
Identification of Novel Molecular Panel as Potential Biomarkers of PAN-Gastrointestinal Cancer Screening: Bioinformatics and Experimental Analysis

 , , ,
, , , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Ethical Code
2.3. Integrative Analysis of Microarray and RNA-Seq Data: A Pathway to Biomarker Discovery in Gastrointestinal Malignancies
2.3.1. Microarray
2.3.2. RNA-Seq
2.4. Participating in the Experimental Wet Lab
2.5. RNA Isolation, cDNA Synthesis, and Optimizing qPCR Performance
2.6. Statistical Analysis
3. Results
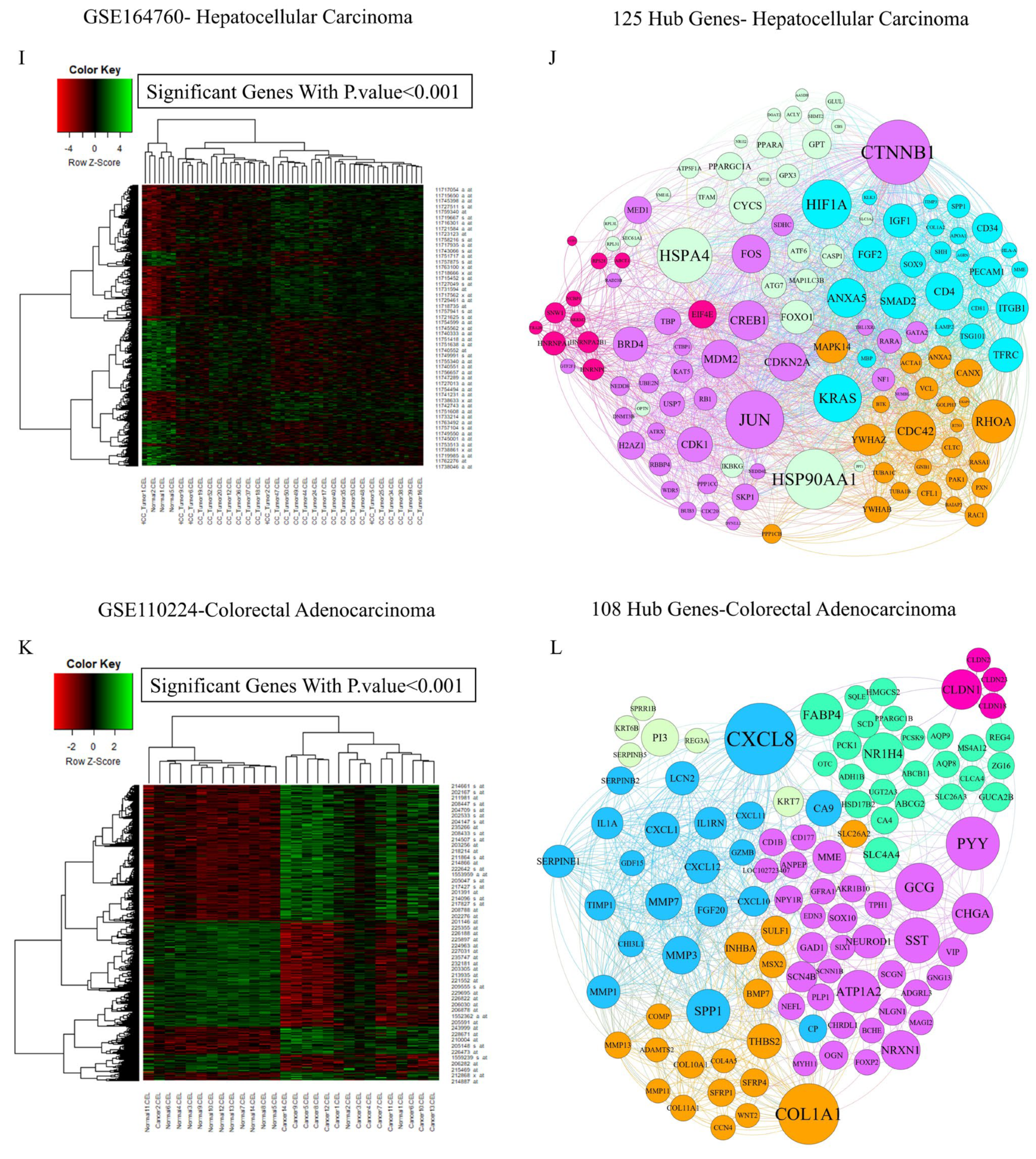
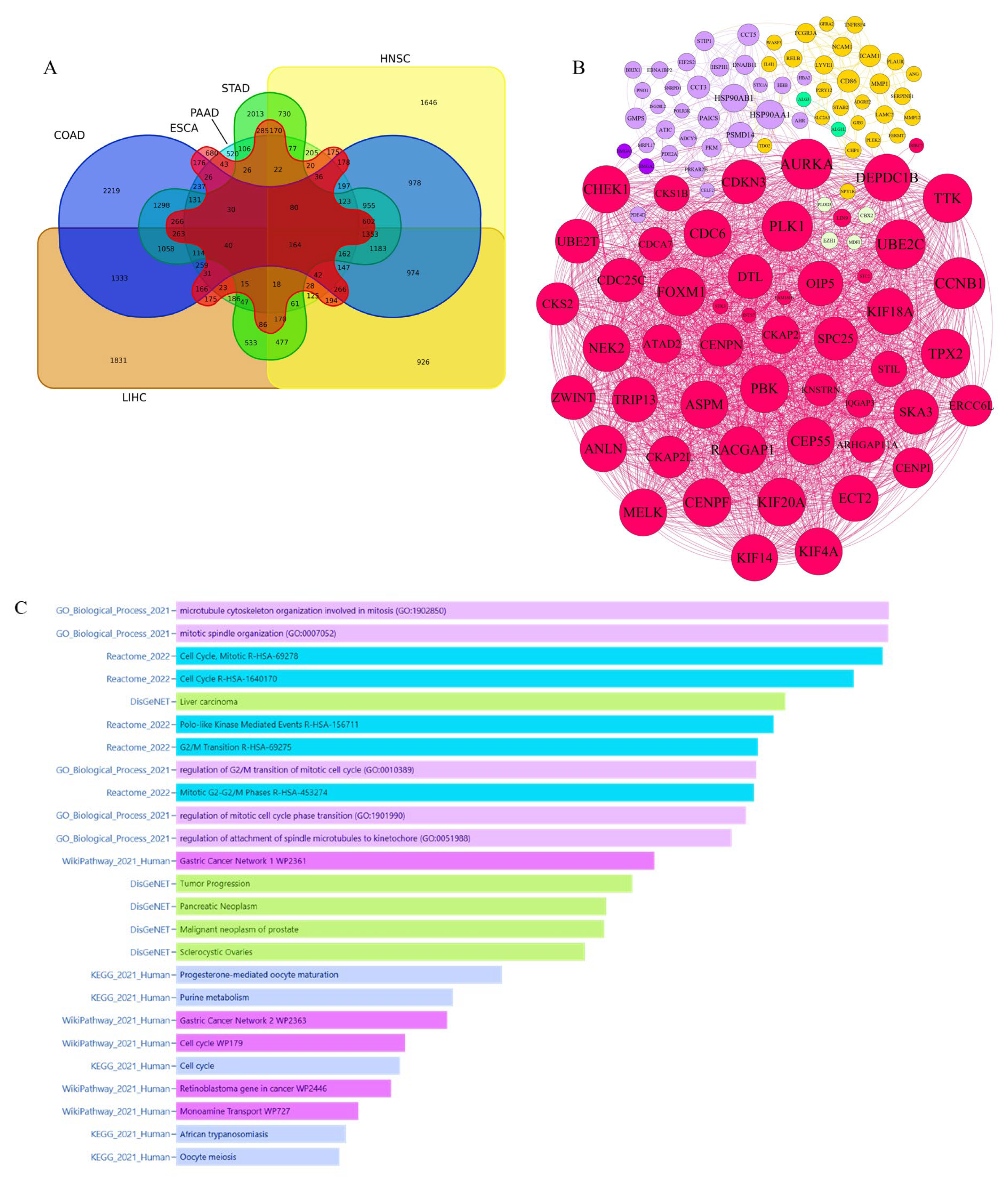
3.1. Integrated Biomarker Discovery in PAN-Gastrointestinal Cancers
3.1.1. Pinpointing Key Drivers of Gastrointestinal Malignancy Through Integrated Microarray Data Analysis
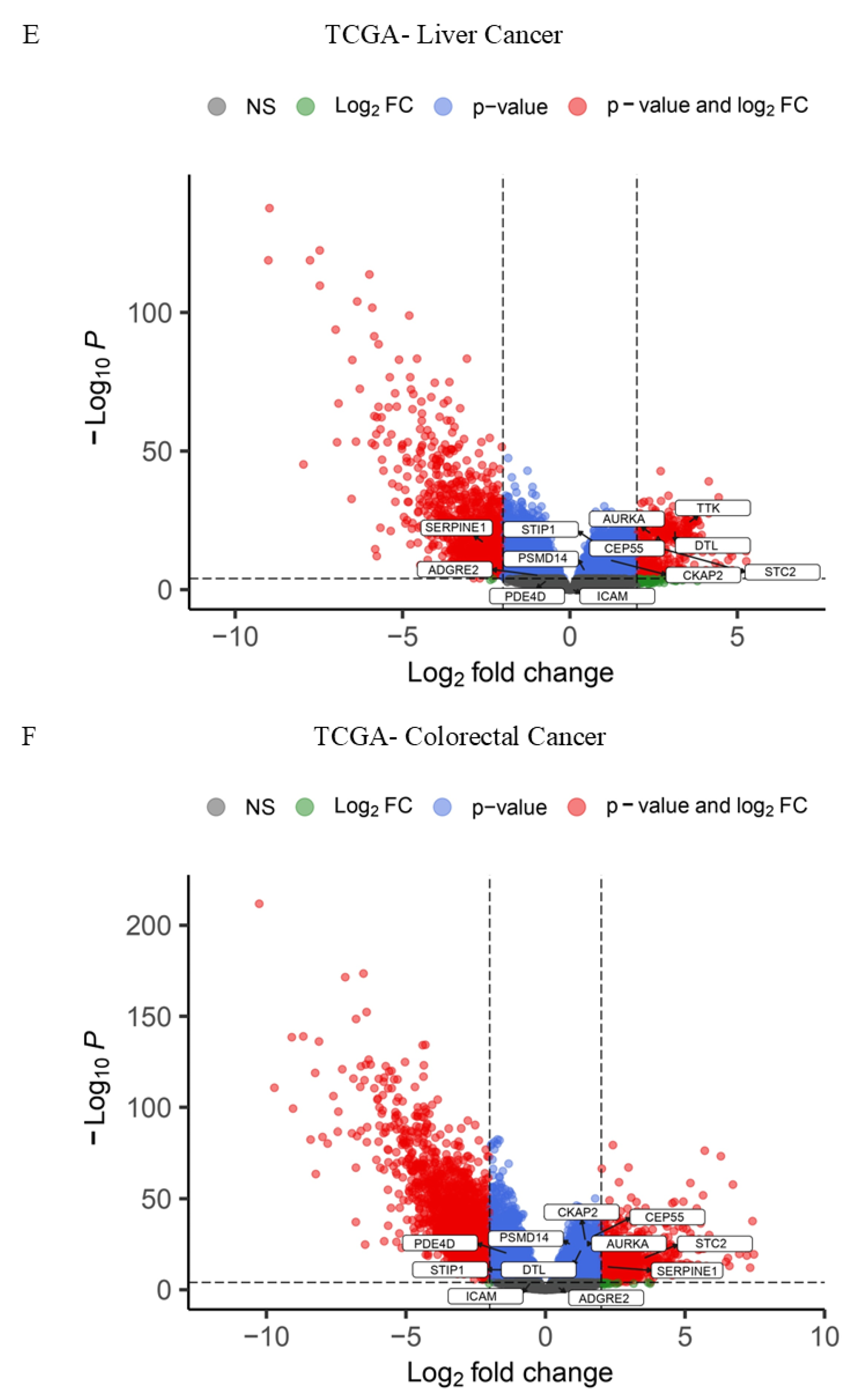
3.1.2. Pinpointing Key Drivers of Gastrointestinal Malignancy Through Integrated TCGA RNA-Seq Data Analysis
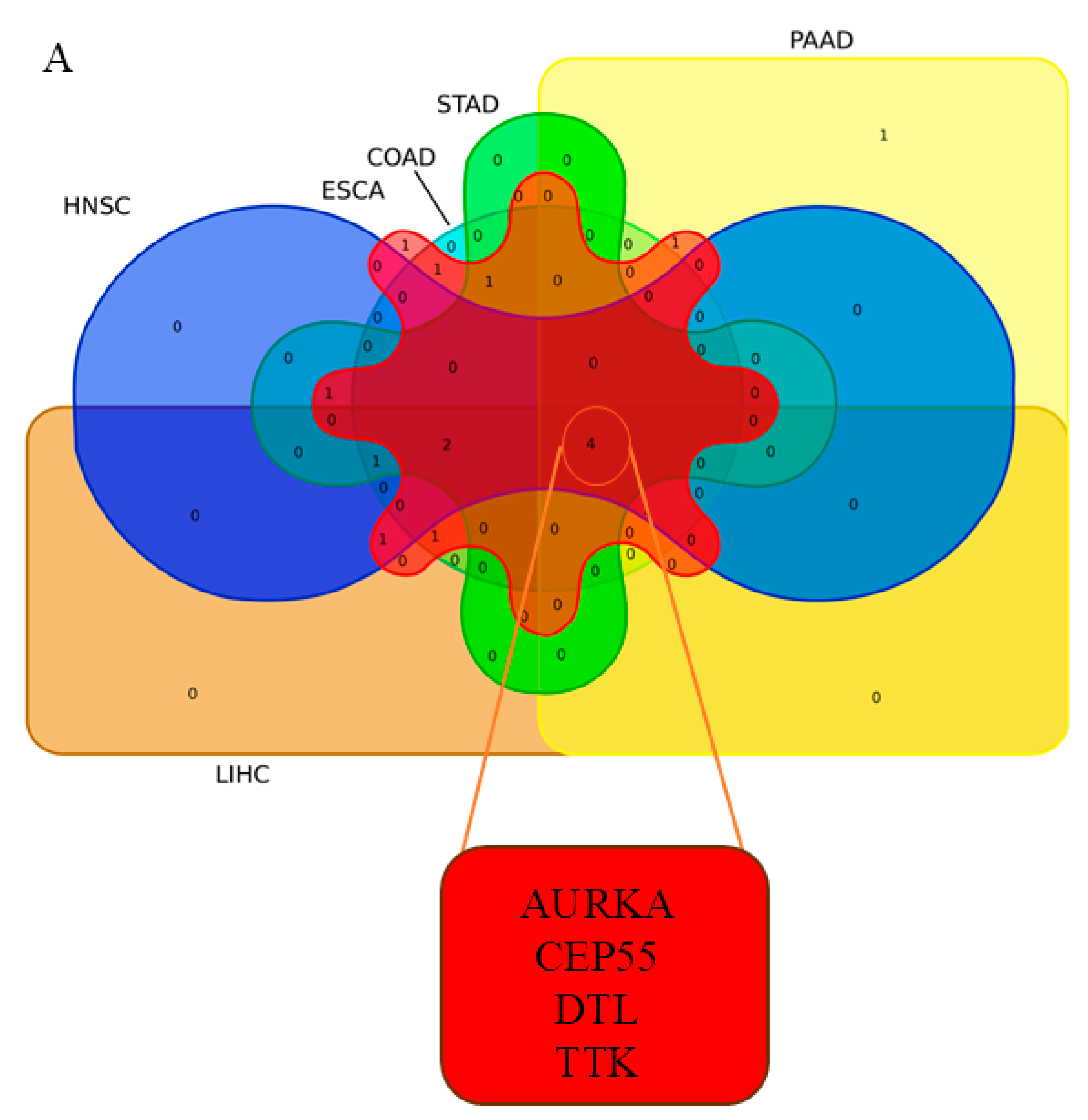
3.1.3. Integrative Analysis of Microarray and RNA-Seq Data Reveals Key Pathways and a 16-Gene Biomarker Panel for Gastrointestinal Cancers
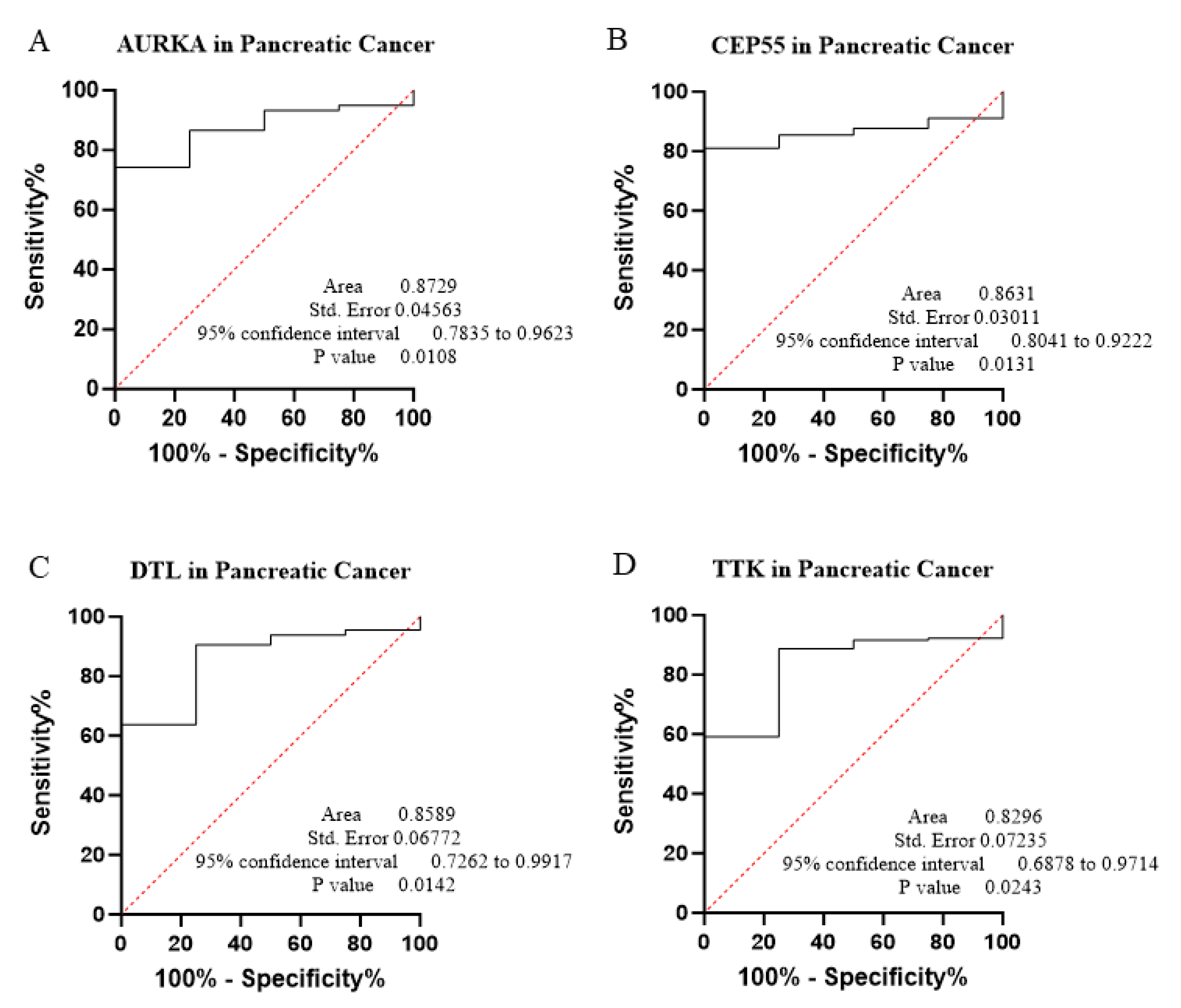
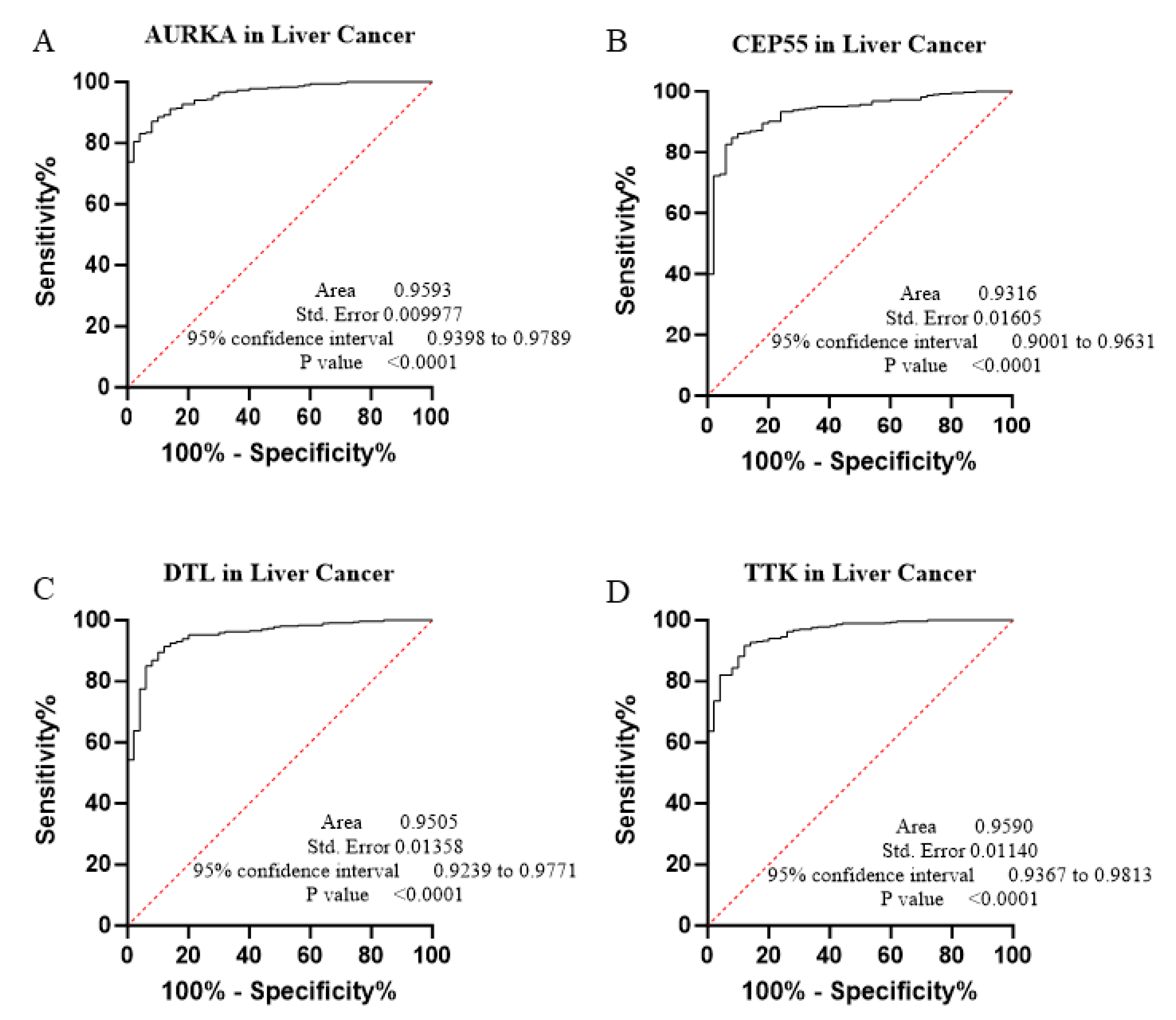
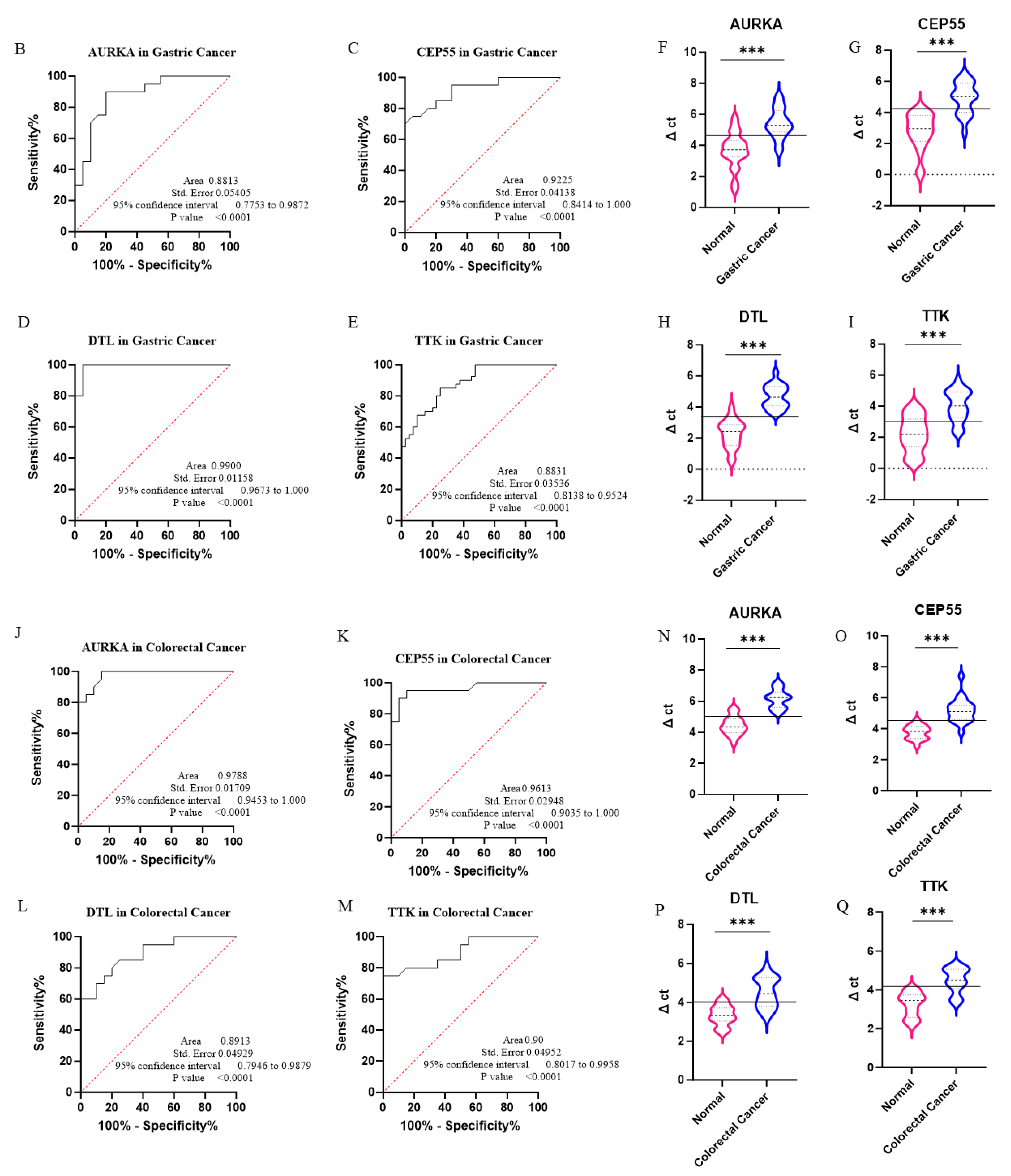
3.2. Diagnostic Performance and ROC Curve Analysis
3.3. Experimental Validation via qPCR
4. Discussion
5. Limitations and Future Perspectives
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chattopadhyay, M.; Gupta, S. Drugs Affecting Gastrointestinal Functions. In Essentials of Pharmacodynamics and Drug Action; Springer: Berlin/Heidelberg, Germany, 2024; pp. 145–168. [Google Scholar]
- Moravveji, S.S.; Khoshbakht, S.; Mokhtari, M.; Salimi, M.; Masoudi-Nejad, A. Pan-cancer analysis of biological events on cell cycle instability in gastrointestinal cancers with integrative scoring method. Genomics 2022, 114, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Bijlsma, M.F.; Sadanandam, A.; Tan, P.; Vermeulen, L. Molecular subtypes in cancers of the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 333–342. [Google Scholar] [CrossRef]
- Adam, R.S.; Blomberg, I.; Ten Hoorn, S.; Bijlsma, M.F.; Vermeulen, L. The recurring features of molecular subtypes in distinct gastrointestinal malignancies—A systematic review. Crit. Rev. Oncol./Hematol. 2021, 164, 103428. [Google Scholar] [CrossRef]
- Ning, Y.; Lin, K.; Fang, J.; Ding, Y.; Zhang, Z.; Chen, X.; Zhao, Q.; Wang, H.; Wang, F. Gastrointestinal pan-cancer landscape of tumor matrix heterogeneity identifies biologically distinct matrix stiffness subtypes predicting prognosis and chemotherapy efficacy. Comput. Struct. Biotechnol. J. 2023, 21, 2744–2758. [Google Scholar] [CrossRef]
- Salem, M.E.; Puccini, A.; Xiu, J.; Raghavan, D.; Lenz, H.J.; Korn, W.M.; Shields, A.F.; Philip, P.A.; Marshall, J.L.; Goldberg, R.M. Comparative molecular analyses of esophageal squamous cell carcinoma, esophageal adenocarcinoma, and gastric adenocarcinoma. Oncologist 2018, 23, 1319–1327. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, J.; Chen, Y.; Xu, R.; Zhao, Q.; Guo, W. MUC4, MUC16, and TTN genes mutation correlated with prognosis, and predicted tumor mutation burden and immunotherapy efficacy in gastric cancer and pan-cancer. Clin. Transl. Med. 2020, 10, e155. [Google Scholar] [CrossRef]
