
Full-Length Transcriptome Sequencing and hsp Gene Family Analysis Provide New Insights into the Stress Response Mechanisms of Mystus guttatus

 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Total RNA Extraction
2.3. PacBio Library Construction and Sequencing
2.4. SMRT Sequencing Data Processing
2.5. Full-Length Transcriptome Annotations Analysis
2.6. Basic Information About the HSP Family
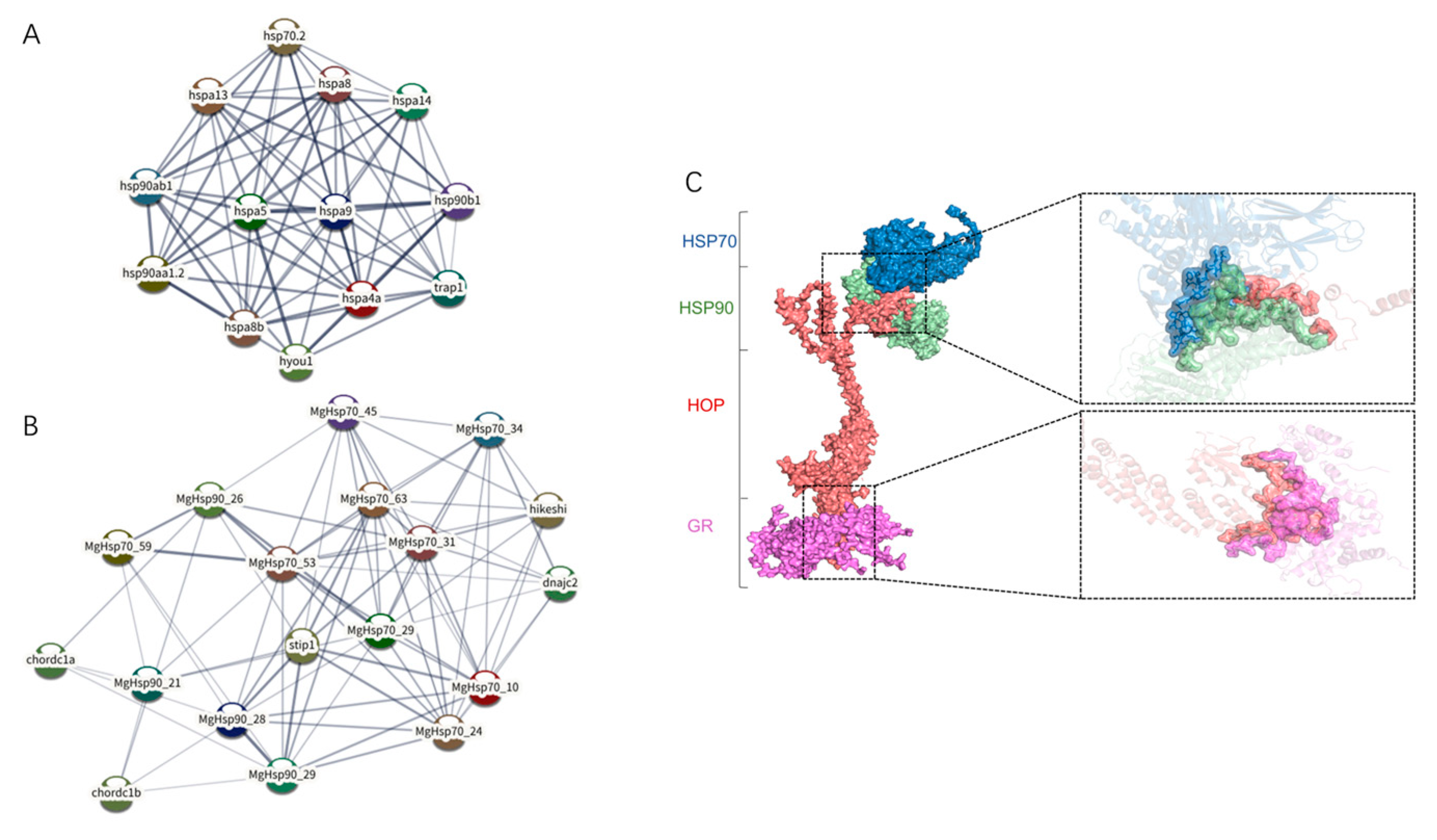
2.7. Interaction Mechanism Analysis of the HSP Family
2.8. Evolutionary and Motif Analysis of hsp Gene Family
3. Results
