
Phylogenetic Reassessment of Murinae Inferred from the Mitogenome of the Monotypic Genus Dacnomys Endemic to Southeast Asia: New Insights into Genetic Diversity Erosion


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
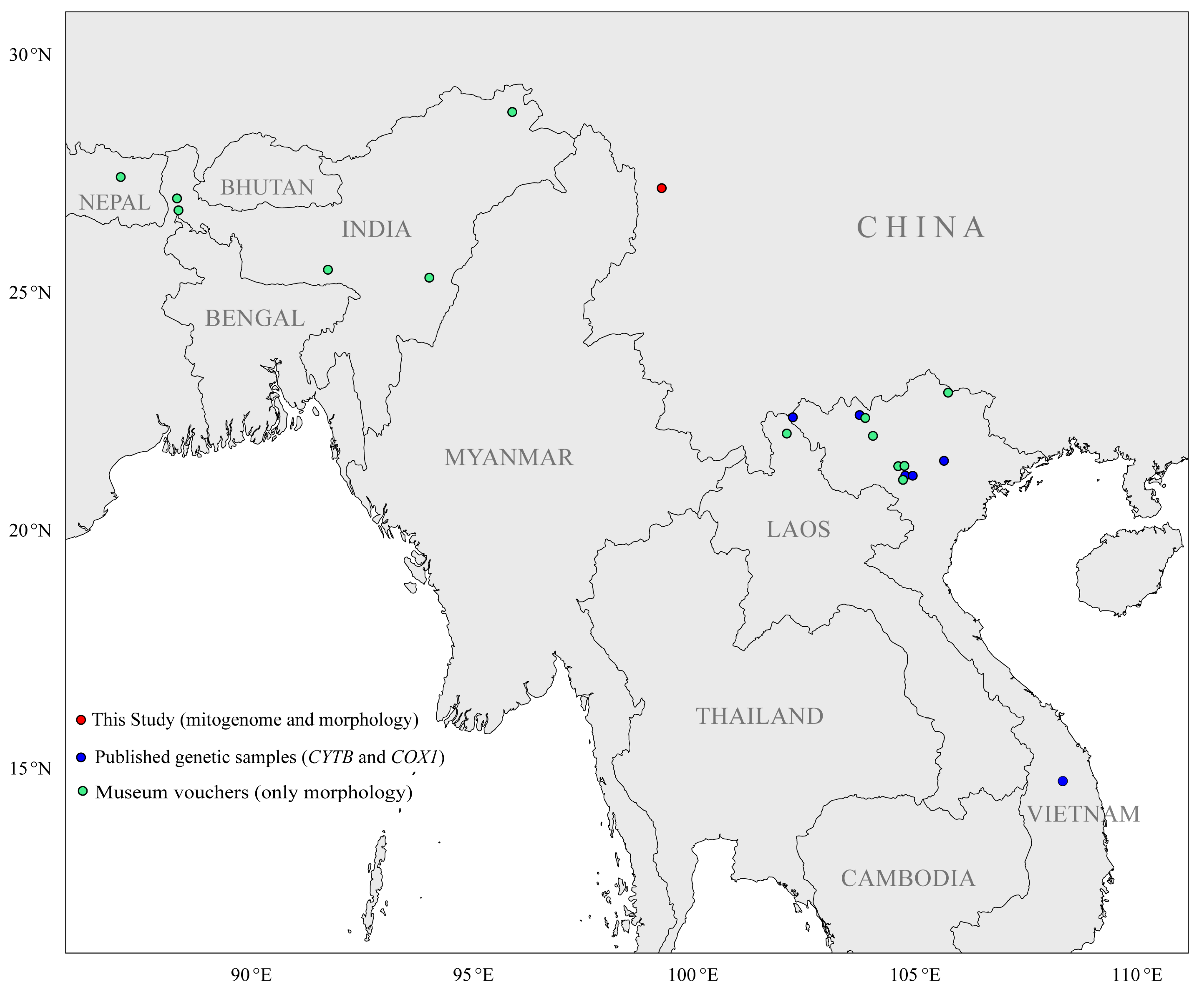
2.1. Specimen Collection and Identification, DNA Extraction and Sequencing
2.2. Sequence Assembly and Annotation
2.3. Sequence Analyses, Genetic Diversity and Morphological Visualization
2.4. Phylogenetic Reconstruction