- Cui, Y.; Guo, W.; Li, Y.; Shi, J.; Ma, S.; Guan, F. Pan-cancer analysis identifies ESM1 as a novel oncogene for esophageal cancer. Esophagus 2021, 18, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Y.; Ren, Y.; Cheng, Z.; Zhang, Y.; Wang, X.; Zhao, H.; Lu, H. Pan-cancer single-cell analysis reveals the core factors and pathway in specific cancer stem cells of upper gastrointestinal cancer. Front. Bioeng. Biotechnol. 2022, 10, 849798. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Mukhtar, S. Protein–Protein Interaction Network Exploration Using Cytoscape. In Protein-Protein Interactions: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2023; pp. 419–427. [Google Scholar]
- Majeed, S.; Uzair, M.; Qamar, U.; Farooq, A. Social Network Analysis Visualization Tools: A Comparative Review. In Proceedings of the 2020 IEEE 23rd International Multitopic Conference (INMIC), Bahawalpur, Pakistan, 5–7 November 2020; pp. 1–6. [Google Scholar]
- Evangelista, J.E.; Xie, Z.; Marino, G.B.; Nguyen, N.; Clarke, D.J.; Ma’ayan, A. Enrichr-KG: Bridging enrichment analysis across multiple libraries. Nucleic Acids Res. 2023, 51, W168–W179. [Google Scholar] [CrossRef]
- Mohamadynejad, P.; Moghanibashi, M.; Bagheri, K. Identification of novel nuclear pore complex associated proteins in esophageal carcinoma by an integrated bioinformatics analysis. J. Biomol. Struct. Dyn. 2024, 42, 7221–7232. [Google Scholar] [CrossRef]
- Khashei Varnamkhasti, K.; Moghanibashi, M.; Naeimi, S. Genes whose expressions in the primary lung squamous cell carcinoma are able to accurately predict the progression of metastasis through lymphatic system, inferred from a bioinformatics analyses. Sci. Rep. 2023, 13, 6733. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Alipour, M.; Moghanibashi, M.; Naeimi, S.; Mohamadynejad, P. LINC00894, YEATS2-AS1, and SUGP2 genes as novel biomarkers for N0 status of lung adenocarcinoma. Sci. Rep. 2025, 15, 10628. [Google Scholar] [CrossRef]
- Chen, Y.; McCarthy, D.; Robinson, M.; Smyth, G.K. edgeR: Differential expression analysis of digital gene expression data User’s Guide. In Bioconductor User’s Guide; Fred Hutchinson Cancer Center: Seattle, WA, USA, 2014. [Google Scholar]
- Pereira, M.B.; Wallroth, M.; Jonsson, V.; Kristiansson, E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genom. 2018, 19, 274. [Google Scholar] [CrossRef]
- Khashei Varnamkhasti, K.; Moghanibashi, M.; Naeimi, S. Implications of ZNF334 gene in lymph node metastasis of lung SCC: Potential bypassing of cellular senescence. J. Transl. Med. 2024, 22, 372. [Google Scholar] [CrossRef]
- Oliveros, J.C.; VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. 2007. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 27 June 2025).
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network analysis and visualization of proteomics data. J. Proteome Res. 2018, 18, 623–632. [Google Scholar] [CrossRef]
- Kang, H. Sample size determination and power analysis using the G* Power software. J. Educ. Eval. Health Prof. 2021, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, Q.; Gao, M.; Li, J.; Pang, B. The role of Cyclin Dependent Kinase Inhibitor 3 (CDKN3) in promoting human tumors: Literature review and pan-cancer analysis. Heliyon 2024, 10, e26061. [Google Scholar] [CrossRef] [PubMed]