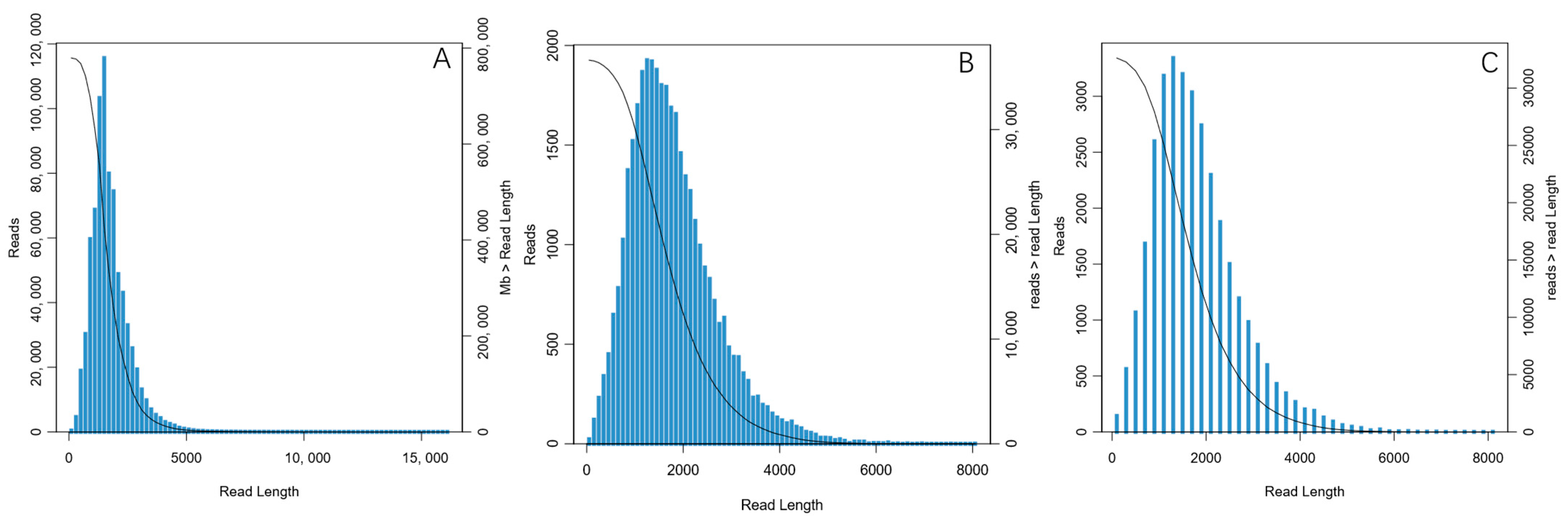
3.1. Full-Length Transcriptome Sequencing Data
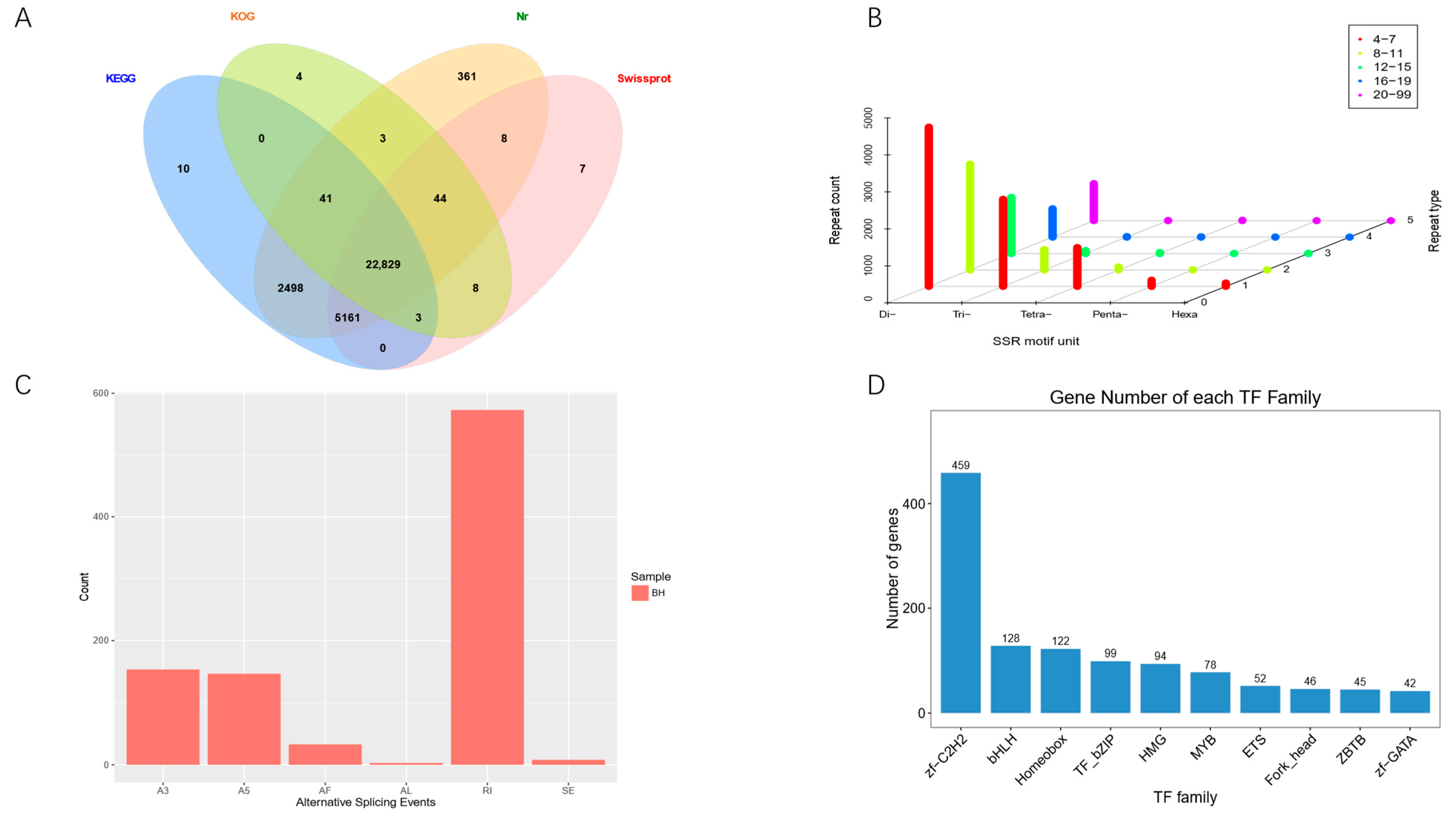
3.2. Annotation and Analysis of Full-Length Transcriptome
3.3. HSP70 and HSP90 Identification and Subcellular Localization
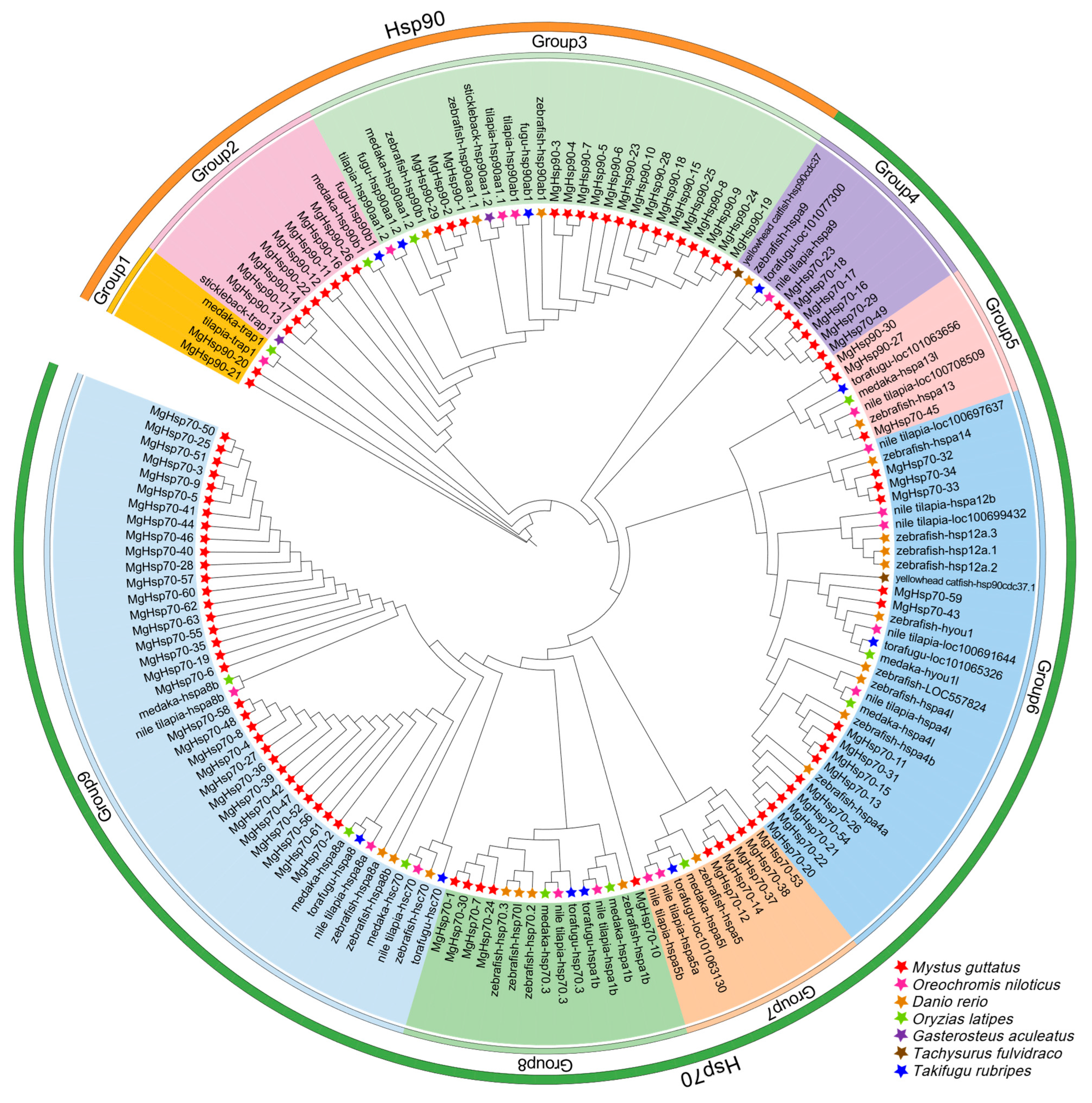
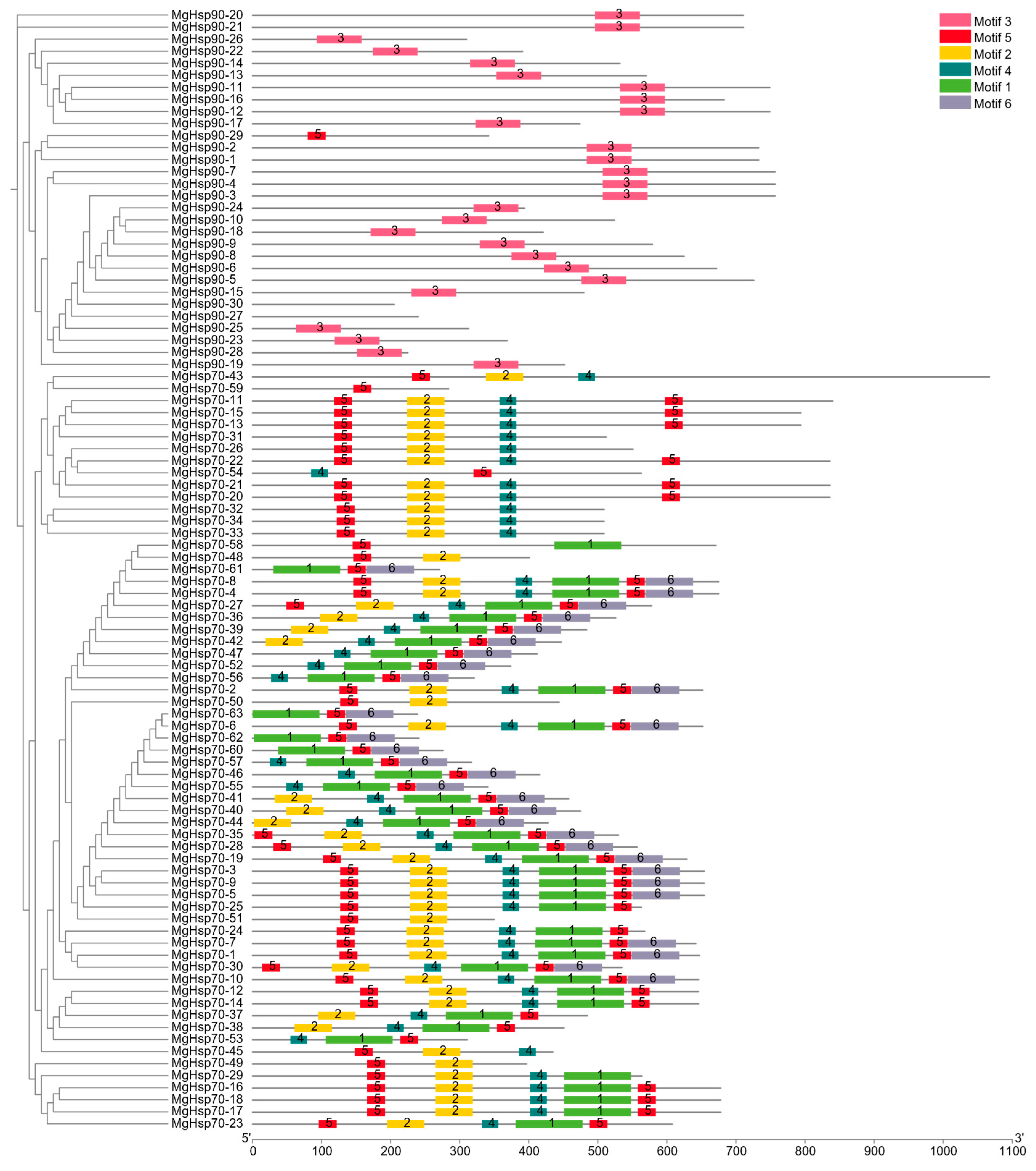
3.4. HSP Family Phylogeny and Motif Analysis
3.5. Interaction Mechanism Analysis of HSP Family
3.6. Selective Pressure Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.C. Study on the Age and Growth of Mystus guttatus in the Pearl River. Chin. Aquat. Sci. 1999, 4, 62–66. [Google Scholar]
