
Genome-Wide Identification, Evolutionary Expansion, and Expression Analyses of Aux/IAA Gene Family in Castanea mollissima During Seed Kernel Development

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification, Molecular Characteristics, and Phylogenetic Analysis
2.2. Chromosomal Distribution and Collinear Analysis
2.3. GO/KEGG Enrichment, Transcription Factors (TFs) Regulatory and Protein-Protein Interaction (PPI) Network Analysis
2.4. Plant Materials and Phenotypic Determination
2.5. Expression Analysis of CmAux/IAA Genes
2.6. RNA Sequencing and DEG Analysis
2.7. WGCNA Analysis and RT-qPCR Validation
2.8. Statistical Analysis
3. Results
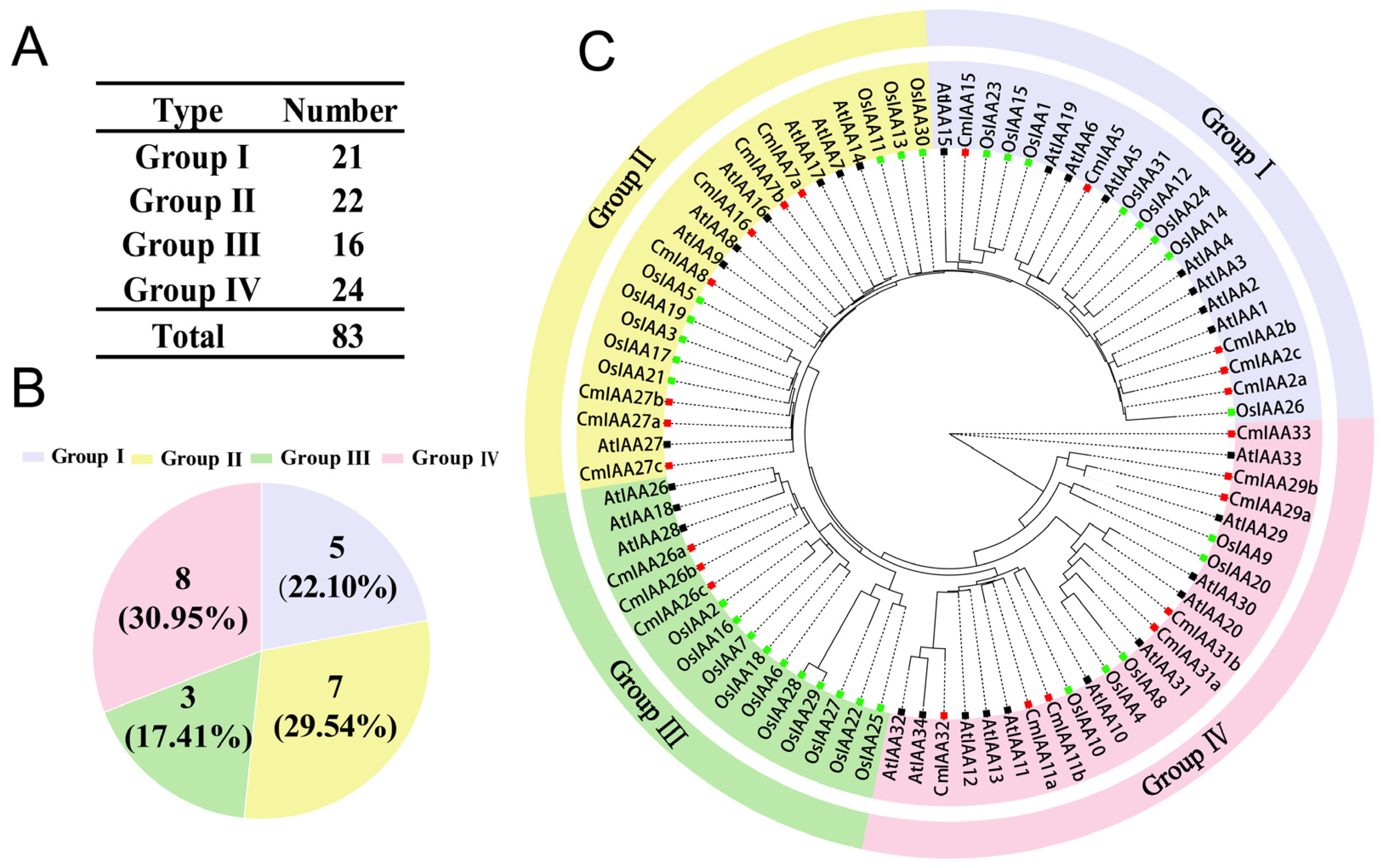
3.1. Identification and Phylogenetic Analysis
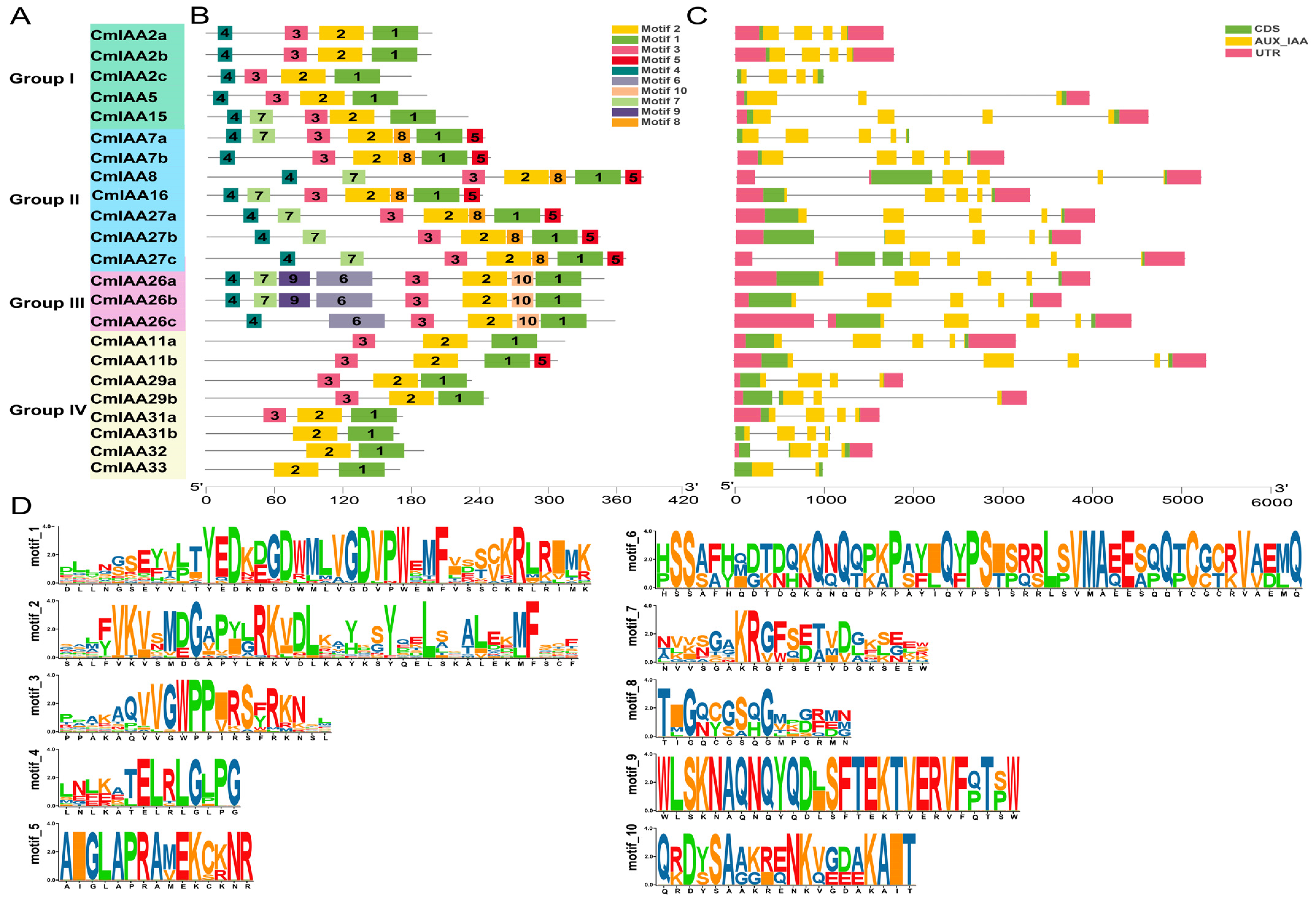
3.2. Gene Structure and Conserved Motif Analysis
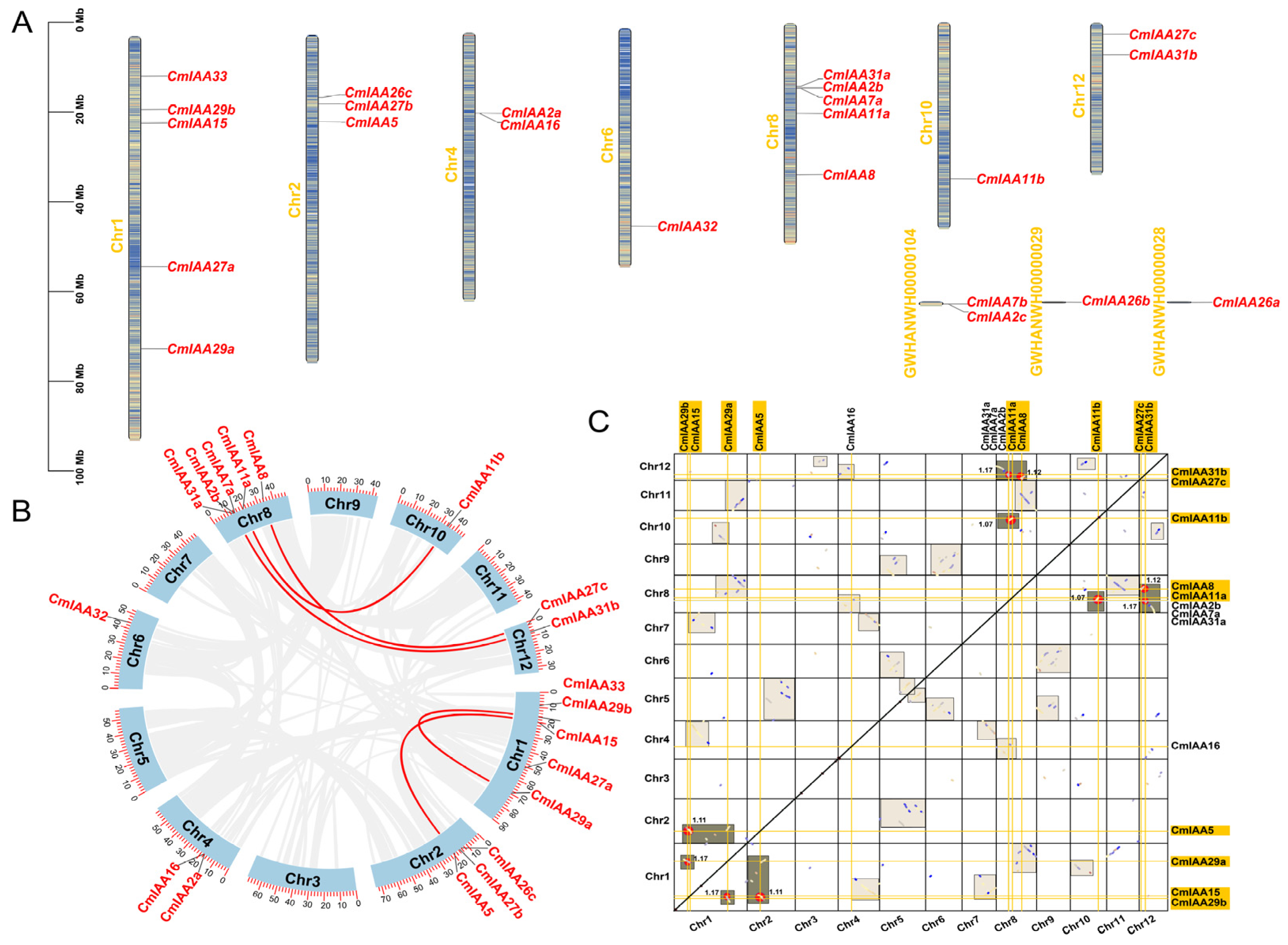
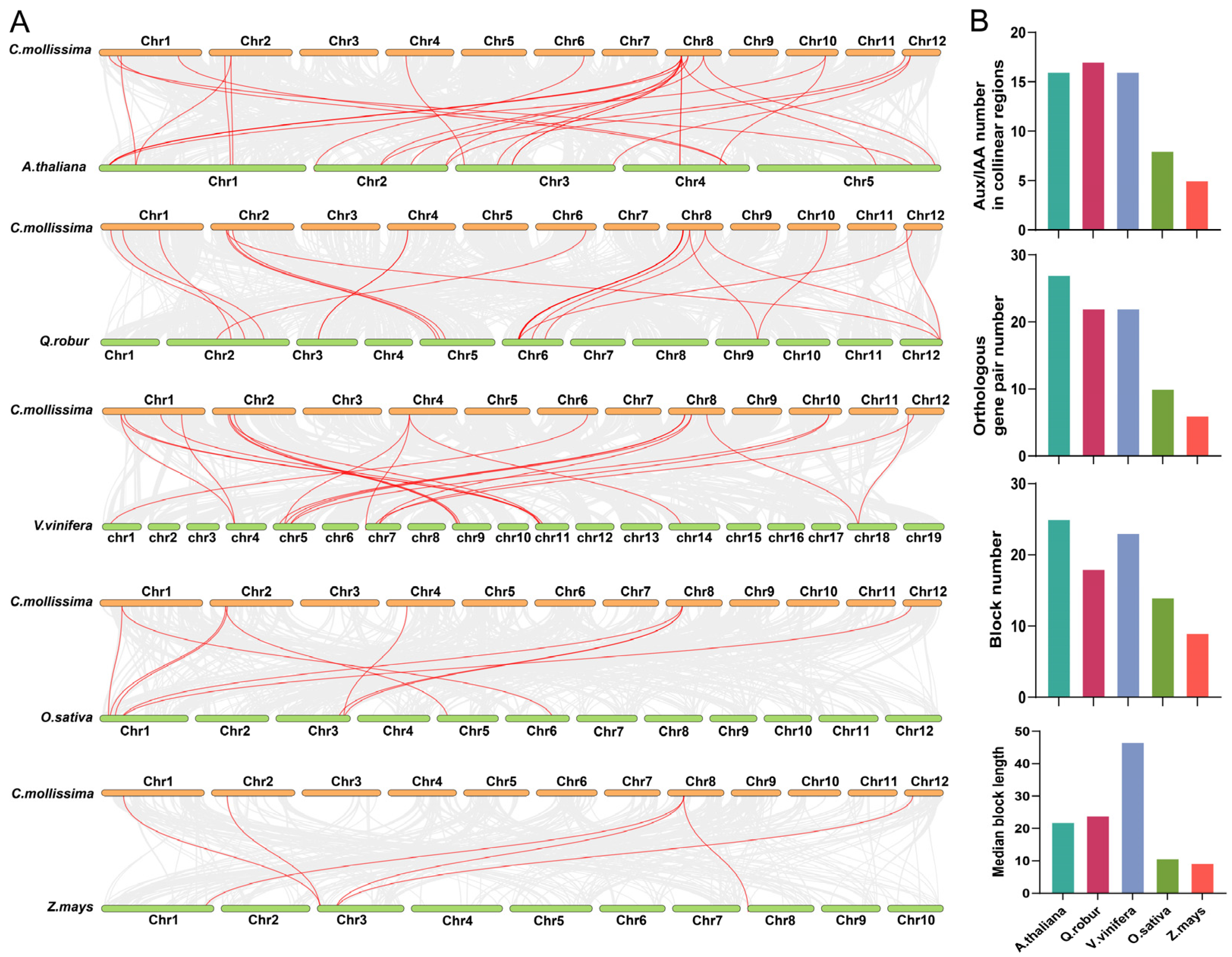
3.3. Chromosome Location and Collinear Analyses
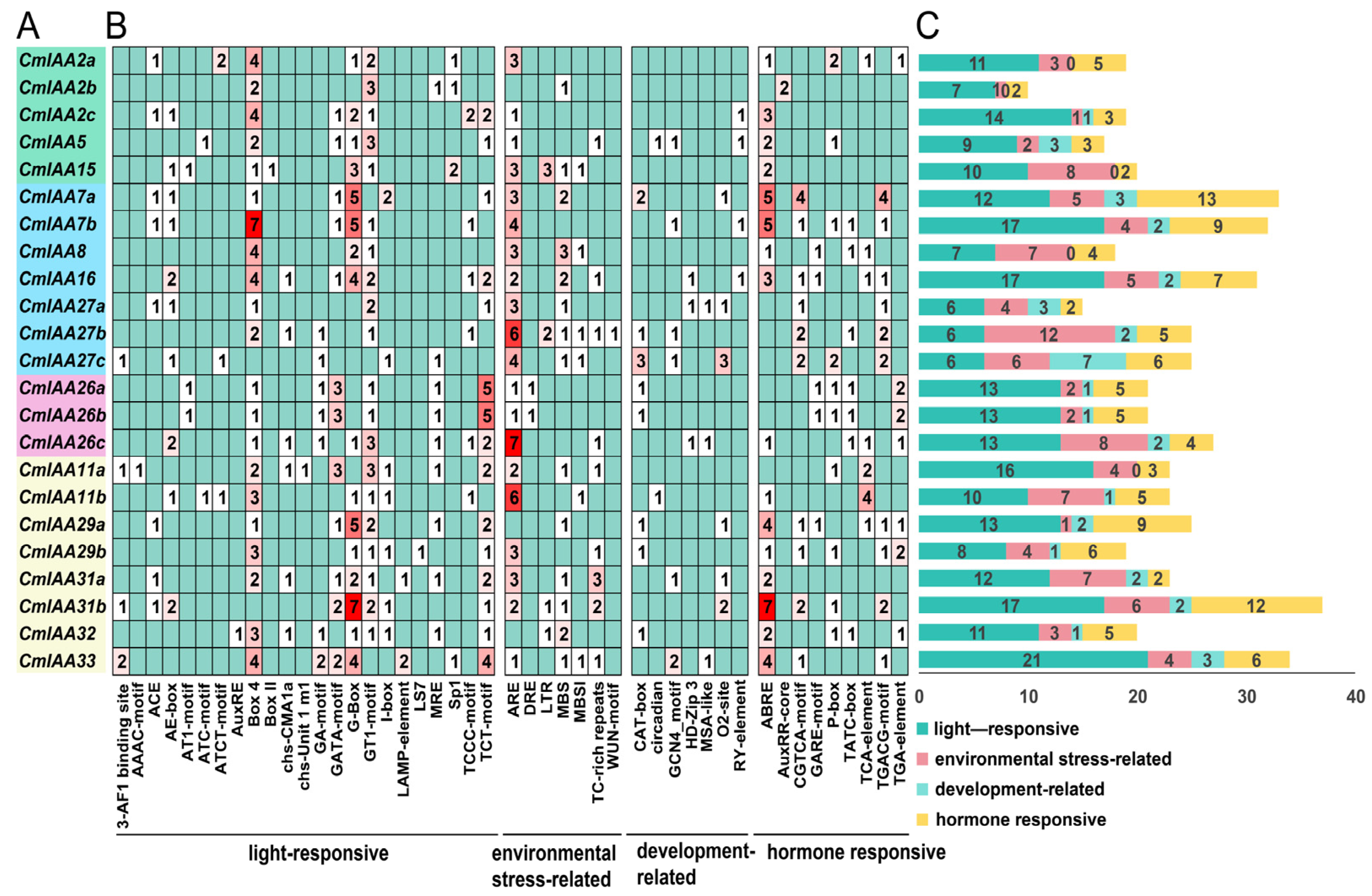
3.4. Cis-Acting Elements Analysis
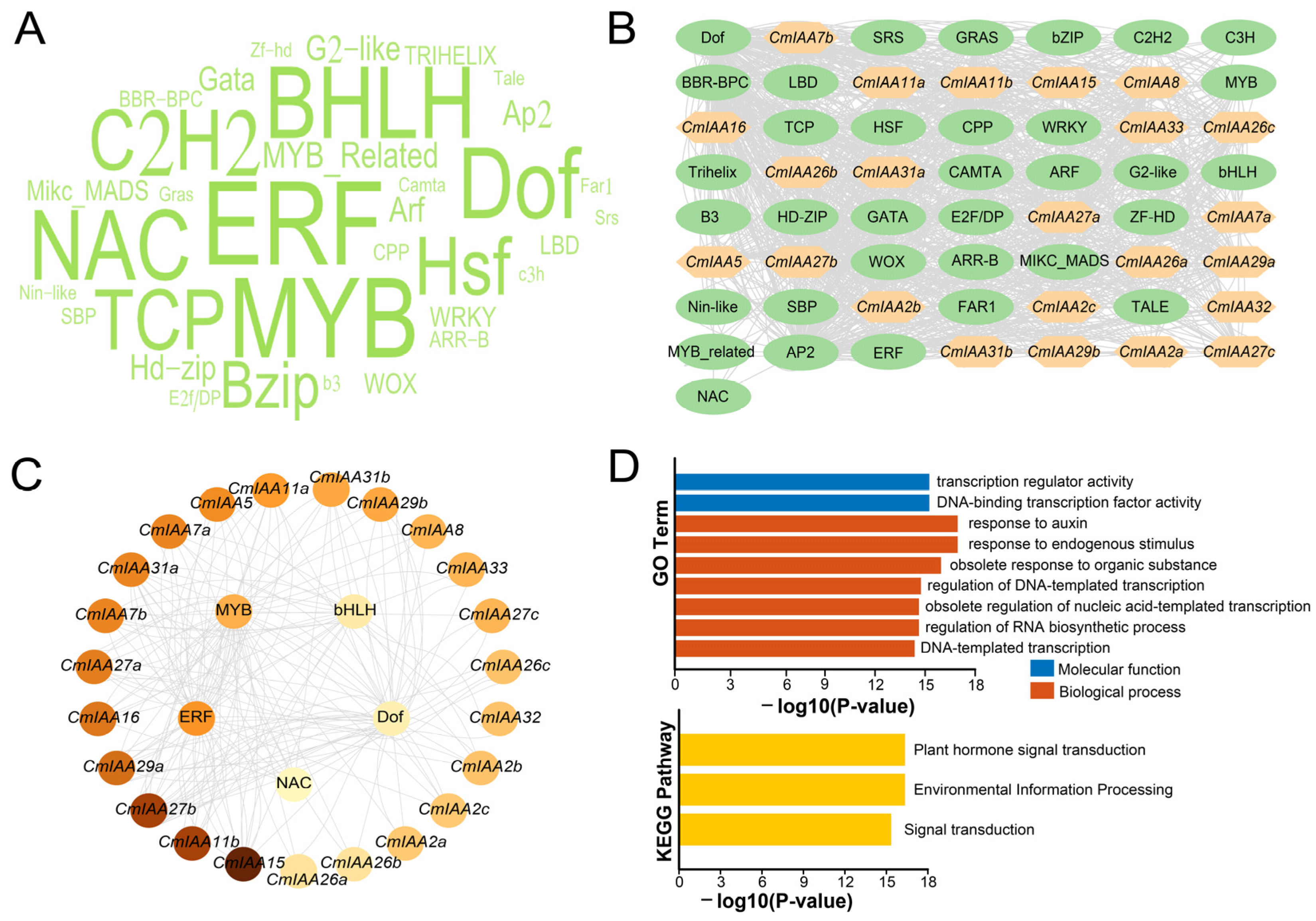
3.5. TFs Regulatory Network Analysis, GO/KEGG Enrichment, and PPI Network Analysis
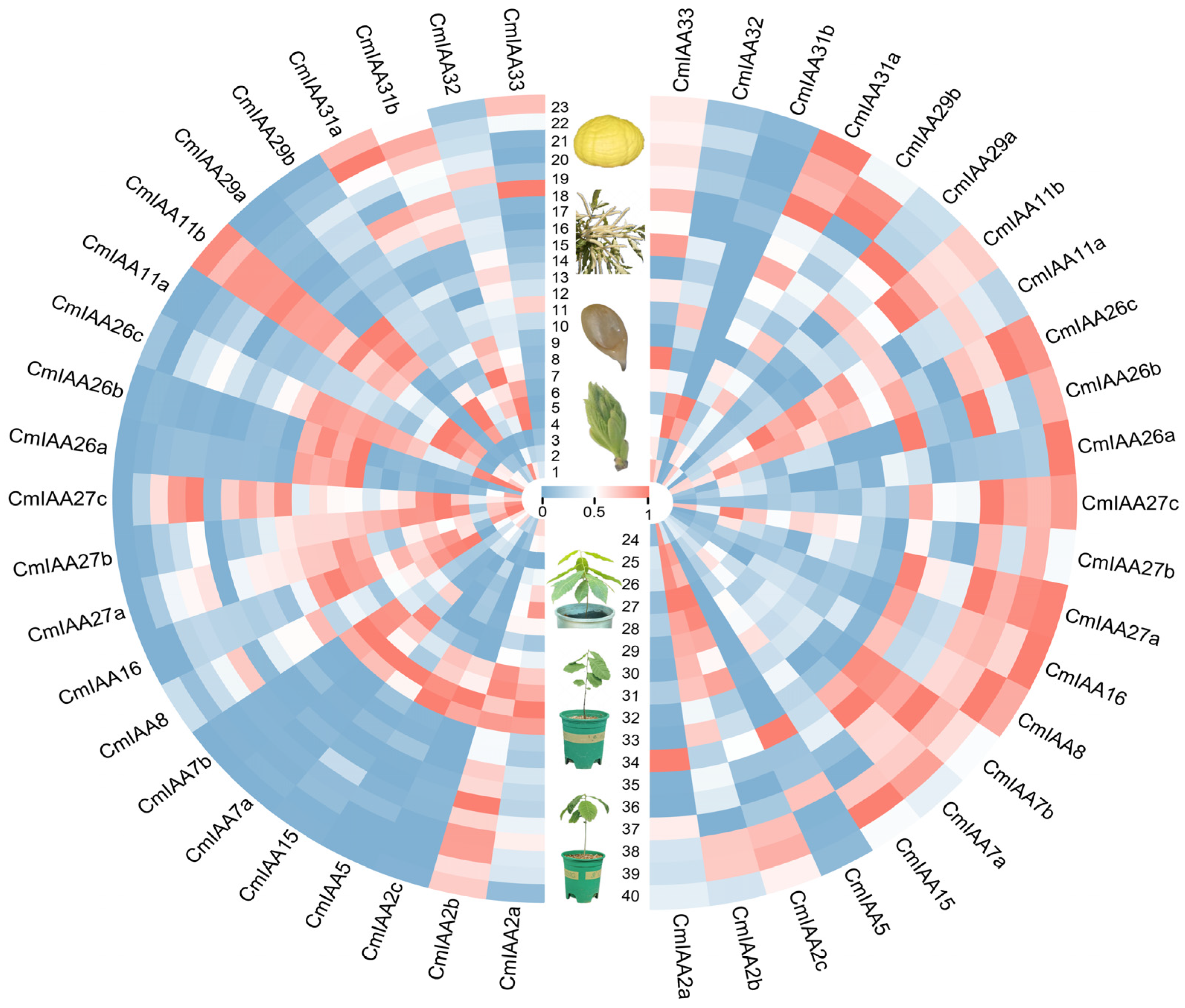
3.6. Expression Patterns of CmAux/IAA Genes in Different Tissues and Under Abiotic Stress
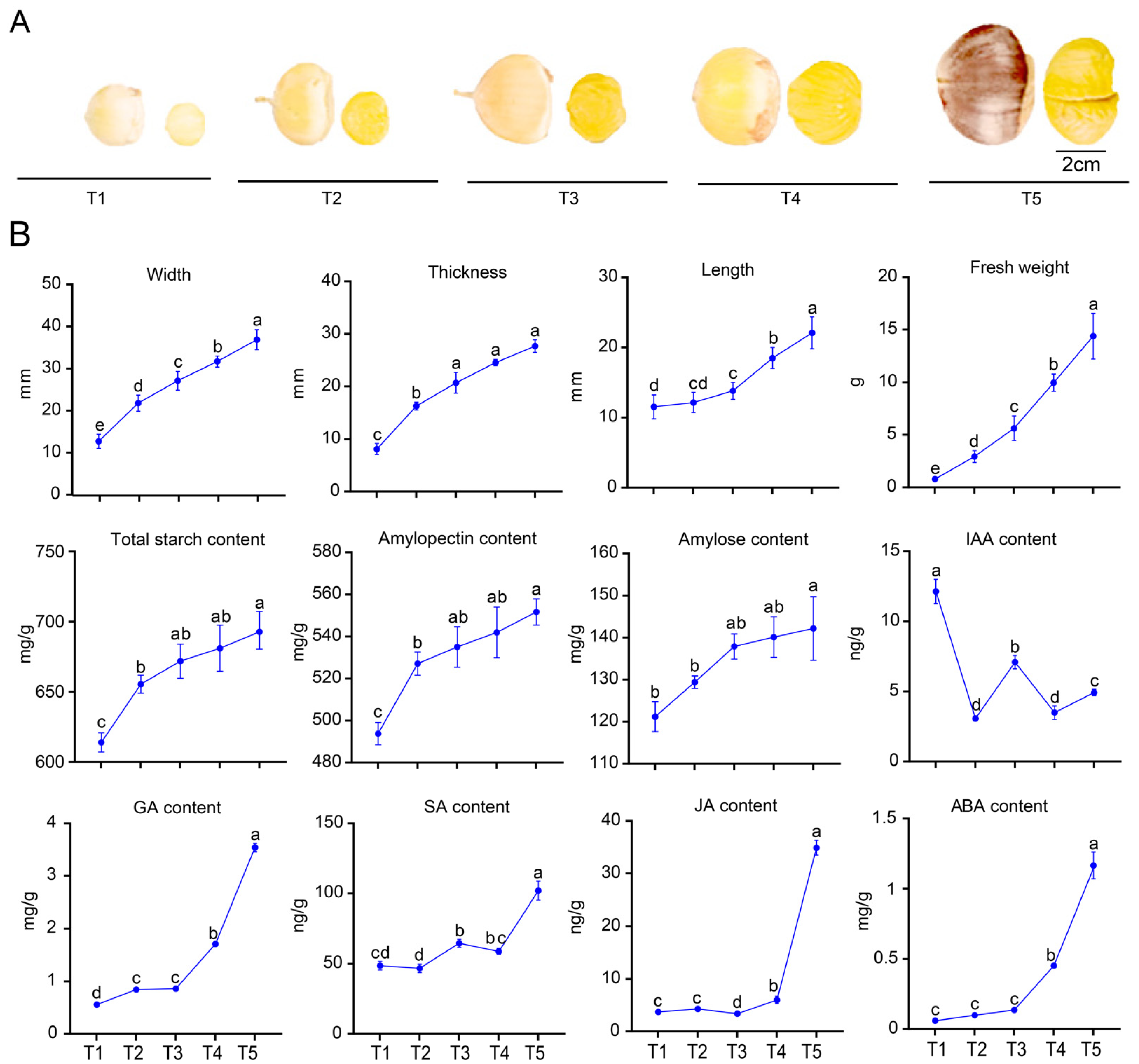
3.7. Phenotypic Changes During the Development
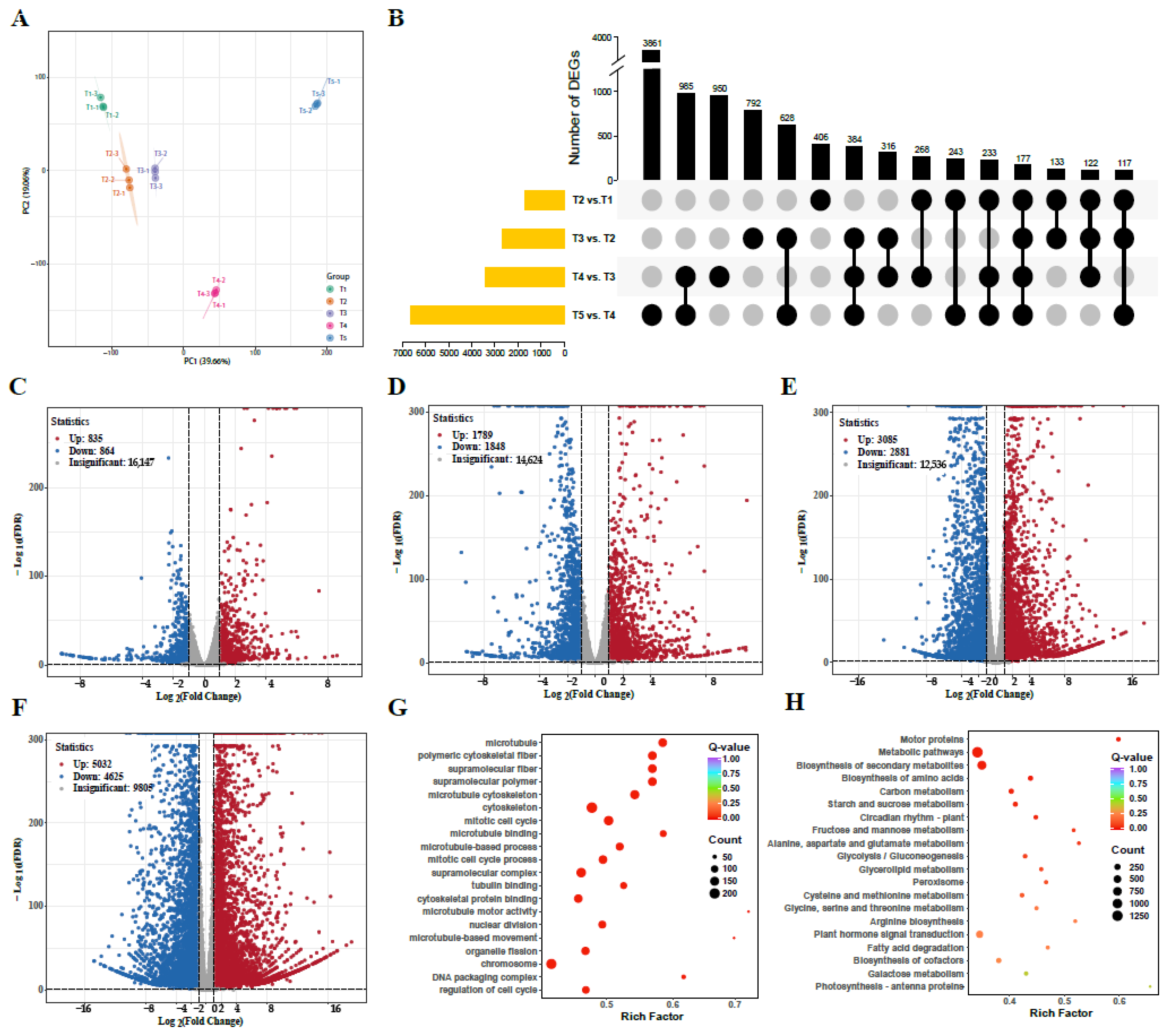
3.8. RNA Sequencing and DEGs Identification
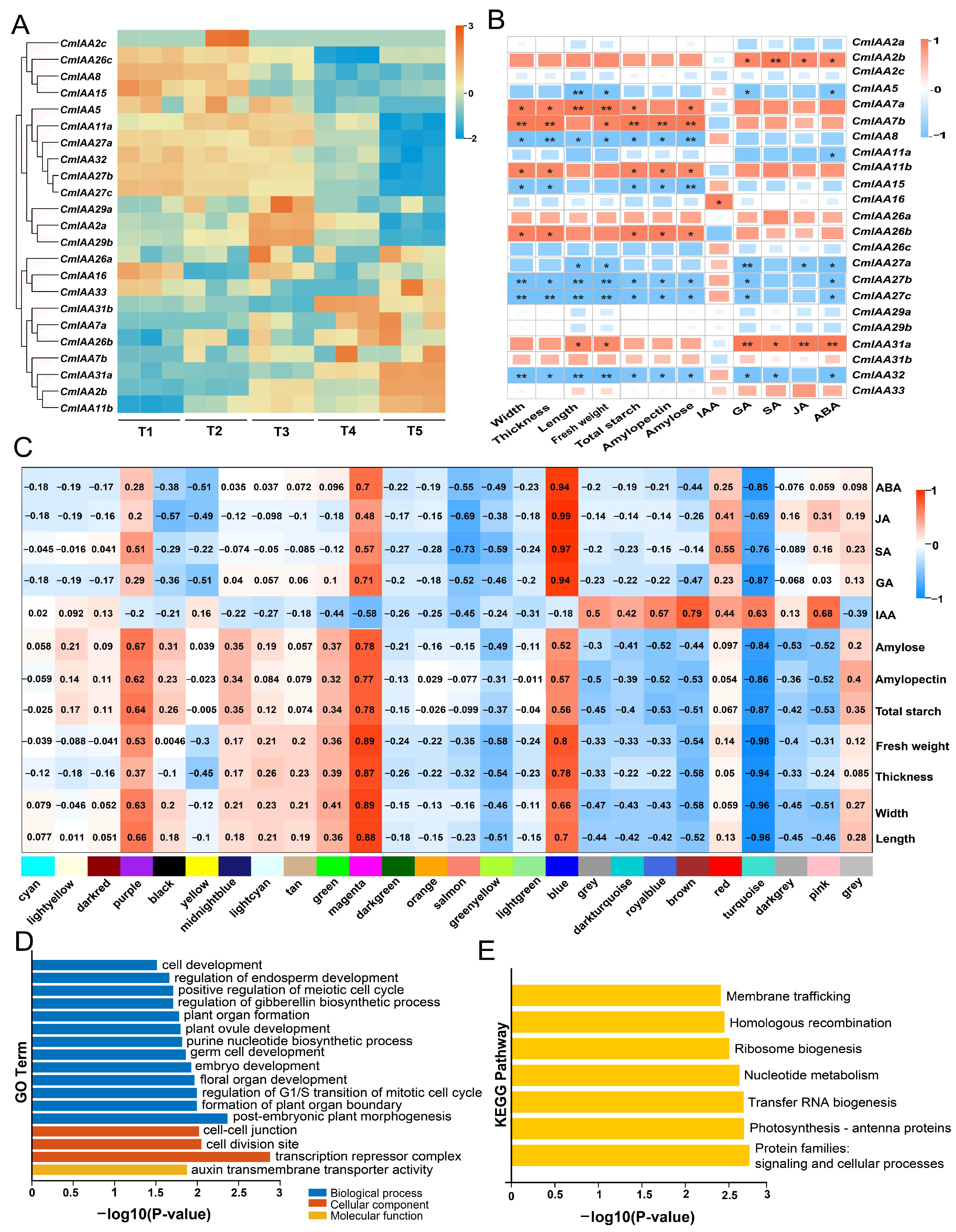
3.9. Expression Patterns of CmAux/IAAs and Their Connections with Phenotypic Indicators
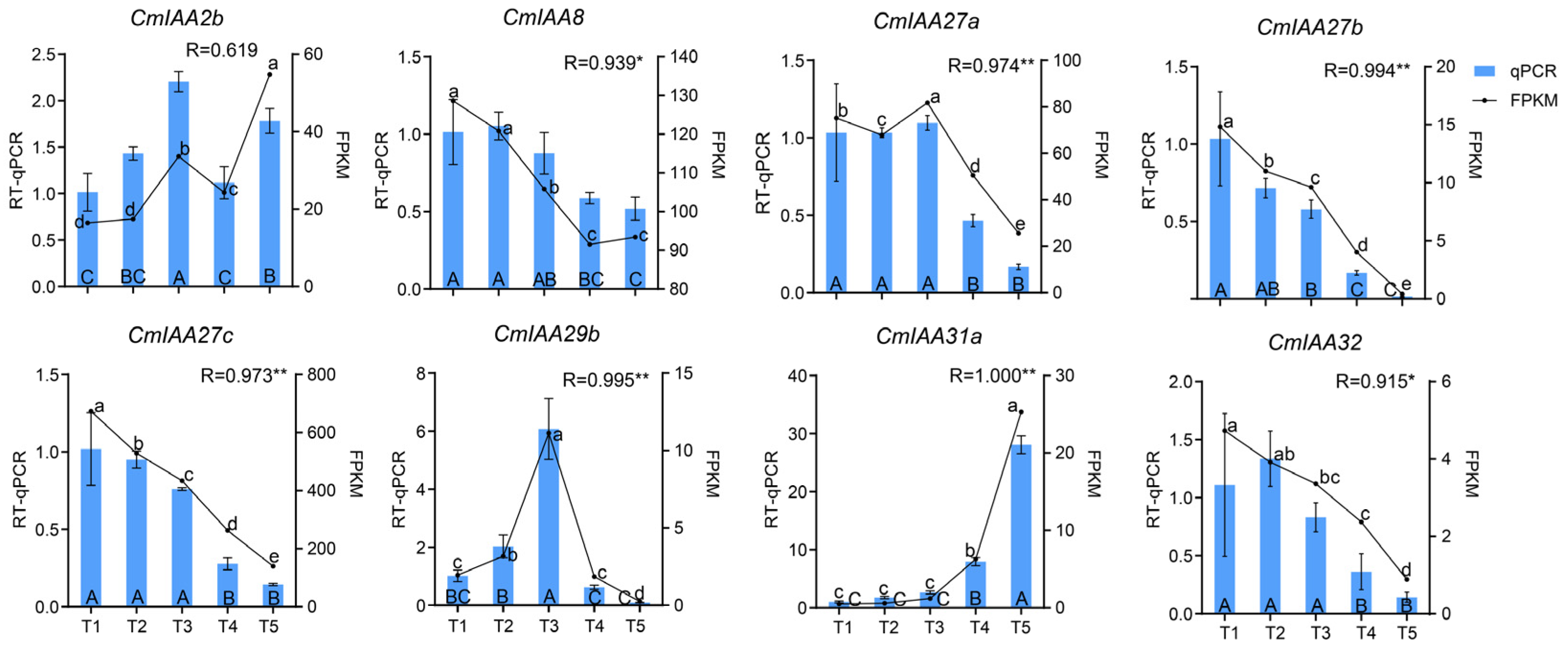
3.10. Expression Analysis of CmAux/IAA Genes During Seed Kernel Development by RT-qPCR
4. Discussion
4.1. The Molecular Characteristics of Aux/IAA Genes
4.2. Analysis of the Evolution and Expansion of the CmAux/IAA Gene Family
4.3. Expression Pattern of CmAux/IAA Genes During Seed Kernel Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, J.; Zhou, J.J.; Zhang, J.Z. Aux/IAA Gene Family in Plants: Molecular Structure, Regulation, and Function. Int. J. Mol. Sci. 2018, 19, 259. [Google Scholar] [CrossRef]
- Kalluri, U.C.; Difazio, S.P.; Brunner, A.M.; Tuskan, G.A. Genome-wide analysis of Aux/IAA and ARF gene families in Populus trichocarpa. BMC Plant Biol. 2007, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y.; Yan, H.; Tian, T.; You, Q.; Zhang, L.; Xu, W.; Su, Z. PlantEAR: Functional Analysis Platform for Plant EAR Motif-Containing Proteins. Front. Genet. 2018, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Mockaitis, K.; Estelle, M. Auxin receptors and plant development: A new signaling paradigm. Annu. Rev. Cell Dev. Biol. 2008, 24, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.W.; Bartel, B. Auxin: Regulation, action, and interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef]