- Petralia, F.; Ma, W.; Yaron, T.M.; Caruso, F.P.; Tignor, N.; Wang, J.M.; Charytonowicz, D.; Johnson, J.L.; Huntsman, E.M.; Marino, G.B. Pan-cancer proteogenomics characterization of tumor immunity. Cell 2024, 187, 1255–1277.e27. [Google Scholar] [CrossRef] [PubMed]
- Kaviani, E.; Hosseini, A.; Mahmoudi Maymand, E.; Ramzi, M.; Ghaderi, A.; Ramezani, A. Triggering of lymphocytes by CD28, 4-1BB, and PD-1 checkpoints to enhance the immune response capacities. PLoS ONE 2022, 17, e0275777. [Google Scholar] [CrossRef]
- Dai, L.; Huang, Z.; Li, W. Analysis of the PD-1 ligands among gastrointestinal cancer patients: Focus on cancer immunity. Front. Oncol. 2021, 11, 637015. [Google Scholar] [CrossRef]
- Dhakras, P.; Uboha, N.; Horner, V.; Reinig, E.; Matkowskyj, K.A. Gastrointestinal cancers: Current biomarkers in esophageal and gastric adenocarcinoma. Transl. Gastroenterol. Hepatol. 2020, 5, 55. [Google Scholar] [CrossRef]
- Li, Z.; Chen, S.; Feng, W.; Luo, Y.; Lai, H.; Li, Q.; Xiu, B.; Li, Y.; Li, Y.; Huang, S. A pan-cancer analysis of HER2 index revealed transcriptional pattern for precise selection of HER2-targeted therapy. eBioMedicine 2020, 62, 103074. [Google Scholar] [CrossRef]
- Westphalen, C.; Preinfalk, A.; Kruger, S.; Haas, M.; Renz, B.; Riener, M.-O.; Weber, A.; Kirchner, T.; Werner, J.; Heinemann, V. Neurotrophic tropomyosin receptor kinase (NTRK) and nerve growth factor (NGF) are not expressed in Caucasian patients with biliary tract cancers: Pooled data from three independent cohorts. Clin. Transl. Oncol. 2019, 21, 1108–1111. [Google Scholar] [CrossRef]
- Saleh, R.R.; Peinado, P.; Fuentes-Antrás, J.; Pérez-Segura, P.; Pandiella, A.; Amir, E.; Ocaña, A. Prognostic value of lymphocyte-activation gene 3 (LAG3) in cancer: A meta-analysis. Front. Oncol. 2019, 9, 1040. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, N.; Zhang, Z.; Zhang, Y.; Zhang, G.; Chen, W. Upregulation of T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) in monocytes/macrophages associates with gastric cancer progression. Immunol. Investig. 2017, 46, 134–148. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, R.; Shen, L. Evaluation and reflection on claudin 18.2 targeting therapy in advanced gastric cancer. Chin. J. Cancer Res. 2020, 32, 263. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Elsner, J.; Cashion, D.; Robinson, D.; Bahmanyar, S.; Tehrani, L.; Fultz, K.E.; Narla, R.K.; Peng, X.; Tran, T.; Apuy, J. Structure-guided optimization provides a series of TTK protein inhibitors with Potent Antitumor Activity. J. Med. Chem. 2021, 64, 12670–12679. [Google Scholar] [CrossRef]
- Chandler, B.C.; Moubadder, L.; Ritter, C.L.; Liu, M.; Cameron, M.; Wilder-Romans, K.; Zhang, A.; Pesch, A.M.; Michmerhuizen, A.R.; Hirsh, N. TTK inhibition radiosensitizes basal-like breast cancer through impaired homologous recombination. J. Clin. Investig. 2020, 130, 958–973. [Google Scholar] [CrossRef]
- Chan, C.Y.-K.; Chiu, D.K.-C.; Yuen, V.W.-H.; Law, C.-T.; Wong, B.P.-Y.; Thu, K.L.; Cescon, D.W.; Soria-Bretones, I.; Cheu, J.W.-S.; Lee, D. CFI-402257, a TTK inhibitor, effectively suppresses hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2119514119. [Google Scholar] [CrossRef]