2.5. Divergence Time Estimation
3. Results
3.1. Morphological Characteristics of D. millardi
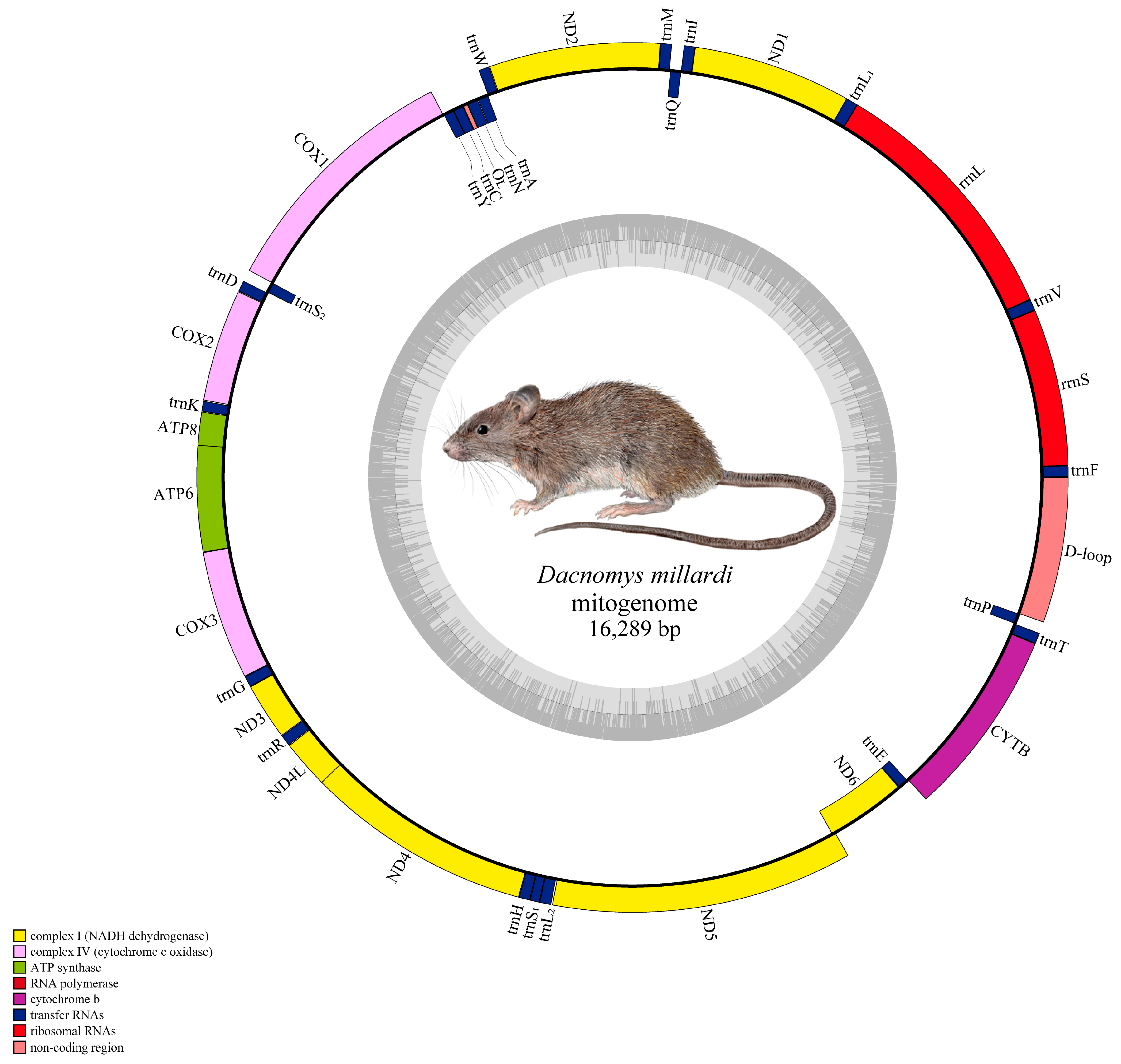
3.2. Mitogenome Architecture of D. millardi
3.3. tRNAs and rRNAs Structural Features
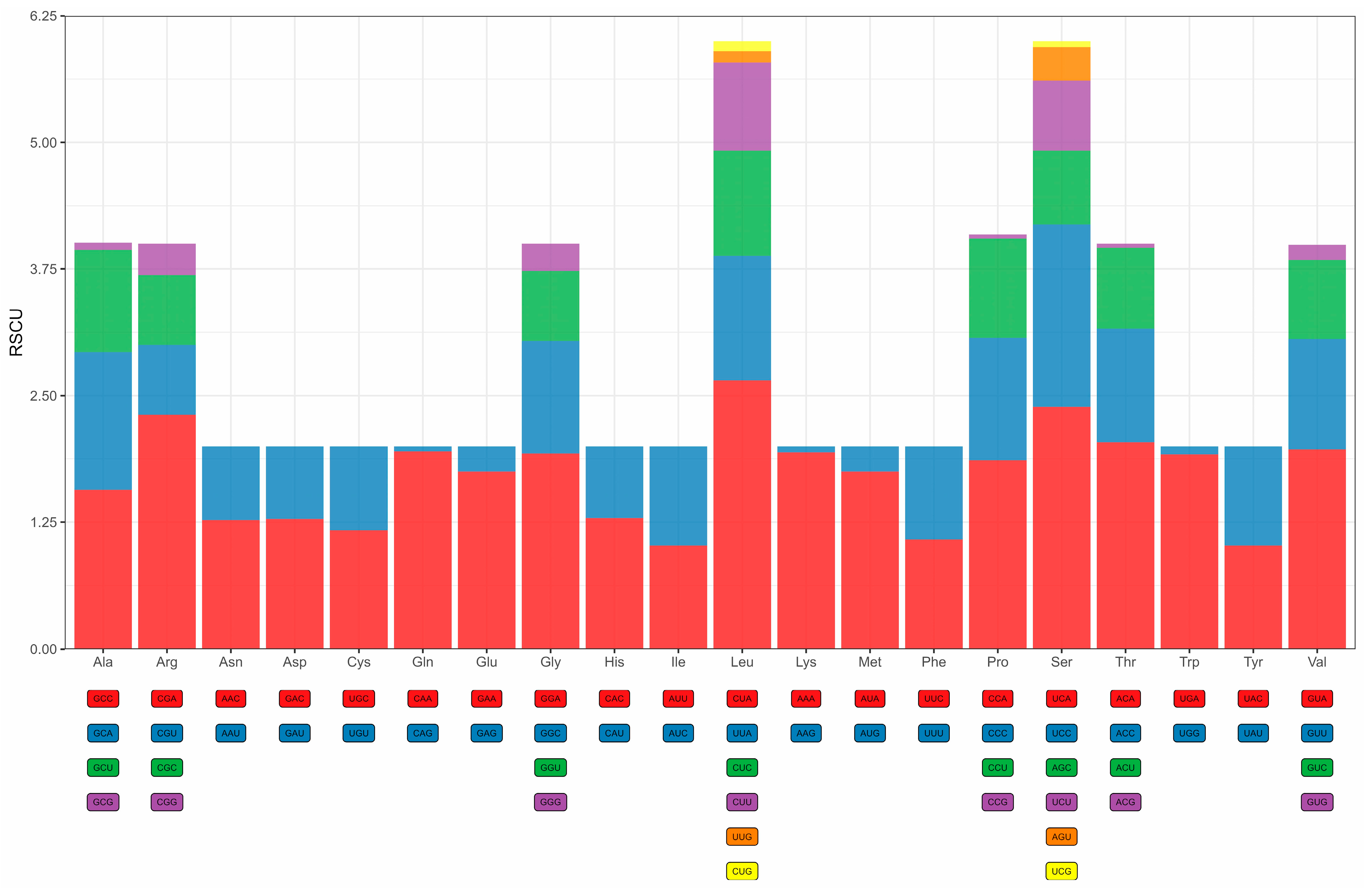
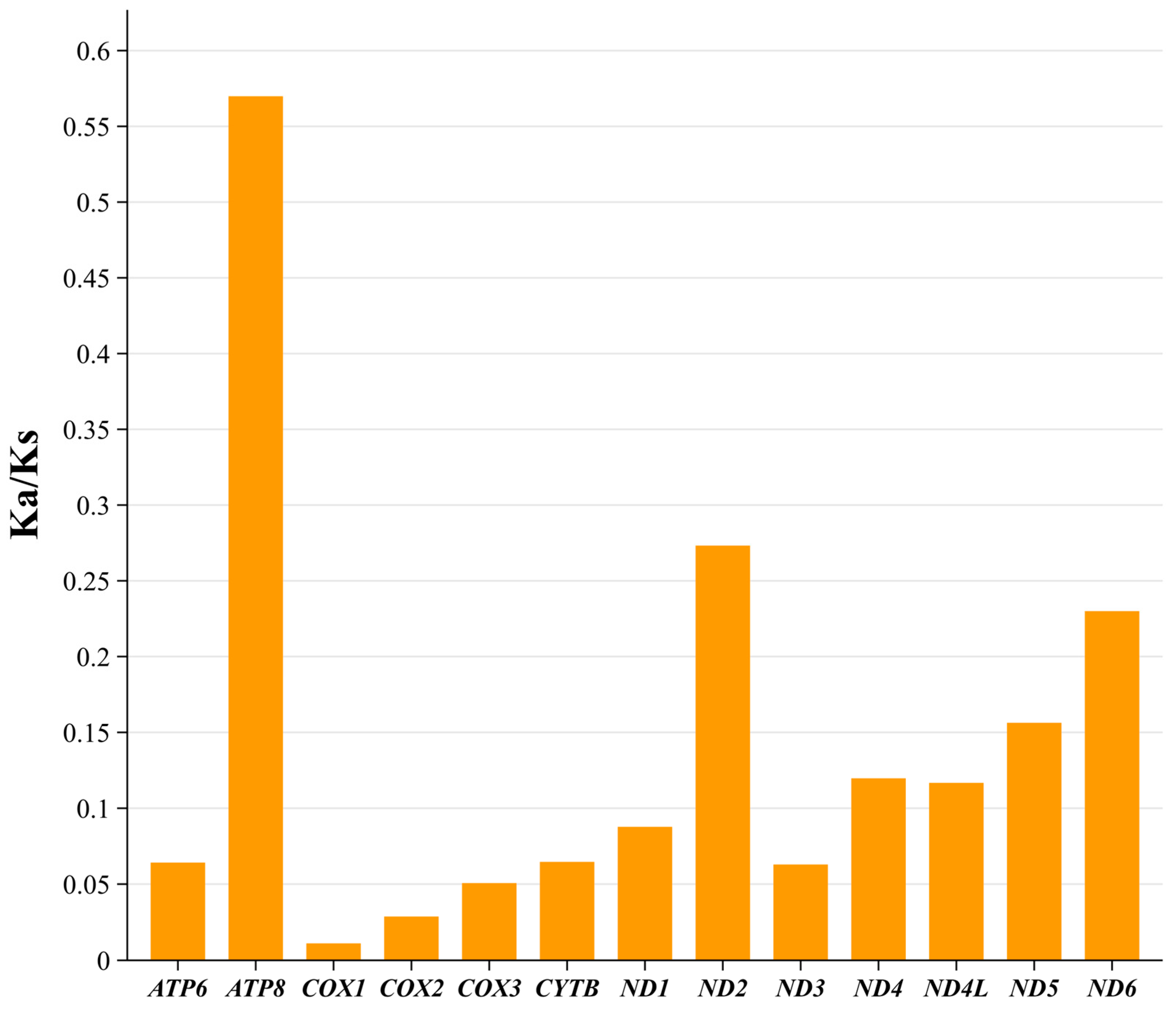
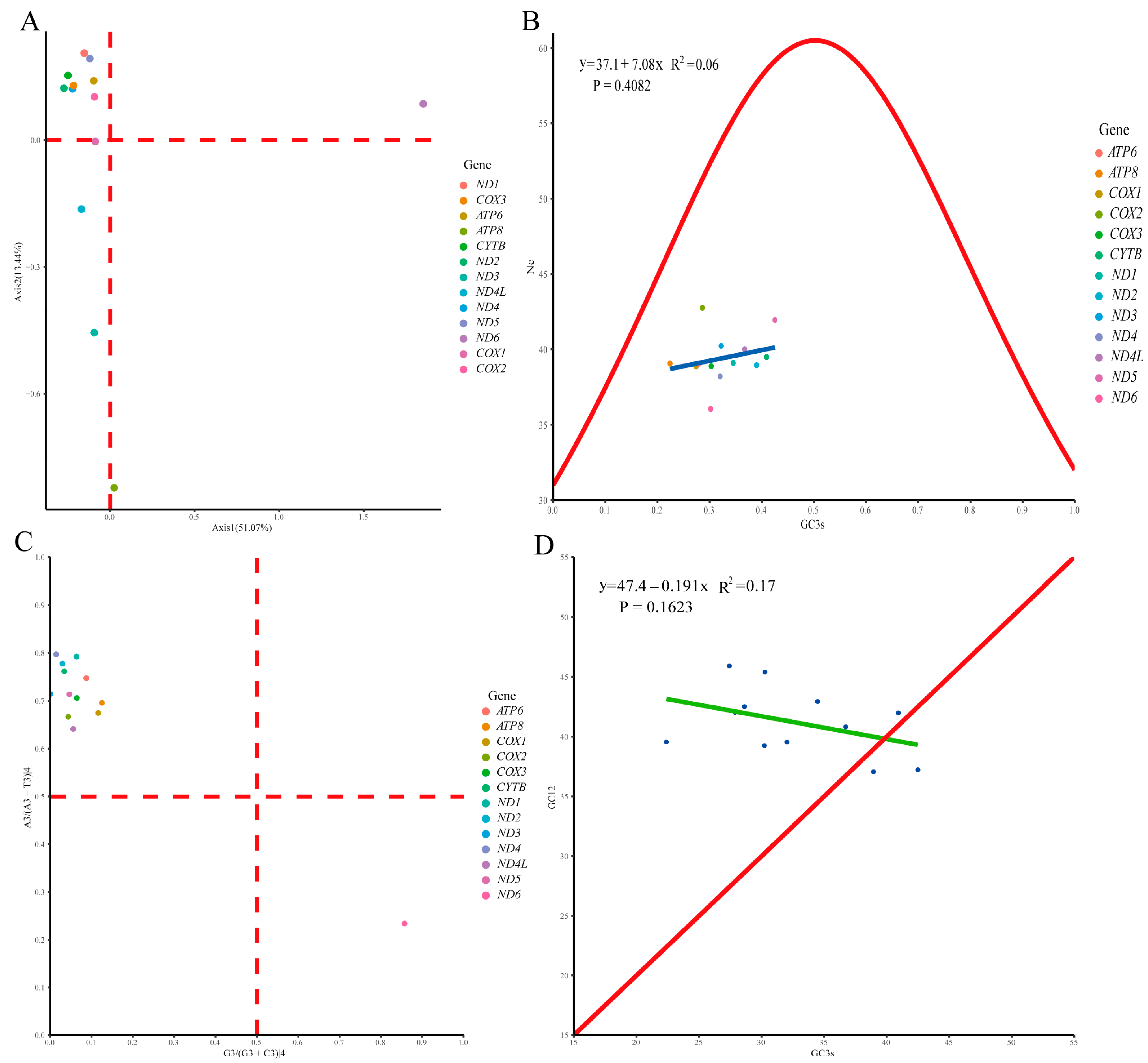
3.4. Protein-Coding Genes, Codon Usage, and Genetic Diversity
3.5. Phylogenetic Analysis and Divergence Time Estimation
4. Discussion
4.1. Evolutionary Conservation and Structural Features in the D. millardi Mitogenome
4.2. Codon Usage Bias and Evolutionary Constraints in the D. millardi Mitogenome
4.3. Phylogenetic Reconstruction and Divergence Time Estimation
4.4. Evolutionary Constraints and Anthropogenic Pressures Drive Genetic Diversity Erosion in the Threatened Murid D. millardi
- Habitats loss and geographic confinement synergistically drive genetic diversity erosion
- 2.