- Morgan, K.E.; Zarembinski, T.I.; Theologis, A.; Abel, S. Biochemical characterization of recombinant polypeptides corresponding to the predicted betaalphaalpha fold in Aux/IAA proteins. FEBS Lett. 1999, 454, 283–287. [Google Scholar] [CrossRef]
- Guilfoyle, T.J.; Hagen, G. Auxin response factors. Curr. Opin. Plant Biol. 2007, 10, 453–460. [Google Scholar] [CrossRef]
- Smalle, J.; Vierstra, R.D. The ubiquitin 26S proteasome proteolytic pathway. Annu. Rev. Plant Biol. 2004, 55, 555–590. [Google Scholar] [CrossRef]
- Cao, M.; Chen, R.; Li, P.; Yu, Y.; Zheng, R.; Ge, D.; Zheng, W.; Wang, X.; Gu, Y.; Gelová, Z.; et al. TMK1-mediated auxin signalling regulates differential growth of the apical hook. Nature 2019, 568, 240–243. [Google Scholar] [CrossRef]
- Xu, C.; Shen, Y.; He, F.; Fu, X.; Yu, H.; Lu, W.; Li, Y.; Li, C.; Fan, D.; Wang, H.C.; et al. Auxin-mediated Aux/IAA-ARF-HB signaling cascade regulates secondary xylem development in Populus. New Phytol. 2019, 222, 752–767. [Google Scholar] [CrossRef]
- Wang, C.K.; Han, P.L.; Zhao, Y.W.; Ji, X.L.; Yu, J.Q.; You, C.X.; Hu, D.G.; Hao, Y.J. Auxin regulates anthocyanin biosynthesis through the auxin repressor protein MdIAA26. Biochem. Biophys. Res. Commun. 2020, 533, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Molesini, B.; Pennisi, F.; Vitulo, N.; Pandolfini, T. MicroRNAs associated with AGL6 and IAA9 function in tomato fruit set. BMC Res. Notes 2023, 16, 242. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.J.; Park, J.Y.; Ha, S.B.; Kim, J. Overexpression of IAA1 with domain II mutation impairs cell elongation and cell division in inflorescences and leaves of Arabidopsis. J. Plant Physiol. 2009, 166, 548–553. [Google Scholar] [CrossRef]
- Chen, H.; Song, Z.; Wang, L.; Lai, X.; Chen, W.; Li, X.; Zhu, X. Auxin-responsive protein MaIAA17-like modulates fruit ripening and ripening disorders induced by cold stress in ‘Fenjiao’ banana. Int. J. Biol. Macromol. 2023, 247, 125750. [Google Scholar] [CrossRef]
- Liu, D.J.; Chen, J.Y.; Lu, W.J. Expression and regulation of the early auxin-responsive Aux/IAA genes during strawberry fruit development. Mol. Biol. Rep. 2011, 38, 1187–1193. [Google Scholar] [CrossRef]