- Maia, A.R.R.; Linder, S.; Song, J.-Y.; Vaarting, C.; Boon, U.; Pritchard, C.E.; Velds, A.; Huijbers, I.J.; Van Tellingen, O.; Jonkers, J. Mps1 inhibitors synergise with low doses of taxanes in promoting tumour cell death by enhancement of errors in cell division. Br. J. Cancer 2018, 118, 1586–1595. [Google Scholar] [CrossRef]
- Du, H.; Zhang, L.; Chen, J.; Chen, X.; Qiang, R.; Ding, X.; Wang, Y.; Yang, X. Upregulation of TTK expression is associated with poor prognosis and immune infiltration in endometrial cancer patients. Cancer Cell Int. 2024, 24, 20. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, A.; Lin, J.; Wu, L.; Zhang, H.; Yang, X.; Wan, X.; Miao, R.; Sang, X.; Zhao, H. Mps1/TTK: A novel target and biomarker for cancer. J. Drug Target. 2017, 25, 112–118. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, B.; Zhu, N.; Tao, M.; Jun, Y.; Chen, X.; Wang, Q.; Luo, C. Mitotic checkpoint kinase Mps1/TTK predicts prognosis of colon cancer patients and regulates tumor proliferation and differentiation via PKCα/ERK1/2 and PI3K/Akt pathway. Med. Oncol. 2020, 37, 5. [Google Scholar] [CrossRef]
- Kaistha, B.; Honstein, T.; Müller, V.; Bielak, S.; Sauer, M.; Kreider, R.; Fassan, M.; Scarpa, A.; Schmees, C.; Volkmer, H. Key role of dual specificity kinase TTK in proliferation and survival of pancreatic cancer cells. Br. J. Cancer 2014, 111, 1780–1787. [Google Scholar] [CrossRef]
- Tsai, Y.-M.; Wu, K.-L.; Chang, Y.-Y.; Hung, J.-Y.; Chang, W.-A.; Chang, C.-Y.; Jian, S.-F.; Tsai, P.-H.; Huang, Y.-C.; Chong, I.-W. Upregulation of Thr/Tyr kinase increases the cancer progression by neurotensin and dihydropyrimidinase-like 3 in lung cancer. Int. J. Mol. Sci. 2020, 21, 1640. [Google Scholar] [CrossRef] [PubMed]
- Basanta, C.D.L.A.C.; Bazzi, M.; Hijazi, M.; Bessant, C.; Cutillas, P.R. Community detection in empirical kinase networks identifies new potential members of signalling pathways. PLoS Comput. Biol. 2023, 19, e1010459. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Gao, J.; Zhu, H.; Cui, B.; Zhang, T.; Zhou, Y.; Liu, Q.; He, H.; Xiao, R. A novel USP9X substrate TTK contributes to tumorigenesis in non-small-cell lung cancer. Theranostics 2018, 8, 2348. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Wu, Y.; Zhang, H.; Zhou, H.; Sun, X.; Csizmadia, E.; He, L.; Zhao, Y.; Jiang, C.; Miksad, R.A. Utility of the dual-specificity protein kinase TTK as a therapeutic target for intrahepatic spread of liver cancer. Sci. Rep. 2016, 6, 33121. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Wang, L.; Peng, H.; Xiao, L.; Li, C.; Lin, D.; Yang, K. Silencing TTK expression inhibits the proliferation and progression of prostate cancer. Exp. Cell Res. 2019, 385, 111669. [Google Scholar] [CrossRef]
- Jeffery, J.; Sinha, D.; Srihari, S.; Kalimutho, M.; Khanna, K. Beyond cytokinesis: The emerging roles of CEP55 in tumorigenesis. Oncogene 2016, 35, 683–690. [Google Scholar] [CrossRef]
- Wang, G.; Chen, B.; Su, Y.; Qu, N.; Zhou, D.; Zhou, W. CEP55 as a Promising Immune Intervention Marker to Regulate Tumor Progression: A Pan-Cancer Analysis with Experimental Verification. Cells 2023, 12, 2457. [Google Scholar] [CrossRef]
- Lin, K.; Zhu, X.; Luo, C.; Bu, F.; Zhu, J.; Zhu, Z. Data mining combined with experiments to validate CEP55 as a prognostic biomarker in colorectal cancer. Immun. Inflamm. Dis. 2021, 9, 167–182. [Google Scholar] [CrossRef]