- Wu, X.; Zhou, L.; Peng, F. Analysis of Meat Content and Muscle Nutrient Composition of Spotted Cattle. J. Fish. Sci. 2010, 29, 425–428. [Google Scholar] [CrossRef]
- Chen, W.; Xiang, D.; Gao, S.; Zhu, S.; Wu, Z.; Li, Y.; Li, J. Whole-genome resequencing confirms the genetic effects of dams on an endangered fish Hemibagrus guttatus (Siluriformes: Bagridae): A case study in a tributary of the Pearl River. Gene 2024, 895, 148000. [Google Scholar] [CrossRef] [PubMed]
- Kuang, T.; Shuai, F.; Li, X.; Chen, W.; Lek, S. Genetic diversity and population structure of Hemibagrus guttatus (Bagridae, Siluriformes) in the larger subtropical Pearl River based on COI and Cyt b genes analysis. Ann. De Limnol. Int. J. Limnol. 2021, 57, 7. [Google Scholar] [CrossRef]
- Yang, S.; Wang, Y.; Zheng, N.; Wang, Z.; Yu, K.; Ye, H.; Zhang, X.; Shao, J. Study on the testis and sperm structure of Mystus guttatus. Chin. J. Fish. 2025, 38, 43–50+81. Available online: https://link.cnki.net/urlid/23.1363.S.20240521.2005.004 (accessed on 10 December 2024).