- Overvoorde, P.J.; Okushima, Y.; Alonso, J.M.; Chan, A.; Chang, C.; Ecker, J.R.; Hughes, B.; Liu, A.; Onodera, C.; Quach, H.; et al. Functional genomic analysis of the AUXIN/INDOLE-3-ACETIC ACID gene family members in Arabidopsis thaliana. Plant Cell 2005, 17, 3282–3300. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Kaur, N.; Garg, R.; Thakur, J.K.; Tyagi, A.K.; Khurana, J.P. Structure and expression analysis of early auxin-responsive Aux/IAA gene family in rice (Oryza sativa). Funct. Integr. Genom. 2006, 6, 47–59. [Google Scholar] [CrossRef]
- Audran-Delalande, C.; Bassa, C.; Mila, I.; Regad, F.; Zouine, M.; Bouzayen, M. Genome-wide identification, functional analysis and expression profiling of the Aux/IAA gene family in tomato. Plant Cell Physiol. 2012, 53, 659–672. [Google Scholar] [CrossRef]
- Wang, L.; Xu, K.; Li, Y.; Cai, W.; Zhao, Y.; Yu, B.; Zhu, Y. Genome-Wide Identification of the Aux/IAA Family Genes (MdIAA) and Functional Analysis of MdIAA18 for Apple Tree Ideotype. Biochem. Genet. 2019, 57, 709–733. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, D.; Bian, Y.; Lv, Y.; Xie, Q. Genome-wide analysis of primary auxin-responsive Aux/IAA gene family in maize (Zea mays L.). Mol. Biol. Rep. 2010, 37, 3991–4001. [Google Scholar] [CrossRef]
- Wu, J.; Peng, Z.; Liu, S.; He, Y.; Cheng, L.; Kong, F.; Wang, J.; Lu, G. Genome-wide analysis of Aux/IAA gene family in Solanaceae species using tomato as a model. Mol. Genet. Genom. 2012, 287, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.S.; Ren, X.Y.; Tong, M.N.; Jiang, S.Y.; Zhang, C.Q.; Liu, Q.Q.; Li, Q.F. OsIAA19, an Aux/IAA Family Gene, Involved in the Regulation of Seed-Specific Traits in Rice. Plants 2024, 13, 3538. [Google Scholar] [CrossRef]
- Huang, R.; Peng, F.; Wang, D.; Cao, F.; Guo, C.; Yu, L.; Zhang, J.; Yang, Y. Transcriptome analysis of differential sugar accumulation in the developing embryo of contrasting two Castanea mollissima cultivars. Front. Plant Sci. 2023, 14, 1206585. [Google Scholar] [CrossRef]
- Santos, M.J.; Pinto, T.; Vilela, A. Sweet Chestnut (Castanea sativa Mill.) Nutritional and Phenolic Composition Interactions with Chestnut Flavor Physiology. Foods 2022, 11, 4052. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, X.; Zhang, C.; Zhang, M.; Huang, C.; Yang, H. Amino acid composition and nutritional value evaluation of Chinese chestnut (Castanea mollissima Blume) and its protein subunit. RSC Adv. 2018, 8, 2653–2659. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Newberg, L.A. Error statistics of hidden Markov model and hidden Boltzmann model results. BMC Bioinform. 2009, 10, 212. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Yu, L.; Tian, Y.; Wang, X.; Cao, F.; Wang, H.; Huang, R.; Guo, C.; Zhang, H.; Zhang, J. Genome-wide identification, phylogeny, evolutionary expansion, and expression analyses of ABC gene family in Castanea mollissima under temperature stress. Plant Physiol. Biochem. 2025, 219, 109450. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, E.K.; Zhang, D.; Reynolds, R.H.; Garcia-Ruiz, S.; Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022, 38, 3844–3846. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Hu, M.; Xie, M.; Cui, X.; Huang, J.; Cheng, X.; Liu, L.; Yan, S.; Liu, S.; Tong, C. Characterization and Potential Function Analysis of the SRS Gene Family in Brassica napus. Genes 2023, 14, 1421. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hadar, N.; Weintraub, G.; Gudes, E.; Dolev, S.; Birk, O.S. GeniePool: Genomic database with corresponding annotated samples based on a cloud data lake architecture. Database 2023, 2023, baad043. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three Differential Expression Analysis Methods for RNA Sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 18, 175. [Google Scholar] [CrossRef]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. BioTechniques 1998, 24, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mitteer, D.R.; Greer, B.D.; Randall, K.R.; Briggs, A.M. Further Evaluation of Teaching Behavior Technicians to Input Data and Graph Using GraphPad Prism. Behav. Anal. 2020, 20, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.; Wuensch, K.L. SPSS and SAS programs for comparing Pearson correlations and OLS regression coefficients. Behav. Res. Methods 2013, 45, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Yu, L.; Diao, S.; Zhang, G.; Yu, J.; Zhang, T.; Luo, H.; Duan, A.; Wang, J.; He, C.; Zhang, J. Genome sequence and population genomics provide insights into chromosomal evolution and phytochemical innovation of Hippophae rhamnoides. Plant Biotechnol. J. 2022, 20, 1257–1273. [Google Scholar] [CrossRef]
- Birchler, J.A.; Veitia, R.A. The gene balance hypothesis: From classical genetics to modern genomics. Plant Cell 2007, 19, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.H.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y.D. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS ONE 2017, 12, e0184129. [Google Scholar] [CrossRef]
