
Dissecting the Cellular Heterogeneity Underlying Liver Diseases Through the Integration of GWASs and Single-Cell RNA Sequencing


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
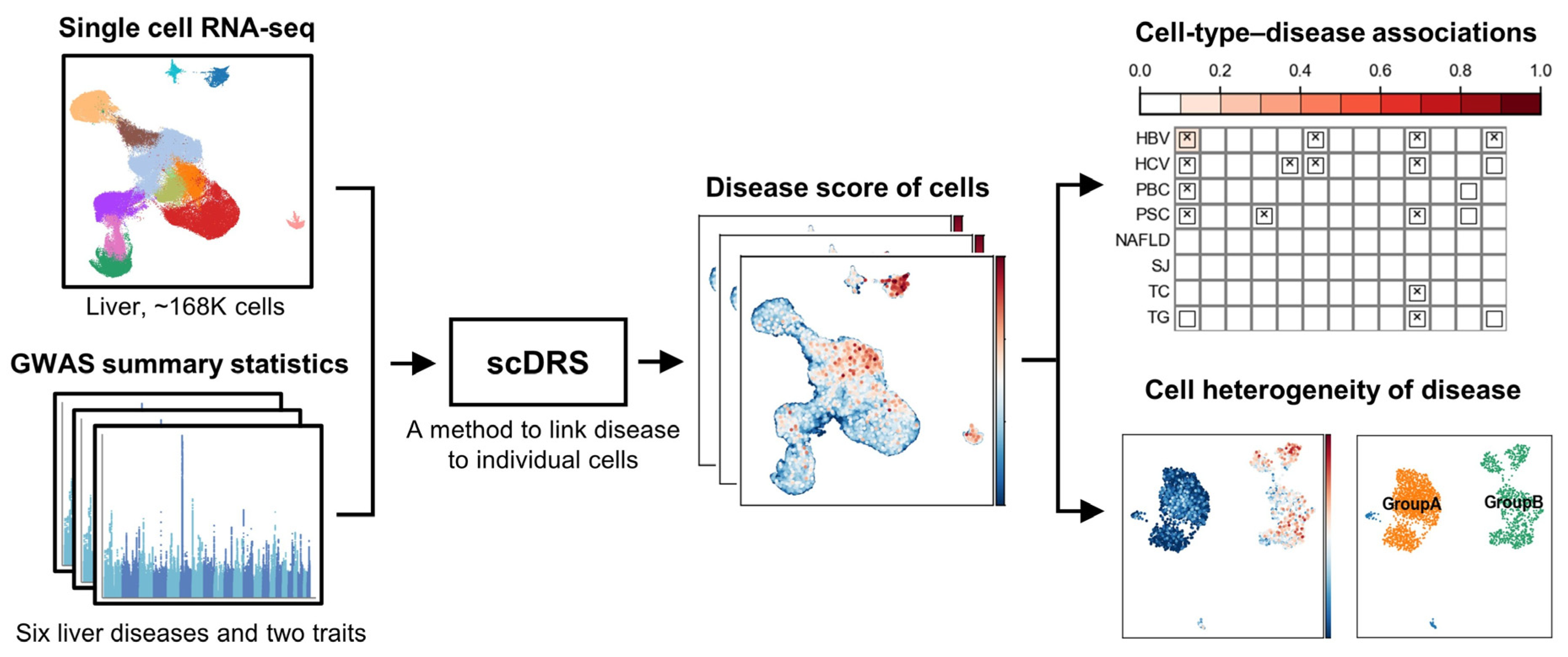
2.1. Overview of Analysis Workflow
2.2. Single-Cell RNA Sequencing Data
2.3. GWAS Summary Statistics