- Shallow evolutionary history
- 3.
- Ecological specialization and adaptive stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, D.E.; Lacher, T.E., Jr.; Mittermeier, R.A. Handbook of the Mammals of the World; Lynx Edicions: Barcelona, Spain, 2017; Volume 7. [Google Scholar]
- Abramov, A.V.; Balakirev, A.E.; Rozhnov, V.V. New data on the distribution and intraspecific variation of the Millard’s giant rat Dacnomys millardi (Mammalia, Rodentia) from Vietnam. Mammal Res. 2017, 62, 307–311. [Google Scholar] [CrossRef]
- Wei, F.W. Taxonomy and Distribution of Mammals in China; Science Press: Beijing, China, 2022. [Google Scholar]
- Pan, Q.H.; Wang, Y.X.; Yan, K. A Field Guild to the Mammals of China; Chinese Forestry Publishing Press: Beijing, China, 2007. [Google Scholar]
- Sunyer, P.; Muñoz, A.; Bonal, R.; Espelta, J.M.; McArthur, C. The ecology of seed dispersal by small rodents: A role for predator and conspecific scents. Funct. Ecol. 2013, 27, 1313–1321. [Google Scholar] [CrossRef]
- Jiang, Z.G. China’s Red List of Biodiversity: Vertebrates; Science Press: Beijing, China, 2021; Volume 1. [Google Scholar]
- Molur, S. Dacnomys millardi. The IUCN Red List of Threatened Species. Available online: https://dx.doi.org/10.2305/IUCN.UK.2016-1.RLTS.T6219A22466747.en (accessed on 4 June 2025).
- Rosa, I.M.D.; Smith, M.J.; Wearn, O.R.; Purves, D.; Ewers, R.M. The Environmental Legacy of Modern Tropical Deforestation. Curr. Biol. 2016, 26, 2161–2166. [Google Scholar] [CrossRef]
- Lynam, A.J.; Tantipisanuh, N.; Chutipong, W.; Ngoprasert, D.; Baker, M.C.; Cutter, P.; Gale, G.; Kitamura, S.; Steinmetz, R.; Sukmasuang, R.; et al. Comparative sensitivity to environmental variation and human disturbance of Asian tapirs (Tapirus indicus) and other wild ungulates in Thailand. Integr. Zool. 2012, 7, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Steppan, S.J.; Schenk, J.J. Muroid rodent phylogenetics: 900-species tree reveals increasing diversification rates. PLoS ONE 2017, 12, e0183070. [Google Scholar] [CrossRef]
- Abreu-Jr, E.F.; Pavan, S.E.; Tsuchiya, M.T.N.; Wilson, D.E.; Percequillo, A.R.; Maldonado, J.E. Museomics of tree squirrels: A dense taxon sampling of mitogenomes reveals hidden diversity, phenotypic convergence, and the need of a taxonomic overhaul. BMC Evol. Biol. 2020, 20, 77. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.L. Mitochondrial genome organization and vertebrate phylogenetics. Genet. Mol. Biol. 2000, 23, 745–752. [Google Scholar] [CrossRef]
- Ladoukakis, E.D.; Zouros, E. Evolution and inheritance of animal mitochondrial DNA: Rules and exceptions. J. Biol. Res. 2017, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.L.; Gretes, W.; Omland, K.E. Late Pleistocene divergence between eastern and western populations of wood ducks (Aix sponsa) inferred by the ‘isolation with migration’ coalescent method. Mol. Ecol. 2005, 14, 3407–3418. [Google Scholar] [CrossRef]
- Yue, H.; Yan, C.; Tu, F.; Yang, C.; Ma, W.; Fan, Z.; Song, Z.; Owens, J.; Liu, S.; Zhang, X. Two novel mitogenomes of Dipodidae species and phylogeny of Rodentia inferred from the complete mitogenomes. Biochem. Syst. Ecol. 2015, 60, 123–130. [Google Scholar] [CrossRef]
- Funk, D.J.; Omland, K.E. Species-Level Paraphyly and Polyphyly: Frequency, Causes, and Consequences, with Insights from Animal Mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 397–423. [Google Scholar] [CrossRef]
- Ding, L.; Li, W.; Liao, J. Characterization of the complete mitochondrial genome of Phodopus roborovskii (Rodentia: Cricetidae) and systematic implications for Cricetinae phylogenetics. Biochem. Syst. Ecol. 2016, 69, 226–235. [Google Scholar] [CrossRef]
- Frank, K.; Barta, E.; Bana, N.Á.; Nagy, J.; Horn, P.; Orosz, L.; Stéger, V. Complete mitochondrial genome sequence of a Hungarian red deer (Cervus elaphus hippelaphus) from high-throughput sequencing data and its phylogenetic position within the family Cervidae. Acta Biol. Hung. 2016, 67, 133–147. [Google Scholar] [CrossRef]
- Sarvani, R.K.; Parmar, D.R.; Tabasum, W.; Thota, N.; Sreenivas, A.; Gaur, A. Characterization of the complete mitogenome of Indian Mouse Deer, Moschiola indica (Artiodactyla: Tragulidae) and its evolutionary significance. Sci. Rep. 2018, 8, 2697. [Google Scholar] [CrossRef]
- Thalmann, O.; Shapiro, B.; Cui, P.; Schuenemann, V.J.; Sawyer, S.K.; Greenfield, D.L.; Germonpré, M.B.; Sablin, M.V.; López-Giráldez, F.; Domingo-Roura, X.; et al. Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs. Science 2013, 342, 871–874. [Google Scholar] [CrossRef]
- Waddell, P.J.; Cao, Y.; Hauf, J.; Hasegawa, M.; Olmstead, R. Using Novel Phylogenetic Methods to Evaluate Mammalian mtDNA, Including Amino Acid-Invariant Sites-LogDet plus Site Stripping, to Detect Internal Conflicts in the Data, with Special Reference to the Positions of Hedgehog, Armadillo, and Elephant. Syst. Biol. 1999, 48, 31–53. [Google Scholar] [CrossRef]