- Wang, T.; Long, C.; Chang, M.; Wu, Y.; Su, S.; Wei, J.; Jiang, S.; Wang, X.; He, J.; Xing, D.; et al. Genome-wide identification of the B3 transcription factor family in pepper (Capsicum annuum) and expression patterns during fruit ripening. Sci. Rep. 2024, 14, 2226. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Yang, W.; Hassan, M.J.; Liu, J.; Yang, Y.; Zhou, Q.; Li, H.; Peng, Y. Genome-wide identification of Aux/IAA gene family in white clover (Trifolium repens L.) and functional verification of TrIAA18 under different abiotic stress. BMC Plant Biol. 2024, 24, 346. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.; Sun, X.; Yue, P.; Qiao, J.; Sun, J.; Wang, A.; Yuan, H.; Yu, W. The MdAux/IAA2 Transcription Repressor Regulates Cell and Fruit Size in Apple Fruit. Int. J. Mol. Sci. 2022, 23, 9454. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Hayat, F.; Mushtaq, N.; Khalil-Ur-Rehman, M.; Khan, U.; Yasoob, T.B.; Khan, M.N.; Ni, Z.; Ting, S.; Gao, Z. Bioinformatics Study of Aux/IAA Family Genes and Their Expression in Response to Different Hormones Treatments during Japanese Apricot Fruit Development and Ripening. Plants 2022, 11, 1898. [Google Scholar] [CrossRef]
- Molesini, B.; Dusi, V.; Pennisi, F.; Pandolfini, T. How Hormones and MADS-Box Transcription Factors Are Involved in Controlling Fruit Set and Parthenocarpy in Tomato. Genes 2020, 11, 1441. [Google Scholar] [CrossRef]
- Li, R.; Huang, X.; Yang, L.; Liao, J.; Wei, X.; Li, J.; Zeng, G.; Liu, D.; Shi, Z.; Zhao, Z. Whole genome sequencing of Castanea mollissima and molecular mechanisms of sugar and starch synthesis. Front. Plant Sci. 2024, 15, 1455885. [Google Scholar] [CrossRef]
- Liu, K.; Yuan, C.; Feng, S.; Zhong, S.; Li, H.; Zhong, J.; Shen, C.; Liu, J. Genome-wide analysis and characterization of Aux/IAA family genes related to fruit ripening in papaya (Carica papaya L.). BMC Genom. 2017, 18, 351. [Google Scholar] [CrossRef]
- Nozawa, R.S.; Gilbert, N. RNA: Nuclear Glue for Folding the Genome. Trends Cell Biol. 2019, 29, 201–211. [Google Scholar] [CrossRef]
- Sato, A.; Yamamoto, K.T. Overexpression of the non-canonical Aux/IAA genes causes auxin-related aberrant phenotypes in Arabidopsis. Physiol. Plant 2008, 133, 397–405. [Google Scholar] [CrossRef]
- Feng, K.; Hou, X.L.; Xing, G.M.; Liu, J.X.; Duan, A.Q.; Xu, Z.S.; Li, M.Y.; Zhuang, J.; Xiong, A.S. Advances in AP2/ERF super-family transcription factors in plant. Crit. Rev. Biotechnol. 2020, 40, 750–776. [Google Scholar] [CrossRef]