- Zaki, M.S.A.; Eldeen, M.A.; Abdulsahib, W.K.; Shati, A.A.; Alqahtani, Y.A.; Al-Qahtani, S.M.; Otifi, H.M.; Asiri, A.; Hassan, H.M.; Emam Mohammed Ahmed, H. A comprehensive pan-cancer analysis identifies CEP55 as a potential oncogene and novel therapeutic target. Diagnostics 2023, 13, 1613. [Google Scholar] [CrossRef]
- Li, G.-S.; Zhang, W.; Huang, W.-Y.; He, R.-Q.; Huang, Z.-G.; Gan, X.-Y.; Yang, Z.; Dang, Y.-W.; Kong, J.-L.; Zhou, H.-F. CEP55: An immune-related predictive and prognostic molecular biomarker for multiple cancers. BMC Pulm. Med. 2023, 23, 166. [Google Scholar] [CrossRef]
- Xie, X.; Liang, H.; Jiangting, W.; Wang, Y.; Ma, X.; Tan, Z.; Cheng, L.; Luo, Z.; Wang, T. Cancer-testis antigen CEP55 serves as a prognostic biomarker and is correlated with immune infiltration and immunotherapy efficacy in pan-cancer. Front. Mol. Biosci. 2023, 10, 1198557. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, A.R.; Sharma, G.; Chakraborty, C.; Kim, J. PLK-1: Angel or devil for cell cycle progression. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2016, 1865, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Milletti, G.; Colicchia, V.; Cecconi, F. Cycl ers’ kinases in cell division: From molecules to cancer therapy. Cell Death Differ. 2023, 30, 2035–2052. [Google Scholar] [CrossRef] [PubMed]
- Frosk, P.; Arts, H.H.; Philippe, J.; Gunn, C.S.; Brown, E.L.; Chodirker, B.; Simard, L.; Majewski, J.; Fahiminiya, S.; Russell, C. A truncating mutation in CEP55 is the likely cause of MARCH, a novel syndrome affecting neuronal mitosis. J. Med. Genet. 2017, 54, 490–501. [Google Scholar] [CrossRef]
- Bondeson, M.L.; Ericson, K.; Gudmundsson, S.; Ameur, A.; Pontén, F.; Wesström, J.; Frykholm, C.; Wilbe, M. A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy. Clin. Genet. 2017, 92, 510–516. [Google Scholar] [CrossRef]
- Sinha, D.; Nag, P.; Nanayakkara, D.; Duijf, P.H.; Burgess, A.; Raninga, P.; Smits, V.A.; Bain, A.L.; Subramanian, G.; Wall, M. Cep55 overexpression promotes genomic instability and tumorigenesis in mice. Commun. Biol. 2020, 3, 593. [Google Scholar] [CrossRef]
- Kalimutho, M.; Sinha, D.; Jeffery, J.; Nones, K.; Srihari, S.; Fernando, W.C.; Duijf, P.H.; Vennin, C.; Raninga, P.; Nanayakkara, D. CEP 55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol. Med. 2018, 10, e8566. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, Y.; Li, Y.; Lu, J.; Huang, Q.; Xu, R.; Feng, Y.; Yan, S. High CEP55 expression is associated with poor prognosis in non-small-cell lung cancer. OncoTargets Ther. 2018, 11, 4979–4990. [Google Scholar] [CrossRef]
- Xu, L.; Xia, C.; Sheng, F.; Sun, Q.; Xiong, J.; Wang, S. CEP55 promotes the proliferation and invasion of tumour cells via the AKT signalling pathway in osteosarcoma. Carcinogenesis 2018, 39, 623–631. [Google Scholar] [CrossRef]
- Chen, C.; Lu, P.; Chen, Y.; Fu, S.; Wu, K.; Tsou, A.; Lee, Y.; Lin, T.; Hsu, S.; Lin, W. FLJ10540-elicited cell transformation is through the activation of PI3-kinase/AKT pathway. Oncogene 2007, 26, 4272–4283. [Google Scholar] [CrossRef]