- Xie, F. The gonadal development and histological observation of cultured zebrafish. J. Dalian Ocean. Univ. 2014, 29, 147–150. [Google Scholar]
- Li, F. The growth, reproduction, feeding habits, and aquaculture techniques of Mystus guttatus. Hubei Agric. Sci. 2006, 6, 807–809. [Google Scholar]
- Xu, S. Artificial breeding technology of Mystus guttatus. Freshw. Fish. 2001, 2, 21–22. [Google Scholar]
- Chang, J.; Wang, S.; Meng, Q.; Meng, X.; Yang, M. Artificial Reproduction Techniques of Spotted Flycatcher in Southwest China. J. Heilongjiang Aquac. 2024, 43, 236–239. [Google Scholar]
- Abd El-Hack, M.E.; El-Saadony, M.T.; Nader, M.M.; Salem, H.M.; El-Tahan, A.M.; Soliman, S.M.; Khafaga, A.F. Effect of environmental factors on growth performance of Nile tilapia (Oreochromis niloticus). Int. J. Biometeorol. 2022, 66, 2183–2194. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Leebens-Mack, J.H.; Barker, M.S.; Carpenter, E.J.; Deyholos, M.K.; Gitzendanner, M.A.; Graham, S.W.; Grosse, I.; Li, Z.; Melkonian, M.; Mirarab, S.; et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef]
- Gupta, R.S. Phylogenetic Analysis of the 90 kd Heat-Shock Family of Protein Sequences and an Examination of the Relationship Among Animals, Plants, and Fungi Species. Mol. Biol. Evol. 1995, 12, 1063–1073. [Google Scholar]
- Johnston, I.A.; Kent, M.P.; Boudinot, P.; Looseley, M.; Bargelloni, L.; Faggion, S.; Merino, G.A.; Ilsley, G.R.; Bobe, J.; Tsigenopoulos, C.S.; et al. Advancing fish breeding in aquaculture through genome functional annotation. Aquaculture 2024, 583, 740589. [Google Scholar] [CrossRef]
- Tiwari, S.R. Knowledge Integration in Government–Industry Project Network. Knowl. Process Manag. 2015, 22, 11–21. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; RÖTh, S.; Schleiff, E.; Scharf, K.-D. Prospects of engineering thermotolerance in crops through modulation of heat stress transcription factor and heat shock protein networks. Plant Cell Environ. 2015, 38, 1881–1895. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Zhang, R.; Duan, Q.; Luo, Q.; Deng, L. PacBio Full-Length Transcriptome of a Tetraploid Sinocyclocheilus multipunctatus Provides Insights into the Evolution of Cavefish. Animals 2023, 13, 3399. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef]
- Fu, Q.; Zhang, P.; Zhao, S.; Li, Y.; Li, X.; Cao, M.; Yang, N.; Li, C. A novel full-length transcriptome resource from multiple immune-related tissues in turbot (Scophthalmus maximus) using Pacbio SMART sequencing. Fish Shellfish Immunol. 2022, 129, 106–113. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef]
- Alamancos, G.P.; Pages, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. Rna 2015, 21, 1521–1531. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]
- Vidal-Limon, A.; Aguilar-Toala, J.E.; Liceaga, A.M. Integration of Molecular Docking Analysis and Molecular Dynamics Simulations for Studying Food Proteins and Bioactive Peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Lam-Tung, N.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.-J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef]
- Tian, Y.; Wen, H.; Qi, X.; Zhang, X.; Sun, Y.; Li, J.; He, F.; Zhang, M.; Zhang, K.; Yang, W.; et al. Alternative splicing (AS) mechanism plays important roles in response to different salinity environments in spotted sea bass. Int. J. Biol. Macromol. 2020, 155, 50–60. [Google Scholar] [CrossRef]
- Cui, J.; Shen, N.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 2020, 16, 85. [Google Scholar] [CrossRef]
- Lebrigand, K.; Magnone, V.; Barbry, P.; Waldmann, R. High throughput error corrected Nanopore single cell transcriptome sequencing. Nat. Commun. 2020, 11, 4025. [Google Scholar] [CrossRef]
- Shumate, A.; Wong, B.; Pertea, G.; Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 2022, 18, e1009730. [Google Scholar] [CrossRef]
- Luo, H.; Liu, H.; Zhang, J.; Hu, B.; Zhou, C.; Xiang, M.; Yang, Y.; Zhou, M.; Jing, T.; Li, Z.; et al. Full-length transcript sequencing accelerates the transcriptome research of Gymnocypris namensis, an iconic fish of the Tibetan Plateau. Sci. Rep. 2020, 10, 9668. [Google Scholar] [CrossRef]