- Yan, H.; Pei, X.; Zhang, H.; Li, X.; Zhang, X.; Zhao, M.; Chiang, V.L.; Sederoff, R.R.; Zhao, X. MYB-Mediated Regulation of Anthocyanin Biosynthesis. Int. J. Mol. Sci. 2021, 22, 3103. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Dubos, C. The arabidopsis bHLH transcription factor family. Trends Plant Sci. 2024, 29, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Christophe Liseron-Monfils, D.W. Revealing gene regulation and associations through biological networks. Curr. Plant Biol. 2015, 3–4, 30–39. [Google Scholar] [CrossRef]
- Wen, S.; Ying, J.; Ye, Y.; Cai, Y.; Li, L.; Qian, R. Genome-wide identification and salt stress-responsive expression profiling of Aux/IAA gene family in Asparagus officinalis. BMC Plant Biol. 2025, 25, 759. [Google Scholar] [CrossRef]
- Alves, S.; Braga, Â.; Parreira, D.; Alhinho, A.T.; Silva, H.; Ramos, M.J.N.; Costa, M.M.R.; Morais-Cecílio, L. Genome-wide identification, phylogeny, and gene duplication of the epigenetic regulators in Fagaceae. Physiol. Plant 2022, 174, e13788. [Google Scholar] [CrossRef]
- Street, N.R. Structural Genomics of Angiosperm Trees: Genome Duplications, Ploidy, and Repeat Sequences. In Comparative and Evolutionary Genomics of Angiosperm Trees; Groover, A., Cronk, Q., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 101–120. [Google Scholar]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef]
- Huang, Z.; Duan, W.; Song, X.; Tang, J.; Wu, P.; Zhang, B.; Hou, X. Retention, Molecular Evolution, and Expression Divergence of the Auxin/Indole Acetic Acid and Auxin Response Factor Gene Families in Brassica Rapa Shed Light on Their Evolution Patterns in Plants. Genome Biol. Evol. 2015, 8, 302–316. [Google Scholar] [CrossRef]
- Hou, Y.; Li, H.; Zhai, L.; Xie, X.; Li, X.; Bian, S. Identification and functional characterization of the Aux/IAA gene VcIAA27 in blueberry. Plant Signal Behav. 2020, 15, 1700327. [Google Scholar] [CrossRef] [PubMed]
- Rusak, G.; Cerni, S.; Stupin Polancec, D.; Ludwig-Müller, J. The responsiveness of the IAA2 promoter to IAA and IBA is differentially affected in Arabidopsis roots and shoots by flavonoids. Biol. Plant. 2010, 54, 403–414. [Google Scholar] [CrossRef]
- Wang, J.; Yan, D.W.; Yuan, T.T.; Gao, X.; Lu, Y.T. A gain-of-function mutation in IAA8 alters Arabidopsis floral organ development by change of jasmonic acid level. Plant Mol. Biol. 2013, 82, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, Z.; Eaglesfield, R.; Carr, C.; Amtmann, A. Cryptic variation in RNA-directed DNA-methylation controls lateral root development when auxin signalling is perturbed. Nat. Commun. 2020, 11, 218. [Google Scholar] [CrossRef]