- Yang, C.; Yang, Y.; Wang, W.; Zhou, W.; Zhang, X.; Xiao, Y.; Zhang, H. CEP55 3’-UTR promotes epithelial–mesenchymal transition and enhances tumorigenicity of bladder cancer cells by acting as a ceRNA regulating miR-497-5p. Cell. Oncol. 2022, 45, 1217–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, Q.; Li, E.; Shi, T.; Chen, H. CEP55 predicts the poor prognosis and promotes tumorigenesis in endometrial cancer by regulating the Foxo1 signaling. Mol. Cell. Biochem. 2023, 478, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.-M.; Liu, L.; Gu, W.-Y.; Huang, L.-Y.; Yang, Y.; Huang, Y.-H.; Luo, R.-Z. CEP55 positively affects tumorigenesis of esophageal squamous cell carcinoma and is correlated with poor prognosis. J. Oncol. 2021, 2021, 8890715. [Google Scholar] [CrossRef]
- Wu, S.; Wu, D.; Pan, Y.; Liu, H.; Shao, Z.; Wang, M. Correlation between EZH2 and CEP55 and lung adenocarcinoma prognosis. Pathol. -Res. Pract. 2019, 215, 292–301. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, P.; Zhao, H. The association between AURKA gene rs2273535 polymorphism and gastric cancer risk in a Chinese population. Front. Physiol. 2018, 9, 1124. [Google Scholar] [CrossRef]
- Qi, J.; Gao, X.; Zhong, X.; Zhang, N.; Wang, R.; Zhang, H.; Pan, T.; Liu, X.; Yao, Y.; Wu, Q. Selective inhibition of Aurora A and B kinases effectively induces cell cycle arrest in t (8; 21) acute myeloid leukemia. Biomed. Pharmacother. 2019, 117, 109113. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, Y.; Hill, C.; Chen, K.; Cheng, C.; Liu, X.; Duan, P.; Gu, Y.; Wu, Y.; Ewing, R.M. Identification of MYCN non-amplified neuroblastoma subgroups points towards molecular signatures for precision prognosis and therapy stratification. Br. J. Cancer 2024, 130, 1841–1854. [Google Scholar] [CrossRef]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef]
- Lu, H.; Gomaa, A.; Wang-Bishop, L.; Ballout, F.; Hu, T.; McDonald, O.; Washington, M.K.; Livingstone, A.S.; Wang, T.C.; Peng, D. Unfolded protein response is activated by aurora kinase A in esophageal adenocarcinoma. Cancers 2022, 14, 1401. [Google Scholar] [CrossRef]
- Mendiola, M.; Barriuso, J.; Mariño-Enríquez, A.; Redondo, A.; Domínguez-Cáceres, A.; Hernández-Cortés, G.; Pérez-Fernández, E.; Sánchez-Navarro, I.; Vara, J.Á.F.; Suárez, A. Aurora kinases as prognostic biomarkers in ovarian carcinoma. Hum. Pathol. 2009, 40, 631–638. [Google Scholar] [CrossRef]
- Dong, B.; Chai, M.; Chen, H.; Feng, Q.; Jin, R.; Hu, S. Screening and verifying key genes with poor prognosis in colon cancer through bioinformatics analysis. Transl. Cancer Res. 2020, 9, 6720. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.-P.; Wang, J.-Y.; Jao, S.-W.; Wu, C.-C.; Chen, J.-H.; Hsiao, K.-H.; Lin, C.-Y.; Chen, S.-H.; Su, S.-Y.; Chen, Y.-J. Over-expression of AURKA, SKA3 and DSN1 contributes to colorectal adenoma to carcinoma progression. Oncotarget 2016, 7, 45803. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, A.; Taraviras, S.; Nishitani, H.; Lygerou, Z. CRL4Cdt2: Coupling genome stability to ubiquitination. Trends Cell Biol. 2020, 30, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Kiran, S.; Dar, A.; Singh, S.K.; Lee, K.Y.; Dutta, A. The deubiquitinase USP46 is essential for proliferation and tumor growth of HPV-transformed cancers. Mol. Cell 2018, 72, 823–835.e5. [Google Scholar] [CrossRef]