- Diao, J.; Yu, X.; Wang, X.; Fan, Y.; Wang, S.; Li, L.; Wang, Y.; Xu, L.; Gai, C.; Ye, H.; et al. Full-length transcriptome sequencing combined with RNA-seq analysis revealed the immune response of fat greenling (Hexagrammos otakii) to Vibrio harveyi in early infection. Microb. Pathog. 2020, 149, 104527. [Google Scholar] [CrossRef] [PubMed]
- Todd, L.; Hooper, M.J.; Haugan, A.K.; Finkbeiner, C.; Jorstad, N.; Radulovich, N.; Wong, C.K.; Donaldson, P.C.; Jenkins, W.; Chen, Q.; et al. Efficient stimulation of retinal regeneration from Müller glia in adult mice using combinations of proneural bHLH transcription factors. Cell Rep. 2021, 37, 109857. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Zhou, Y.; Wang, J.; Yu, X.; Tong, J. Identifying Candidate Genes Involved in the Regulation of Early Growth Using Full-Length Transcriptome and RNA-Seq Analyses of Frontal and Parietal Bones and Vertebral Bones in Bighead Carp (Hypophthalmichthys nobilis). Front. Genet. 2021, 11, 603454. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Burguete, T.; Peña-Marin, E.S.; García-Gasca, A.; Alvarez-González, C.A.; Llera-Herrera, R. Nutrigenomic marker discovery by de novo transcriptomic sequencing during early development of the tropical gar (Atractosteus tropicus). Aquac. Res. 2021, 52, 3829–3842. [Google Scholar] [CrossRef]
- Baillo, E.H.; Kimotho, R.N.; Zhang, Z.; Xu, P. Transcription Factors Associated with Abiotic and Biotic Stress Tolerance and Their Potential for Crops Improvement. Genes 2019, 10, 771. [Google Scholar] [CrossRef]
- Najafabadi, H.S.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.N.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef]
- Assmann, S.M.; Chou, H.-L.; Bevilacqua, P.C. Rock, scissors, paper: How RNA structure informs function. Plant Cell 2023, 35, 1671–1707. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Pan, J.; Wang, M.; Zhong, L.; Wang, J.; Qin, Q.; Liu, H.; Shao, J.; Chen, X.; et al. Use of comparative transcriptome analysis to identify candidate genes related to albinism in channel catfish (Ictalurus punctatus). Aquaculture 2019, 500, 75–81. [Google Scholar] [CrossRef]
- Healy, T.M.; Schulte, P.M. Patterns of alternative splicing in response to cold acclimation in fish. J. Exp. Biol. 2019, 222, jeb193516. [Google Scholar] [CrossRef]
- Sun, J.; Liu, Z.; Quan, J.; Li, L.; Zhao, G.; Lu, J. RNA-seq Analysis Reveals Alternative Splicing Under Heat Stress in Rainbow Trout (Oncorhynchus mykiss). Mar. Biotechnol. 2022, 24, 5–17. [Google Scholar] [CrossRef]
- Carneiro Vieira, M.L.; Santini, L.; Diniz, A.L.; Munhoz, C.d.F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-Y.; Guo, H.-Y.; Liu, B.-S.; Zhang, N.; Zhu, K.-C.; Xian, L.; Zhao, P.-H.; Yang, H.-Y.; Zhang, D.-C. Genome-wide identification of heat shock protein gene family and their responses to pathogen challenge in Trachinotus ovatus. Fish Shellfish Immunol. 2024, 145, 109309. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Wu, X.; Li, T.; Jia, M.; Liu, X.; Li, P.; Zhou, X.; Ji, X.; Yue, X. Genome-wide survey of heat shock factors and heat shock protein 70s and their regulatory network under abiotic stresses in Brachypodium distachyon. PLoS ONE 2017, 12, e0180352. [Google Scholar] [CrossRef]
- Murphy, M.E. The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef]
- Nakamoto, H.; Fujita, K.; Ohtaki, A.; Watanabe, S.; Narumi, S.; Maruyama, T.; Suenaga, E.; Misono, T.S.; Kumar, P.K.R.; Goloubinoff, P.; et al. Physical Interaction between Bacterial Heat Shock Protein (Hsp) 90 and Hsp70 Chaperones Mediates Their Cooperative Action to Refold Denatured Proteins*. J. Biol. Chem. 2014, 289, 6110–6119. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef]
- Jia, S.; Ding, G.; Wang, C.; Feng, B.; Wang, Z.; Wang, L.; Jiang, Y.; Cui, W.; Qiao, X.; Tang, L.; et al. N-linked glycosylation sites in G protein of infectious hematopoietic necrosis virus (IHNV) affect its virulence and immunogenicity in rainbow trout. Fish Shellfish Immunol. 2019, 89, 537–547. [Google Scholar] [CrossRef]