- Padmanabhan, M.S.; Goregaoker, S.P.; Golem, S.; Shiferaw, H.; Culver, J.N. Interaction of the tobacco mosaic virus replicase protein with the Aux/IAA protein PAP1/IAA26 is associated with disease development. J. Virol. 2005, 79, 2549–2558. [Google Scholar] [CrossRef]
- Takahashi, N.; Ogita, N.; Koike, T.; Nishimura, K.; Inagaki, S.; Umeda, M. Local induction of IAA5 and IAA29 promotes DNA damage-triggered stem cell death in Arabidopsis roots. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zimmermann, P.; Hirsch-Hoffmann, M.; Hennig, L.; Gruissem, W. GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 2004, 136, 2621–2632. [Google Scholar] [CrossRef]
- De Grassi, A.; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene families. Gene 2008, 421, 1–6. [Google Scholar] [CrossRef]
- Lallemand, T.; Leduc, M.; Desmazières, A.; Aubourg, S.; Rizzon, C.; Landès, C.; Celton, J.M. Insights into the Evolution of Ohnologous Sequences and Their Epigenetic Marks Post-WGD in Malus Domestica. Genome Biol. Evol. 2023, 15, evad178. [Google Scholar] [CrossRef]
- Schranz, M.E.; Mohammadin, S.; Edger, P.P. Ancient whole genome duplications, novelty and diversification: The WGD Radiation Lag-Time Model. Curr. Opin. Plant Biol. 2012, 15, 147–153. [Google Scholar] [CrossRef]
- Abdullaev, E.T.; Haridoss, D.A.; Arndt, P.F. Reconstruction of Segmental Duplication Rates and Associated Genomic Features by Network Analysis. Genome Biol. Evol. 2025, 17, evaf011. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ficklin, S.P.; Wang, X.; Feltus, F.A.; Paterson, A.H. Large-Scale Gene Relocations following an Ancient Genome Triplication Associated with the Diversification of Core Eudicots. PLoS ONE 2016, 11, e0155637. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Bassa, C.; Audran, C.; Mila, I.; Cheniclet, C.; Chevalier, C.; Bouzayen, M.; Roustan, J.P.; Chervin, C. The auxin Sl-IAA17 transcriptional repressor controls fruit size via the regulation of endoreduplication-related cell expansion. Plant Cell Physiol. 2014, 55, 1969–1976. [Google Scholar] [CrossRef]