- Luo, Y.; He, Z.; Liu, W.; Zhou, F.; Liu, T.; Wang, G. DTL is a prognostic biomarker and promotes bladder cancer progression through regulating the AKT/mTOR axis. Oxidative Med. Cell. Longev. 2022, 2022, 3369858. [Google Scholar] [CrossRef]
- Li, Z.; Wang, R.; Qiu, C.; Cao, C.; Zhang, J.; Ge, J.; Shi, Y. Role of DTL in hepatocellular carcinoma and its impact on the tumor microenvironment. Front. Immunol. 2022, 13, 834606. [Google Scholar] [CrossRef]
- Lu, J.-J.; Chen, F.-J.; Li, Y.; Xu, X.; Peng, C.; Yu, N.; Su, L.-N.; Tang, L. DTL promotes melanoma progression through rewiring cell glucose metabolism. Ann. Transl. Med. 2022, 10, 68. [Google Scholar] [CrossRef]
- Chang, X.; Jian, L. LncRNA ZFPM2-AS1 drives the progression of nasopharyngeal carcinoma via modulating the downstream miR-3612/DTL signaling. Anti-Cancer Drugs 2022, 33, 523–533. [Google Scholar] [CrossRef]
- Cui, H.; Wang, Q.; Lei, Z.; Feng, M.; Zhao, Z.; Wang, Y.; Wei, G. DTL promotes cancer progression by PDCD4 ubiquitin-dependent degradation. J. Exp. Clin. Cancer Res. 2019, 38, 350. [Google Scholar] [CrossRef]
- Tang, Y.; Lei, Y.; Gao, P.; Jia, J.; Du, H.; Wang, Q.; Yan, Z.; Zhang, C.; Liang, G.; Wang, Y. Pan-cancer analysis and experimental validation of DTL as a potential diagnosis, prognosis and immunotherapy biomarker. BMC Cancer 2023, 23, 328. [Google Scholar] [CrossRef]
- Wu, C.; Tan, J.; Wang, X.; Qin, C.; Long, W.; Pan, Y.; Li, Y.; Liu, Q. Pan-cancer analyses reveal molecular and clinical characteristics of cuproptosis regulators. Imeta 2023, 2, e68. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HNSC * | ESCA * | STAD * | PAAD * | LIHC * | COAD * | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Quantity of specimens | Normal | Cancer | Normal | Cancer | Normal | Cancer | Normal | Cancer | Normal | Cancer | Normal | Cancer |
| GEO | 12 | 6 | 16 | 16 | 20 | 30 | 5 | 10 | 6 | 53 | 14 | 14 |
| TCGA | 32 | 297 | 13 | 185 | 36 | 412 | 4 | 179 | 51 | 373 | 41 | 483 |
| Characteristic | Colorectal Adenocarcinoma (n = 20) | Gastric Adenocarcinoma (n = 20) | p Value |
|---|---|---|---|
| Age | |||
| <50 | 4 (19.0%) | 3 (14.3%) | 0.693 |
| ≥50 | 17 (81.0%) | 18 (85.7%) | |
| Gender | |||
| Male | 15 (71.4%) | 12 (57.1%) | 0.476 |
| Female | 6 (28.6%) | 9 (42.9%) | |
| TNM Stage | |||
| Stage I | 11 (52.4%) | 10 (47.6%) | 0.827 |
| Stage II | 10 (47.6%) | 11 (52.4%) | |
| Tumor Size (cm) | |||
| <5 | NA | ||
| Gene | Sequence of Primers | Annealing Temperature |
|---|---|---|
| AURKA | Forward: 5′-GCAACCAGTGTACCTCATCCTG-3′ | 60 °C |
| Reverse: 5′-AAGTCTTCCAAAGCCCACTGCC-3′ | ||
| CEP55 | Forward: 5′-TCGACCGTCAACATGTGCAGCA-3′ | 60 °C |
| Reverse: 5′- GGCTCTGTGATGGCAAACTCATG-3′ | ||
| DTL | Forward: 5′-CCAGCCTTAGTCCAGATGACCA-3′ | 60 °C |
| Reverse: 5′-GAGAATGACCCAGGAGCACAGT-3′ | ||
| TTK | Forward: 5′-CCGAGATTTGGTTGTGCCTGGA-3′ | 60 °C |
| Reverse: 5′-CATCTGACACCAGAGGTTCCTTG-3′ | ||
| B2M | Forward: 5′-CCACTGAAAAAGATGAGTATGCCT-3′ | 60 °C |
| Reverse: 5′-CCAATCCAAATGCGGCATCTTCA-3′ |
| Gene | Cancer Type | AUC * | 95% CI ** | p Value | Sensitivity (%) | Specificity (%) | Cut-Off Value |
|---|---|---|---|---|---|---|---|
| AURKA | HNSC | 0.9370 | 0.9096–0.9643 | <0.0001 | 83.14 | 90.91 | 4.266 |
| ESCA | 0.9717 | 0.9423–1.000 | <0.0001 | 91.35 | 92.31 | 3.991 | |
| STAD | 0.8713 | 0.8271–0.9154 | <0.0001 | 81.07 | 80.56 | 4.312 | |
| PAAD | 0.8729 | 0.7835–0.9623 | <0.0108 | 74.30 | 100.00 | 3.008 | |