- Song, L.; Li, C.; Xie, Y.; Liu, S.; Zhang, J.; Yao, J.; Jiang, C.; Li, Y.; Liu, Z. Genome-wide identification of Hsp70 genes in channel catfish and their regulated expression after bacterial infection. Fish Shellfish Immunol. 2016, 49, 154–162. [Google Scholar] [CrossRef]
- Xie, Y.; Song, L.; Weng, Z.; Liu, S.; Liu, Z. Hsp90, Hsp60 and sHsp families of heat shock protein genes in channel catfish and their expression after bacterial infections. Fish Shellfish Immunol. 2015, 44, 642–651. [Google Scholar] [CrossRef]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noel, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, G.; Shao, C.; Huang, Q.; Liu, G.; Zhang, P.; Song, W.; An, N.; Chalopin, D.; Volff, J.-N.; et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet. 2014, 46, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, X.; Bai, J.; Fang, D.; Qiu, Y.; Jiang, W.; Yuan, H.; Bian, C.; Lu, J.; He, S.; et al. The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Maynard, G.A.; Kinnison, M.T.; Zydlewski, J.D. Size selection from fishways and potential evolutionary responses in a threatened Atlantic salmon population. River Res. Appl. 2017, 33, 1004–1015. [Google Scholar] [CrossRef]
- van Velzen, R.; Holmer, R.; Bu, F.; Rutten, L.; van Zeijl, A.; Liu, W.; Santuari, L.; Cao, Q.; Sharma, T.; Shen, D.; et al. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proc. Natl. Acad. Sci. USA 2018, 115, E4700–E4709. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Marks, M.E.; Peichel, C.L.; Blackman, B.K.; Nereng, K.S.; Jónsson, B.; Schluter, D.; Kingsley, D.M. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 2004, 428, 717–723. [Google Scholar] [CrossRef]
- Shiffman, M.E.; Soo, R.M.; Dennis, P.G.; Morrison, M.; Tyson, G.W.; Hugenholtz, P. Gene and genome-centric analyses of koala and wombat fecal microbiomes point to metabolic specialization for Eucalyptus digestion. PeerJ 2017, 5, e4075. [Google Scholar] [CrossRef]
- Whisson, D.A.; Dixon, V.; Taylor, M.L.; Melzer, A. Failure to Respond to Food Resource Decline Has Catastrophic Consequences for Koalas in a High-Density Population in Southern Australia. PLoS ONE 2016, 11, e0144348. [Google Scholar] [CrossRef]
- Polkinghorne, A.; Hanger, J.; Timms, P. Recent advances in understanding the biology, epidemiology and control of chlamydial infections in koalas. Vet. Microbiol. 2013, 165, 214–223. [Google Scholar] [CrossRef]
- Jahan, K.; Nie, H.; Yan, X. Revealing the potential regulatory relationship between HSP70, HSP90 and HSF genes under temperature stress. Fish Shellfish Immunol. 2023, 134, 108607. [Google Scholar] [CrossRef]
- Huang, X.; Li, S.; Gao, Y.; Zhan, A. Genome-Wide Identification, Characterization and Expression Analyses of Heat Shock Protein-Related Genes in a Highly Invasive Ascidian Ciona savignyi. Front. Physiol. 2018, 9, 1043. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin Patrick, R.; Agard David, A. Glucocorticoid Receptor Function Regulated by Coordinated Action of the Hsp90 and Hsp70 Chaperone Cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.P.; Sullivan, W.P.; Toft, D.O. The Assembly and Intermolecular Properties of the hsp70-Hop-hsp90 Molecular Chaperone Complex*. J. Biol. Chem. 2002, 277, 38294–38304. [Google Scholar] [CrossRef]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.D.; Toft, D.O. Hop Modulates hsp70/hsp90 Interactions in Protein Folding*. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef]
- Wang, R.Y.-R.; Noddings, C.M.; Kirschke, E.; Myasnikov, A.G.; Johnson, J.L.; Agard, D.A. Structure of Hsp90–Hsp70–Hop–GR reveals the Hsp90 client-loading mechanism. Nature 2022, 601, 460–464. [Google Scholar] [CrossRef]
- Bai, G.; Pan, Y.; Zhang, Y.; Li, Y.; Wang, J.; Wang, Y.; Teng, W.; Jin, G.; Geng, F.; Cao, J. Research advances of molecular docking and molecular dynamic simulation in recognizing interaction between muscle proteins and exogenous additives. Food Chem. 2023, 429, 136836. [Google Scholar] [CrossRef]