- Xu, X.; Janke, A.; Arnason, U. The complete mitochondrial DNA sequence of the greater Indian rhinoceros, Rhinoceros unicornis, and the Phylogenetic relationship among Carnivora, Perissodactyla, and Artiodactyla (+Cetacea). Mol. Biol. Evol. 1996, 13, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- İbiş, O.; Selçuk, A.Y.; Sacks, B.N.; Yıldız, B.; Özcan, S.; Kefelioğlu, H.; Tez, C. Whole mitochondrial genome of long-clawed mole vole (Prometheomys schaposchnikowi) from Turkey, with its phylogenetic relationships. Genomics 2020, 112, 3247–3255. [Google Scholar] [CrossRef]
- Yu, H.; Jamieson, A.; Hulme-Beaman, A.; Conroy, C.J.; Knight, B.; Speller, C.; Al-Jarah, H.; Eager, H.; Trinks, A.; Adikari, G.; et al. Palaeogenomic analysis of black rat (Rattus rattus) reveals multiple European introductions associated with human economic history. Nat. Commun. 2022, 13, 2399. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, S.; Wang, J.; Fu, C.; Wang, X.; Liu, S.; Chen, S. New insights into the mitogenomic phylogeny and evolutionary history of Murinae (Rodentia, Muridae) with the description of a new tribe. ZooKeys 2025, 1233, 55–74. [Google Scholar] [CrossRef]
- Barbosa, S.; Paupério, J.; Pavlova, S.V.; Alves, P.C.; Searle, J.B. The Microtus voles: Resolving the phylogeny of one of the most speciose mammalian genera using genomics. Mol. Phylogent. Evol. 2018, 125, 85–92. [Google Scholar] [CrossRef]
- Fenn, J.D.; Song, H.; Cameron, S.L.; Whiting, M.F. A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol. Phylogenet. Evol. 2008, 49, 59–68. [Google Scholar] [CrossRef]
- Jacobsen, M.W.; Hansen, M.M.; Orlando, L.; Bekkevold, D.; Bernatchez, L.; Willerslev, E.; Gilbert, M.T.P. Mitogenome sequencing reveals shallow evolutionary histories and recent divergence time between morphologically and ecologically distinct European whitefish (Coregonus spp.). Mol. Ecol. 2012, 21, 2727–2742. [Google Scholar] [CrossRef]
- Rubinoff, D.; Holland, B.S.; Savolainen, V. Between Two Extremes: Mitochondrial DNA is neither the Panacea nor the Nemesis of Phylogenetic and Taxonomic Inference. Syst. Biol. 2005, 54, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.T.; Xie, Y. A Guide to the Mammals of China; Princeton University Press: Princeton, NJ, USA, 2009. [Google Scholar]
- Balakirev, A.E.; Abramov, A.V.; Tikhonov, A.N.; Rozhnov, V.V. Molecular phylogeny of the Dacnomys division (Rodentia, Muridae): The taxonomic positions of Saxatilomys and Leopoldamys. Dokl. Biol. Sci. 2012, 445, 251–254. [Google Scholar] [CrossRef]
- He, K.; Li, Y.-J.; Brandley, M.C.; Lin, L.-K.; Wang, Y.-X.; Zhang, Y.-P.; Jiang, X.-L. A multi-locus phylogeny of Nectogalini shrews and influences of the paleoclimate on speciation and evolution. Mol. Phylogent. Evol. 2010, 56, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Modi, A.; Vai, S.; Caramelli, D.; Lari, M. The Illumina Sequencing Protocol and the NovaSeq 6000 System. Methods Mol. Biol. 2021, 2242, 15–42. [Google Scholar] [CrossRef]
- O’Halloran, D.M. fastQ_brew: Module for analysis, preprocessing, and reformatting of FASTQ sequence data. BMC Res. Notes 2017, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogent. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Steinke, D.; Nicolas, V.; Schaeffer, B.; Missoup, A.D.; Kennis, J.; Colyn, M.; Denys, C.; Tatard, C.; Cruaud, C.; Laredo, C. Assessment of Three Mitochondrial Genes (16S, Cytb, CO1) for Identifying Species in the Praomyini Tribe (Rodentia: Muridae). PLoS ONE 2012, 7, e36586. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Krutovsky, K.V.; Neale, D.B. Nucleotide Diversity and Linkage Disequilibrium in Cold-Hardiness- and Wood Quality-Related Candidate Genes in Douglas Fir. Genetics 2005, 171, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S.; Battistuzzi, F.U. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Perna, N.; Kocher, T. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.F.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Patil, S.S.; Indrabalan, U.B.; Suresh, K.P.; Shome, B.R. Analysis of codon usage bias of classical swine fever virus. Vet. World 2021, 14, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1996, 42, 323. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogent. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X.; Kumar, S. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Prlic, A.; Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comp. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Aghová, T.; Kimura, Y.; Bryja, J.; Dobigny, G.; Granjon, L.; Kergoat, G.J. Fossils know it best: Using a new set of fossil calibrations to improve the temporal phylogenetic framework of murid rodents (Rodentia: Muridae). Mol. Phylogent. Evol. 2018, 128, 98–111. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A.; Susko, E. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Gelman, A.; Rubin, D.B. Inference from Iterative Simulation Using Multiple Sequences. Stat. Sci. 1992, 7, 457–472. [Google Scholar] [CrossRef]