| LIHC | 0.9593 | 0.9398–0.9789 | <0.0001 | 78.34 | 84.00 | 7.474 | |
| COAD | 0.9435 | 0.9167–0.9704 | <0.0001 | 83.64 | 95.12 | 5.010 | |
| CEP55 | HNSC | 0.9317 | 0.9037–0.9598 | <0.0001 | 82.18 | 90.91 | 4.851 |
| ESCA | 0.9422 | 0.8832–1.000 | <0.0001 | 83.78 | 92.31 | 4.622 | |
| STAD | 0.9055 | 0.8676–0.9433 | <0.0001 | 83.25 | 86.11 | 4.230 | |
| PAAD | 0.8631 | 0.8041–0.9222 | <0.0131 | 81.01 | 100.00 | 2.952 | |
| LIHC | 0.9316 | 0.9001–0.9631 | <0.0001 | 84.76 | 92.00 | 0.8585 | |
| COAD | 0.9391 | 0.9128–0.9654 | <0.0001 | 89.86 | 87.80 | 4.374 | |
| DTL | HNSC | 0.9360 | 0.9115–0.9605 | <0.0001 | 78.74 | 97.73 | 4.068 |
| ESCA | 0.9389 | 0.8664–1.000 | <0.0001 | 96.22 | 84.62 | 3.420 | |
| STAD | 0.9088 | 0.8755–0.9421 | <0.0001 | 80.10 | 94.44 | 3.701 | |
| PAAD | 0.8589 | 0.7262–0.9917 | <0.0142 | 81.01 | 100.00 | 2.952 | |
| LIHC | 0.9505 | 0.9239–0.9771 | <0.0001 | 78.34 | 84.00 | 7.474 | |
| COAD | 0.9119 | 0.8787–0.9451 | <0.0001 | 87.37 | 85.37 | 3.943 | |
| TTK | HNSC | 0.8360 | 0.7845–0.8876 | <0.0001 | 57.09 | 93.18 | 4.303 |
| ESCA | 0.9493 | 0.9076–0.9909 | <0.0001 | 78.92 | 100.00 | 4.173 | |
| STAD | 0.8693 | 0.8222–0.9164 | <0.0001 | 63.83 | 97.22 | 3.984 | |
| PAAD | 0.8296 | 0.6878–0.9714 | <0.0243 | 88.83 | 75.00 | 1.444 | |
| LIHC | 0.9590 | 0.9367–0.9813 | <0.0001 | 91.71 | 88.00 | 0.7411 | |
| COAD | 0.9000 | 0.8614–0.9355 | <0.0001 | 87.37 | 87.80 | 3.773 |
| Gene | Cancer Type | AUC * | 95% CI ** | p Value | Youden Index Cut-Off |
|---|---|---|---|---|---|
| AURKA | Colorectal AD | 0.9788 | 0.9453–1.000 | <0.0001 | ≥5.055 |
| Gastric AD | 0.8813 | 0.7753–0.9872 | 0.0001 | ≥4.355 | |
| CEP55 | Colorectal AD | 0.9613 | 0.9035–1.000 | <0.0001 | ≥4.305 |
| Gastric AD | 0.9225 | 0.8414–1.000 | <0.0001 | ≥4.100 | |
| DTL | Colorectal AD | 0.8913 | 0.7946–0.9879 | <0.0001 | ≥3.705 |
| Gastric AD | 0.9900 | 0.9673–1.000 | <0.0001 | ≥3.525 | |
| TTK | Colorectal AD | ~0.90 | 0.8017–0.9956 | <0.0001 | ≥4.140 |
| Gastric AD | 0.8831 | 0.8138–0.9524 | 0.0001 | ≥3.074 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajibabaie, F.; Mohamadynejad, P.; Shariati, L.; Safavi, K.; Abedpoor, N. Identification of Novel Molecular Panel as Potential Biomarkers of PAN-Gastrointestinal Cancer Screening: Bioinformatics and Experimental Analysis. Biology 2025, 14, 803. https://doi.org/10.3390/biology14070803
Hajibabaie F, Mohamadynejad P, Shariati L, Safavi K, Abedpoor N. Identification of Novel Molecular Panel as Potential Biomarkers of PAN-Gastrointestinal Cancer Screening: Bioinformatics and Experimental Analysis. Biology. 2025; 14(7):803. https://doi.org/10.3390/biology14070803
Chicago/Turabian StyleHajibabaie, Fatemeh, Parisa Mohamadynejad, Laleh Shariati, Kamran Safavi, and Navid Abedpoor. 2025. "Identification of Novel Molecular Panel as Potential Biomarkers of PAN-Gastrointestinal Cancer Screening: Bioinformatics and Experimental Analysis" Biology 14, no. 7: 803. https://doi.org/10.3390/biology14070803
APA StyleHajibabaie, F., Mohamadynejad, P., Shariati, L., Safavi, K., & Abedpoor, N. (2025). Identification of Novel Molecular Panel as Potential Biomarkers of PAN-Gastrointestinal Cancer Screening: Bioinformatics and Experimental Analysis. Biology, 14(7), 803. https://doi.org/10.3390/biology14070803