- Zhao, X.; Muhammad, N.; Zhao, Z.; Yin, K.; Liu, Z.; Wang, L.; Luo, Z.; Wang, L.; Liu, M. Molecular regulation of fruit size in horticultural plants: A review. Sci. Hortic. 2021, 288. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, Q.; Feng, Y.; Fan, X.; Zou, F.; Yuan, D.Y.; Zeng, X.; Cao, H. Transcriptomic identification and expression of starch and sucrose metabolism genes in the seeds of Chinese chestnut (Castanea mollissima). J. Agric. Food Chem. 2015, 63, 929–942. [Google Scholar] [CrossRef]
- Bassa, C.; Mila, I.; Bouzayen, M.; Audran-Delalande, C. Phenotypes associated with down-regulation of Sl-IAA27 support functional diversity among Aux/IAA family members in tomato. Plant Cell Physiol. 2012, 53, 1583–1595. [Google Scholar] [CrossRef]
- Venturini, L.; Ferrarini, A.; Zenoni, S.; Tornielli, G.B.; Fasoli, M.; Dal Santo, S.; Minio, A.; Buson, G.; Tononi, P.; Zago, E.D.; et al. De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genom. 2013, 14, 41. [Google Scholar] [CrossRef]
- Hu, J.; Israeli, A.; Ori, N.; Sun, T.P. The Interaction between DELLA and ARF/IAA Mediates Crosstalk between Gibberellin and Auxin Signaling to Control Fruit Initiation in Tomato. Plant Cell 2018, 30, 1710–1728. [Google Scholar] [CrossRef]
- Pattison, R.J.; Csukasi, F.; Catalá, C. Mechanisms regulating auxin action during fruit development. Physiol. Plant 2014, 151, 62–72. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Huang, J.; Wang, J.; Wang, D.; Huang, R.; Liu, X.; Zhang, H.; Zhang, J.; Wang, X.; Yu, L. Genome-Wide Identification, Evolutionary Expansion, and Expression Analyses of Aux/IAA Gene Family in Castanea mollissima During Seed Kernel Development. Biology 2025, 14, 806. https://doi.org/10.3390/biology14070806
Tian Y, Huang J, Wang J, Wang D, Huang R, Liu X, Zhang H, Zhang J, Wang X, Yu L. Genome-Wide Identification, Evolutionary Expansion, and Expression Analyses of Aux/IAA Gene Family in Castanea mollissima During Seed Kernel Development. Biology. 2025; 14(7):806. https://doi.org/10.3390/biology14070806
Chicago/Turabian StyleTian, Yujuan, Jingmiao Huang, Jinxin Wang, Dongsheng Wang, Ruimin Huang, Xia Liu, Haie Zhang, Jingzheng Zhang, Xiangyu Wang, and Liyang Yu. 2025. "Genome-Wide Identification, Evolutionary Expansion, and Expression Analyses of Aux/IAA Gene Family in Castanea mollissima During Seed Kernel Development" Biology 14, no. 7: 806. https://doi.org/10.3390/biology14070806
APA StyleTian, Y., Huang, J., Wang, J., Wang, D., Huang, R., Liu, X., Zhang, H., Zhang, J., Wang, X., & Yu, L. (2025). Genome-Wide Identification, Evolutionary Expansion, and Expression Analyses of Aux/IAA Gene Family in Castanea mollissima During Seed Kernel Development. Biology, 14(7), 806. https://doi.org/10.3390/biology14070806