- Alekseyenko, A.V.; Chen, S.-C.; Ogata, A. MixtureTree Annotator: A Program for Automatic Colorization and Visual Annotation of MixtureTree. PLoS ONE 2015, 10, e0118893. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Rannala, B.; Buckley, T. Frequentist Properties of Bayesian Posterior Probabilities of Phylogenetic Trees Under Simple and Complex Substitution Models. Syst. Biol. 2004, 53, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.Q.; Wang, Y.-Y.; Yang, J.-H.; Lehmann, T.; Le, M.D.; Ziegler, T.; Bonkowski, M. A new species of the Gekko japonicus group (Squamata: Sauria: Gekkonidae) from the border region between China and Vietnam. Zootaxa 2013, 3652, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Prada, C.F.; Boore, J.L. Gene annotation errors are common in the mammalian mitochondrial genomes database. BMC Genom. 2019, 20, 73. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Gissi, C.; Pesole, G.; Saccone, C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 1998, 15, 957–966. [Google Scholar] [CrossRef]
- Zardoya, R.; Garrido-Pertierra, A.; Bautista, J.M. The complete nucleotide sequence of the mitochondrial DNA genome of the rainbow trout, Oncorhynchus mykiss. J. Mol. Evol. 1995, 41, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Ding, L.; Luo, G.; Zhou, Q.; Sun, Y.; Liao, J. Comparative Mitogenome Analysis of Gerbils and the Mitogenome Phylogeny of Gerbillinae (Rodentia: Muridae). Biochem. Genet. 2022, 60, 2226–2249. [Google Scholar] [CrossRef]
- Jiang, X.; Gao, J.; Ni, L.; Hu, J.; Li, K.; Sun, F.; Xie, J.; Bo, X.; Gao, C.; Xiao, J.; et al. The complete mitochondrial genome of Microtus fortis calamorum (Arvicolinae, Rodentia) and its phylogenetic analysis. Gene 2012, 498, 288–295. [Google Scholar] [CrossRef]
- Wu, D.; Zhou, L.; Xue, J.; Xia, Q.; Meng, L. Characterization of Two New Apodemus Mitogenomes (Rodentia: Muridae) and Mitochondrial Phylogeny of Muridae. Diversity 2022, 14, 1089. [Google Scholar] [CrossRef]
- Petersheim, M.; Turner, D.H. Base-stacking and base-pairing contributions to helix stability: Thermodynamics of double-helix formation with CCGG, CCGGp, CCGGAp, ACCGGp, CCGGUp, and ACCGGUp. Biochemistry 1983, 22, 256–263. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Marková, S.; Filipi, K.; Searle, J.B.; Kotlík, P. Mapping 3′ transcript ends in the bank vole (Clethrionomys glareolus) mitochondrial genome with RNA-Seq. BMC Genom. 2015, 16, 870. [Google Scholar] [CrossRef]
- Dorner, M.; Altmann, M.; Paabo, S.; Morl, M. Evidence for import of a lysyl-tRNA into marsupial mitochondria. Mol. Biol. Cell 2001, 12, 2688–2698. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Li, W.; Yuan, B.; Dong, W. The complete mitochondrial genome of Echinolaelaps fukienensis provide insights into phylogeny and rearrangement in the superfamily Dermanyssoidea. PLoS ONE 2023, 18, e0288991. [Google Scholar] [CrossRef]
- Lin, X.; Pu, J.; Dong, W. The first mitogenome of the subfamily Stenoponiinae (Siphonaptera: Ctenophthalmidae) and implications for its phylogenetic position. Sci. Rep. 2024, 14, 18179. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, D.; Ma, G.; Liu, M.; Wang, M.; Jia, R.; Chen, S.; Sun, K.; Yang, Q.; Wu, Y.; et al. Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Riemerella anatipestifer. Int. J. Mol. Sci. 2016, 17, 1304. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Yang, Z.H.; Ren, T.G.; Dong, W.G. The complete mitochondrial genome of Eulaelaps huzhuensis (Mesostigmata: Haemogamasidae). Exp. Appl. Acarol. 2023, 90, 301–316. [Google Scholar] [CrossRef]
- Petrov, D.A.; Charneski, C.A.; Honti, F.; Bryant, J.M.; Hurst, L.D.; Feil, E.J. Atypical AT Skew in Firmicute Genomes Results from Selection and Not from Mutation. PLoS Genet. 2011, 7, e1002283. [Google Scholar] [CrossRef]
- Dorus, S.; Busby, S.A.; Gerike, U.; Shabanowitz, J.; Hunt, D.F.; Karr, T.L. Genomic and functional evolution of the Drosophila melanogaster sperm proteome. Nat. Genet. 2006, 38, 1440–1445. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486. [Google Scholar] [CrossRef]
- Chen, W.; Qian, W.; Miao, K.; Qian, R.; Yuan, S.; Liu, W.; Dai, J.; Hu, C.; Chang, Q. Comparative Mitogenomics of True Frogs (Ranidae, Anura), and Its Implications for the Phylogeny and Evolutionary History of Rana. Animals 2022, 12, 1250. [Google Scholar] [CrossRef]
- Yang, H.; Li, T.; Dang, K.; Bu, W. Compositional and mutational rate heterogeneity in mitochondrial genomes and its effect on the phylogenetic inferences of Cimicomorpha (Hemiptera: Heteroptera). BMC Genom. 2018, 19, 264. [Google Scholar] [CrossRef]
- Yue, B.-S.; Hu, C.; Zhang, C.; Sun, L.; Zhang, Y.; Xie, W.; Zhang, B.; Chang, Q. The mitochondrial genome of pin-tailed snipe Gallinago stenura, and its implications for the phylogeny of Charadriiformes. PLoS ONE 2017, 12, e0175244. [Google Scholar] [CrossRef]
