. Gastroenterology 1996, 111, 1206–1211. [Google Scholar] [CrossRef]
- Lindén, S.; Nordman, H.; Hedenbro, J.; Hurtig, M.; Borén, T.; Carlstedt, I. Strain- and blood group-dependent binding of Helicobacter pylori to human gastric MUC5AC glycoforms. Gastroenterology 2002, 123, 1923–1930. [Google Scholar] [CrossRef]
- Van den Brink, G.R.; Tytgat, K.; van der Hulst, R.; van der Loos, C.M.; Einerhand, A.; Buller, H.; Dekker, J. H. pylori colocalises with MUC5AC in the human stomach. Gut 2000, 46, 601–607. [Google Scholar] [CrossRef]
- Nordman, H.; Davies, J.R.; Lindell, G.; de Bolós, C.; Real, F.; Carlstedt, I. Gastric MUC5AC and MUC6 are large oligomeric mucins that differ in size, glycosylation and tissue distribution. Biochem. J. 2002, 364, 191–200. [Google Scholar]
- Kawakubo, M.; Ito, Y.; Okimura, Y.; Kobayashi, M.; Sakura, K.; Kasama, S.; Fukuda, M.N.; Katsuyama, T.; Nakayama, J. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science 2004, 305, 1003–1006. [Google Scholar]
- Van de Bovenkamp, J.H.; Mahdavi, J.; Korteland-Van Male, A.M.; Büller, H.A.; Einerhand, A.W.; Borén, T.; Dekker, J. The MUC5AC glycoprotein is the primary receptor for Helicobacter pylori in the human stomach. Helicobacter 2003, 8, 521–532. [Google Scholar] [CrossRef]
- Magalhães, A.; Gomes, J.; Ismail, M.N.; Haslam, S.M.; Mendes, N.; Osório, H.; David, L.; Le Pendu, J.; Haas, R.; Dell, A.; et al. Fut2-null mice display an altered glycosylation profile and impaired BabA-mediated Helicobacter pylori adhesion to gastric mucosa. Glycobiology 2009, 19, 1525–1536. [Google Scholar] [CrossRef]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process-First american cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Rad, R.; Gerhard, M.; Lang, R.; Schöniger, M.; Rösch, T.; Schepp, W.; Becker, I.; Wagner, H.; Prinz, C. The Helicobacter pylori blood group antigen-binding adhesin facilitates bacterial colonization and augments a nonspecific immune response. J. Immunol. 2002, 168, 3033–3041. [Google Scholar]
- Sheu, B.S.; Sheu, S.M.; Yang, H.B.; Huang, A.H.; Wu, J. Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut 2003, 52, 927–932. [Google Scholar] [CrossRef]
- Marionneau, S.; Cailleau-Thomas, A.; Rocher, J.; Le Moullac-Vaidye, B.; Ruvoën, N.; Clément, M.; Le Pendu, J. ABH and Lewis histo-blood group antigens, a model for the meaning of oligosaccharide diversity in the face of a changing world. Biochimie 2001, 83, 565–573. [Google Scholar] [CrossRef]
- Lindén, S.; Mahdavi, J.; Semino-Mora, C.; Oslen, C.; Carlstedt, I.; Borén, T.; Dubois, A. Role of ABO secretor status in mucosal innate immunity and H. pylori infection. PLoS Pathog. 2008, 4, e2. [Google Scholar] [CrossRef]
- Murata-Kamiya, N. Pathophysiological functions of the CagA oncoprotein during infection by Helicobacter pylori. Microbes Infect. 2011, 13, 799–807. [Google Scholar] [CrossRef]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef]
- Zambon, C.F.; Navaglia, F.; Basso, D.; Rugge, M.; Plebani, M. Helicobacter pylori babA2, cagA, and s1 vacA genes work synergistically in causing intestinal metaplasia. J. Clin. Pathol. 2003, 56, 287–291. [Google Scholar] [CrossRef]
- Olfat, F.O.; Zheng, Q.; Oleastro, M.; Voland, P.; Borén, T.; Karttunen, R.; Engstrand, L.; Rad, R.; Prinz, C.; Gerhard, M. Correlation of the Helicobacter pylori adherence factor BabA with duodenal ulcer disease in four European countries. Fems Immunol. Med. Microbiol. 2005, 44, 151–156. [Google Scholar] [CrossRef]
- Skoog, E.; Sjöling, Å.; Navabi, N.; Holgersson, J.; Lundin, S.B.; Lindén, S.K. Human gastric mucins differently regulate Helicobacter pylori proliferation, gene expression and interactions with host cells. PLoS One 2012, 7, e36378. [Google Scholar]
- Guruge, J.L.; Falk, P.G.; Lorenz, R.G.; Dans, M.; Wirth, H.P.; Blaser, M.J.; Berg, D.E.; Gordon, J.I. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 1998, 95, 3925–3930. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oleastro, M.; Ménard, A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology 2013, 2, 1110-1134. https://doi.org/10.3390/biology2031110
Oleastro M, Ménard A. The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology. 2013; 2(3):1110-1134. https://doi.org/10.3390/biology2031110
Chicago/Turabian StyleOleastro, Mónica, and Armelle Ménard. 2013. "The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis" Biology 2, no. 3: 1110-1134. https://doi.org/10.3390/biology2031110
APA StyleOleastro, M., & Ménard, A. (2013). The Role of Helicobacter pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology, 2(3), 1110-1134. https://doi.org/10.3390/biology2031110