- Adao, R.; Zanphorlin, L.M.; Lima, T.B.; Sriranganadane, D.; Dahlstrom, K.M.; Pinheiro, G.M.S.; Gozzo, F.C.; Barbosa, L.R.S.; Ramos, C.H.I. Revealing the interaction mode of the highly flexible Sorghum bicolor Hsp70/Hsp90 organizing protein (Hop): A conserved carboxylate clamp confers high affinity binding to Hsp90. J. Proteom. 2019, 191, 191–201. [Google Scholar] [CrossRef]
- Morán Luengo, T.; Kityk, R.; Mayer, M.P.; Rüdiger, S.G.D. Hsp90 Breaks the Deadlock of the Hsp70 Chaperone System. Mol. Cell 2018, 70, 545–552.e9. [Google Scholar] [CrossRef]
- Kravats, A.N.; Hoskins, J.R.; Reidy, M.; Johnson, J.L.; Doyle, S.M.; Genest, O.; Masison, D.C.; Wickner, S. Functional and physical interaction between yeast Hsp90 and Hsp70. Proc. Natl. Acad. Sci. USA 2018, 115, E2210–E2219. [Google Scholar] [CrossRef]
- Flom, G.; Behal Robert, H.; Rosen, L.; Cole Douglas, G.; Johnson Jill, L. Definition of the minimal fragments of Sti1 required for dimerization, interaction with Hsp70 and Hsp90 and in vivo functions. Biochem. J. 2007, 404, 159–167. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Number |
|---|---|
| Total number | 32,647 |
| Total length (bp) | 58,217,443 |
| Maximum Length (bp) | 8049 |
| Minimum Length (bp) | 54 |
| Average Length (bp) | 1783 |
| N50 Length | 2077 |
| Group of Protein | Docking Score | Confidence Score | Ligand RMSD |
|---|---|---|---|
| HSP70-HSP90 | −227.49 | 0.8249 | 27.59 |
| HOP-GR | −242.92 | 0.8651 | 78.01 |
| HSP70-HSP90-HOP-GR | −229.15 | 0.8296 | 37.02 |
| M. guttatus Gene | D. rerio Gene | Ka | Ks | Ka_Ks | Selection |
|---|---|---|---|---|---|
| MgHsp90-21 | trap1 | 0.11933 | 1.416211 | 0.08426 | Purify |
| MgHsp70-59 | hyou1 | 0.037815 | 1.147255 | 0.032961 | Purify |
| MgHsp70-29 | hspa9 | 0.072756 | 1.685386 | 0.043169 | Purify |
| MgHsp70-10 | hspa8b | 0.155257 | 1.116471 | 0.13906 | Purify |
| MgHsp70-63 | hspa8 | 2.510801 | 1.483283 | 1.692732 | Positive |
| MgHsp70-31 | hspa4a | 0.087347 | 1.249427 | 0.06991 | Purify |
| MgHsp70-34 | hspa14 | 0.071319 | 1.10135 | 0.064756 | Purify |
| MgHsp70-45 | hspa13 | 0.112163 | 1.836793 | 0.061065 | Purify |
| MgHsp90-26 | hsp90b1 | 0.045808 | 1.159956 | 0.039491 | Purify |
| MgHsp90-28 | hsp90ab1 | 0.031203 | 1.340752 | 0.023273 | Purify |
| MgHsp90-29 | hsp90aa1.2 | 0.023897 | 2.474259 | 0.009658 | Purify |
| MgHsp70-24 | hsp70.2 | 0.03791 | 0.880748 | 0.043043 | Purify |
| MgHsp70-53 | hspa5 | 0.03482 | 1.330795 | 0.026165 | Purify |
| Fish Species | Data Sources | Isoform Number | N50 Length | References |
|---|---|---|---|---|
| Mystus guttatus | PacBio | 32,647 | 2077 | This paper |
| Gymnocypris namensis | PacBio | 125,396 | 2044 | [41] |
| Hexagrammos otakii | PacBio and Illumina | 42,225 | 2482 | [42] |
| Acipenser dabryanus | PacBio and Illumina | 155,348 | 3365 | [43] |
| Hypophthalmichthys nobilis | PacBio, Illumina and Reference genome | 63,873 | 1741 | [44] |
| Atractosteus tropicus | PacBio and Illumina | 80,065 | 1664 | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, L.; Zhang, X.; Li, Y.; Shi, J.; Li, Y.; Han, Y.; Luo, H.; Wang, D.; Lin, Y.; Ye, H. Full-Length Transcriptome Sequencing and hsp Gene Family Analysis Provide New Insights into the Stress Response Mechanisms of Mystus guttatus. Biology 2025, 14, 840. https://doi.org/10.3390/biology14070840
Qin L, Zhang X, Li Y, Shi J, Li Y, Han Y, Luo H, Wang D, Lin Y, Ye H. Full-Length Transcriptome Sequencing and hsp Gene Family Analysis Provide New Insights into the Stress Response Mechanisms of Mystus guttatus. Biology. 2025; 14(7):840. https://doi.org/10.3390/biology14070840
Chicago/Turabian StyleQin, Lang, Xueling Zhang, Yusen Li, Jun Shi, Yu Li, Yaoquan Han, Hui Luo, Dapeng Wang, Yong Lin, and Hua Ye. 2025. "Full-Length Transcriptome Sequencing and hsp Gene Family Analysis Provide New Insights into the Stress Response Mechanisms of Mystus guttatus" Biology 14, no. 7: 840. https://doi.org/10.3390/biology14070840
APA StyleQin, L., Zhang, X., Li, Y., Shi, J., Li, Y., Han, Y., Luo, H., Wang, D., Lin, Y., & Ye, H. (2025). Full-Length Transcriptome Sequencing and hsp Gene Family Analysis Provide New Insights into the Stress Response Mechanisms of Mystus guttatus. Biology, 14(7), 840. https://doi.org/10.3390/biology14070840