- Zheng, C.G.; Zhu, X.X.; Yan, L.P.; Yao, Y.; Bu, W.J.; Wang, X.H.; Lin, X.L. First complete mitogenomes of Diamesinae, Orthocladiinae, Prodiamesinae, Tanypodinae (Diptera: Chironomidae) and their implication in phylogenetics. PeerJ 2021, 9, e11294. [Google Scholar] [CrossRef]
- Kretzschmar, C.; Roolf, C.; Timmer, K.; Sekora, A.; Knubel, G.; Murua Escobar, H.; Fuellen, G.; Ibrahim, S.M.; Tiedge, M.; Baltrusch, S.; et al. Polymorphisms of the murine mitochondrial ND4, CYTB and COX3 genes impact hematopoiesis during aging. Oncotarget 2016, 7, 74460–74472. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, X.; Ting, N.; Zhang, Y. Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 2011, 11, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tsuchiya, K.; Takezaki, N. A Molecular Phylogenetic Framework for the Ryukyu Endemic Rodents Tokudaia osimensis and Diplothrix legata. Mol. Phylogent. Evol. 2000, 15, 15–24. [Google Scholar] [CrossRef]
- Martin, Y.; Gerlach, G.; Schlötterer, C.; Meyer, A. Molecular Phylogeny of European Muroid Rodents Based on Complete Cytochrome b Sequences. Mol. Phylogent. Evol. 2000, 16, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Song, X.; Yan, C.; Lu, T.; Zhang, X.; Yue, B. Phylogenetic analyses of the harvest mouse, Micromys minutus (Rodentia: Muridae) based on the complete mitogenome sequences. Biochem. Syst. Ecol. 2015, 62, 121–127. [Google Scholar] [CrossRef]
- Minwer-Barakat, R.; García-Alix, A.; Martín-Suárez, E.; Freudenthal, M. Micromys caesaris, a new murid (Rodentia, Mammalia) from the Late Pliocene of the Guadix Basin, southeastern Spain. J. Paleontol. 2016, 82, 436–441. [Google Scholar] [CrossRef]
- Fabre, P.-H.; Upham, N.S.; Emmons, L.H.; Justy, F.; Leite, Y.L.R.; Loss, A.C.; Orlando, L.; Tilak, M.-K.; Patterson, B.D.; Douzery, E.J.P. Mitogenomic phylogeny, diversification, and biogeography of South American spiny rats. Mol. Biol. Evol. 2016, 34, 613–633. [Google Scholar] [CrossRef]
- Tonini, J.; Moore, A.; Stern, D.; Shcheglovitova, M.; Ortí, G. Concatenation and Species Tree Methods Exhibit Statistically Indistinguishable Accuracy under a Range of Simulated conditions. PLoS Curr. 2015, 7, 1. [Google Scholar] [CrossRef]
- Pagès, M.; Fabre, P.H.; Chaval, Y.; Mortelliti, A.; Nicolas, V.; Wells, K.; Michaux, J.R.; Lazzari, V. Molecular phylogeny of South-East Asian arboreal murine rodents. Zool. Scr. 2015, 45, 349–364. [Google Scholar] [CrossRef]
- Peters, J.L.; Zhuravlev, Y.N.; Fefelov, I.; Humphries, E.M.; Omland, K.E. Multilocus Phylogeography of a Holarctic Duck: Colonization of North America from Eurasia by Gadwall (Anas Strepera). Evolution 2008, 62, 1469–1483. [Google Scholar] [CrossRef]
- Shen, Y.; Gong, Y.; Solovyeva, D.; Wang, L.; Li, M.; Hu, M.; Jiang, Y.; Vartanyan, S.; Wang, H. Genetic diversity and shallow genetic differentiation of the endangered scaly-sided merganser Mergus squamatus. Ecol. Evol. 2024, 14, e70011. [Google Scholar] [CrossRef]
- Brook, B.W.; Sodhi, N.S.; Ng, P.K.L. Catastrophic extinctions follow deforestation in Singapore. Nature 2003, 424, 420–423. [Google Scholar] [CrossRef]
- Zang, R.; Connette, G.M.; Oswald, P.; Thura, M.K.; LaJeunesse Connette, K.J.; Grindley, M.E.; Songer, M.; Zug, G.R.; Mulcahy, D.G. Rapid forest clearing in a Myanmar proposed national park threatens two newly discovered species of geckos (Gekkonidae: Cyrtodactylus). PLoS ONE 2017, 12, e0174432. [Google Scholar] [CrossRef]
- Frankham, R. Relationship of Genetic Variation to Population Size in Wildlife. Conserv. Biol. 2002, 10, 1500–1508. [Google Scholar] [CrossRef]
- Soule, M. Allozyme variation: Its determinants in space and time. Mol. Evol. 1976, 60–77. [Google Scholar]
- Knapp, M.; Latinne, A.; Waengsothorn, S.; Rojanadilok, P.; Eiamampai, K.; Sribuarod, K.; Michaux, J.R. Combined Mitochondrial and Nuclear Markers Revealed a Deep Vicariant History for Leopoldamys neilli, a Cave-Dwelling Rodent of Thailand. PLoS ONE 2012, 7, e47670. [Google Scholar] [CrossRef]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C. Defining ‘Evolutionarily Significant Units’ for conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, Y.; Zhao, J.; Yao, M. Low genetic diversity in a critically endangered primate: Shallow evolutionary history or recent population bottleneck? BMC Evol. Biol. 2019, 19, 134. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: London, UK, 2000. [Google Scholar]
- Losos, J. Lizards in an Evolutionary Tree: Ecology and Adaptive Radiation of Anoles; University of California Press: Oakland, CA, USA, 2009. [Google Scholar]
- Nevo, E. Adaptive Convergence and Divergence of Subterranean Mammals. Annu. Rev. Ecol. Syst. 1979, 10, 269–308. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Position (bp) | Size (bp) | Start Codon | Stop Codon | Anticodon | Intergenic Nucleotides |
|---|---|---|---|---|---|---|---|
| trnF | H | 1–69 | 69 | GAA | 0 | ||
| rrnS | H | 70–1023 | 954 | 0 | |||
| trnV | H | 1024–1092 | 69 | UAC | 0 | ||
| rrnL | H | 1093–2668 | 1576 | 0 | |||
| trnL1 | H | 2667–2741 | 75 | UAG | −2 | ||
| ND1 | H | 2739–3698 | 960 | ATA | TAG | −3 | |
| trnI | H | 3697–3765 | 69 | GAU | −2 | ||
| trnQ | L | 3763–3833 | 71 | UUG | −3 | ||
| trnM | H | 3837–3905 | 69 | CAU | 3 | ||
| ND2 | H | 3906–4940 | 1035 | ATT | AGA | 0 | |
| trnW | H | 4942–5007 | 66 | UCA | 1 | ||
| trnA | L | 5009–5077 | 69 | UGC | 1 | ||
| trnN | L | 5079–5149 | 71 | GUU | 1 | ||
| OL | L | 5150–5180 | 31 | 0 | |||
| trnC | L | 5181–5247 | 67 | GCA | 0 | ||
| trnY | L | 5248–5314 | 67 | GUA | 0 | ||
| COX1 | H | 5316–6860 | 1545 | ATG | TAA | 1 | |
| trnS2 | L | 6858–6926 | 69 | UGA | −3 | ||
| trnD | H | 6930–6998 | 69 | GUC | 3 | ||
| COX2 | H | 7000–7683 | 684 | ATG | TAA | 1 | |
| trnK | H | 7687–7750 | 64 | UUU | 3 | ||
| ATP8 | H | 7752–7955 | 204 | ATG | TAA | 1 | |
| ATP6 | H | 7913–8593 | 681 | ATG | TAA | −43 | |
| COX3 | H | 8593–9376 | 784 | ATG | T-- | −1 | |
| trnG | H | 9377–9444 | 68 | UCC | 0 | ||
| ND3 | H | 9445–9792 | 348 | ATC | TAA | 0 | |
| trnR | H | 9794–9862 | 69 | UCG | 1 | ||
| ND4L | H | 9865–10,161 | 297 | ATG | TAA | 2 | |
| ND4 | H | 10,155–11,532 | 1378 | ATG | T-- | −7 | |
| trnH | H | 11,533–11,600 | 68 | GUG | 0 | ||
| trnS1 | H | 11,601–11,659 | 59 | GCU | 0 | ||
| trnL2 | H | 11,659–11,729 | 71 | UAA | −1 | ||
| ND5 | H | 11,736–13,559 | 1824 | ATA | TAA | 6 | |
| ND6 | L | 13,537–14,055 | 519 | ATG | TAA | −23 | |
| trnE | L | 14,056–14,124 | 69 | UUC | 0 | ||
| CYTB | H | 14,130–15,273 | 1144 | ATG | T-- | 5 | |
| trnT | H | 15,274–15,340 | 67 | UGU | 0 | ||
| trnP | L | 15,341–15,408 | 68 | UGG | 0 | ||
| D-loop | H | 15,409–16,289 | 881 | 0 |
| Dacnomys millardi | Size (bp) | A% | T% | G% | C% | A + T% | G + C% | AT Skew | GC Skew |
|---|---|---|---|---|---|---|---|---|---|
| Mitogenome | 16,289 | 33.9 | 28.1 | 12.1 | 25.9 | 62.0 | 38.0 | 0.094 | −0.363 |
| PCGs | 11,400 | 31.6 | 29.7 | 11.7 | 27.0 | 61.3 | 38.7 | 0.031 | −0.396 |
| rRNAs | 2530 | 38.2 | 24.9 | 16.6 | 20.3 | 63.1 | 36.9 | 0.211 | −0.099 |
| tRNAs | 1503 | 34.4 | 31.3 | 17.8 | 16.6 | 65.7 | 34.4 | 0.048 | 0.035 |
| D-loop | 881 | 33.6 | 29.3 | 11.6 | 25.5 | 62.9 | 37.1 | 0.068 | −0.375 |
| OL | 31 | 25.8 | 16.1 | 29.0 | 29.1 | 41.9 | 58.1 | 0.232 | −0.002 |
| Codon | No. | RSCU | Codon | No. | RSCU | Codon | No. | RSCU | Codon | No. | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 107 | 0.92 | GUG(V) | 6 | 0.15 | CAU(H) | 37 | 0.71 | UGG(W) | 4 | 0.08 |
| UUC(F) | 126 | 1.08 | CCU(P) | 44 | 0.98 | CAC(H) | 67 | 1.29 | CGU(R) | 11 | 0.69 |
| UUA(L) | 122 | 1.23 | CCC(P) | 60 | 1.21 | CAA(Q) | 80 | 1.95 | CGC(R) | 11 | 0.69 |
| UUG(L) | 11 | 0.11 | CCA(P) | 92 | 1.86 | CAG(Q) | 2 | 0.05 | CGA(R) | 37 | 2.31 |
| CUU(L) | 86 | 0.87 | CCG(P) | 2 | 0.04 | AAU(N) | 60 | 0.73 | CGG(R) | 5 | 0.31 |
| CUC(L) | 103 | 1.04 | ACU(T) | 60 | 0.80 | AAC(N) | 104 | 1.27 | GGU(G) | 39 | 0.69 |
| CUA(L) | 263 | 2.65 | ACC(T) | 85 | 1.12 | AAA(K) | 96 | 1.94 | GGC(G) | 58 | 1.11 |
| CUG(L) | 10 | 0.10 | ACA(T) | 155 | 2.04 | AAG(K) | 3 | 0.06 | GGA(G) | 101 | 1.93 |
| AUU(I) | 201 | 1.02 | ACG(T) | 3 | 0.04 | GAU(D) | 26 | 0.72 | GGG(G) | 14 | 0.27 |
| AUC(I) | 195 | 0.98 | GCU(A) | 58 | 1.01 | GAC(D) | 46 | 1.28 | UCU(S) | 35 | 0.69 |
| AUA(M) | 193 | 1.75 | GCC(A) | 90 | 1.57 | GAA(E) | 84 | 1.75 | UCC(S) | 92 | 1.80 |
| AUG(M) | 28 | 0.25 | GCA(A) | 78 | 1.36 | GAG(E) | 12 | 0.25 | UCA(S) | 122 | 2.39 |
| GUU(V) | 43 | 1.09 | GCG(A) | 4 | 0.07 | UGU(C) | 12 | 0.83 | UCG(S) | 3 | 0.06 |
| GUC(V) | 31 | 0.78 | UAU(Y) | 62 | 0.98 | UGC(C) | 17 | 1.17 | AGU(S) | 17 | 0.33 |
| GUA(V) | 78 | 1.97 | UAC(Y) | 64 | 1.02 | UGA(W) | 100 | 1.92 | AGC(S) | 37 | 0.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Zhao, D.; Song, W.; Dong, W. Phylogenetic Reassessment of Murinae Inferred from the Mitogenome of the Monotypic Genus Dacnomys Endemic to Southeast Asia: New Insights into Genetic Diversity Erosion. Biology 2025, 14, 948. https://doi.org/10.3390/biology14080948
Wang Z, Zhao D, Song W, Dong W. Phylogenetic Reassessment of Murinae Inferred from the Mitogenome of the Monotypic Genus Dacnomys Endemic to Southeast Asia: New Insights into Genetic Diversity Erosion. Biology. 2025; 14(8):948. https://doi.org/10.3390/biology14080948
Chicago/Turabian StyleWang, Zhongsong, Di Zhao, Wenyu Song, and Wenge Dong. 2025. "Phylogenetic Reassessment of Murinae Inferred from the Mitogenome of the Monotypic Genus Dacnomys Endemic to Southeast Asia: New Insights into Genetic Diversity Erosion" Biology 14, no. 8: 948. https://doi.org/10.3390/biology14080948
APA StyleWang, Z., Zhao, D., Song, W., & Dong, W. (2025). Phylogenetic Reassessment of Murinae Inferred from the Mitogenome of the Monotypic Genus Dacnomys Endemic to Southeast Asia: New Insights into Genetic Diversity Erosion. Biology, 14(8), 948. https://doi.org/10.3390/biology14080948