2.4. Identification of Disease-Associated Cells Using scDRS
2.5. Cell Type-Level Association and Heterogeneity Testing
2.6. Analysis of Cell-Type Heterogeneity
2.7. Code Availability
3. Results
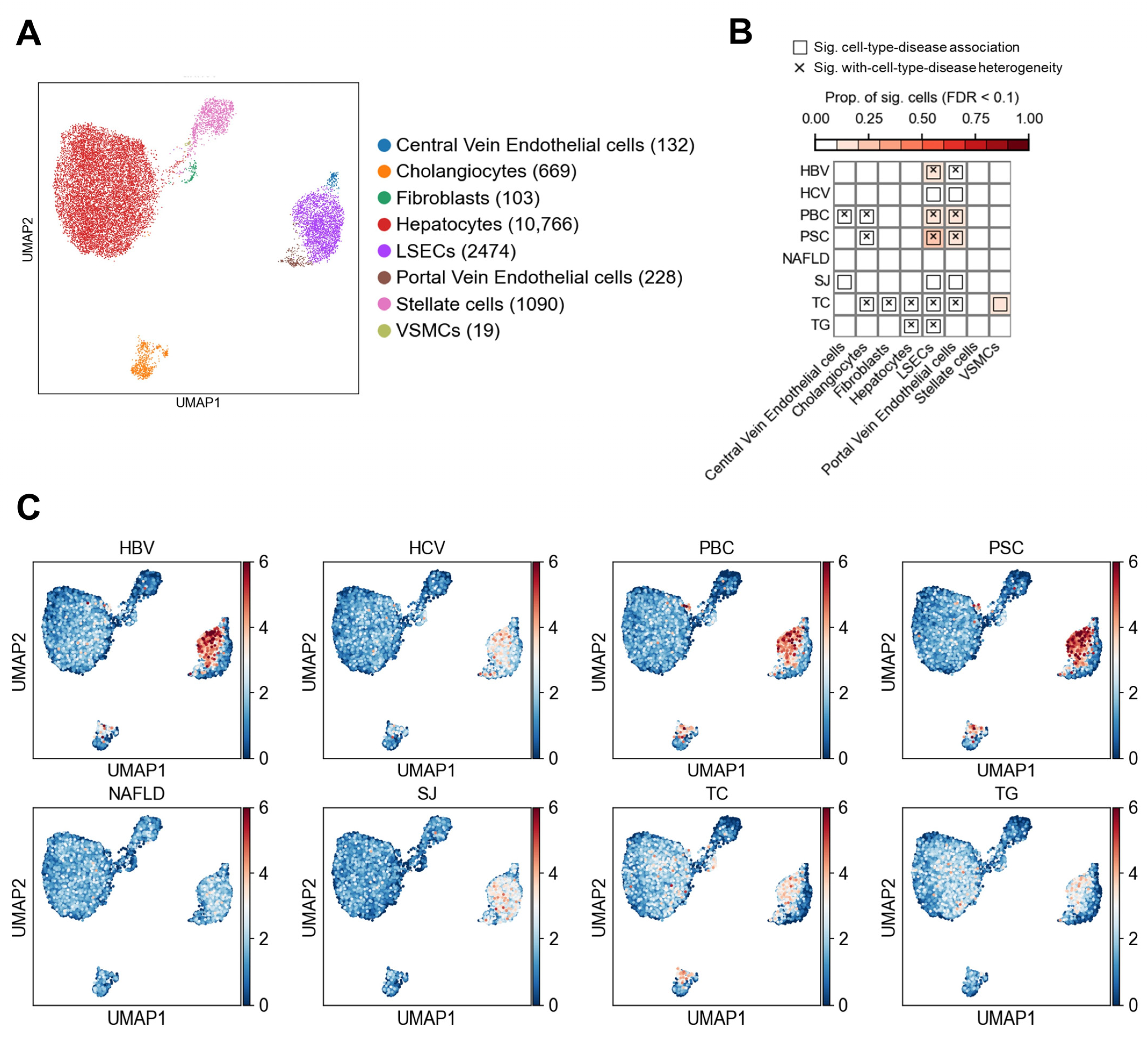
3.1. Association Results in Non-Immune Cells
3.1.1. Association of Non-Immune Cells at the Cell Type Level with Liver Diseases and Traits
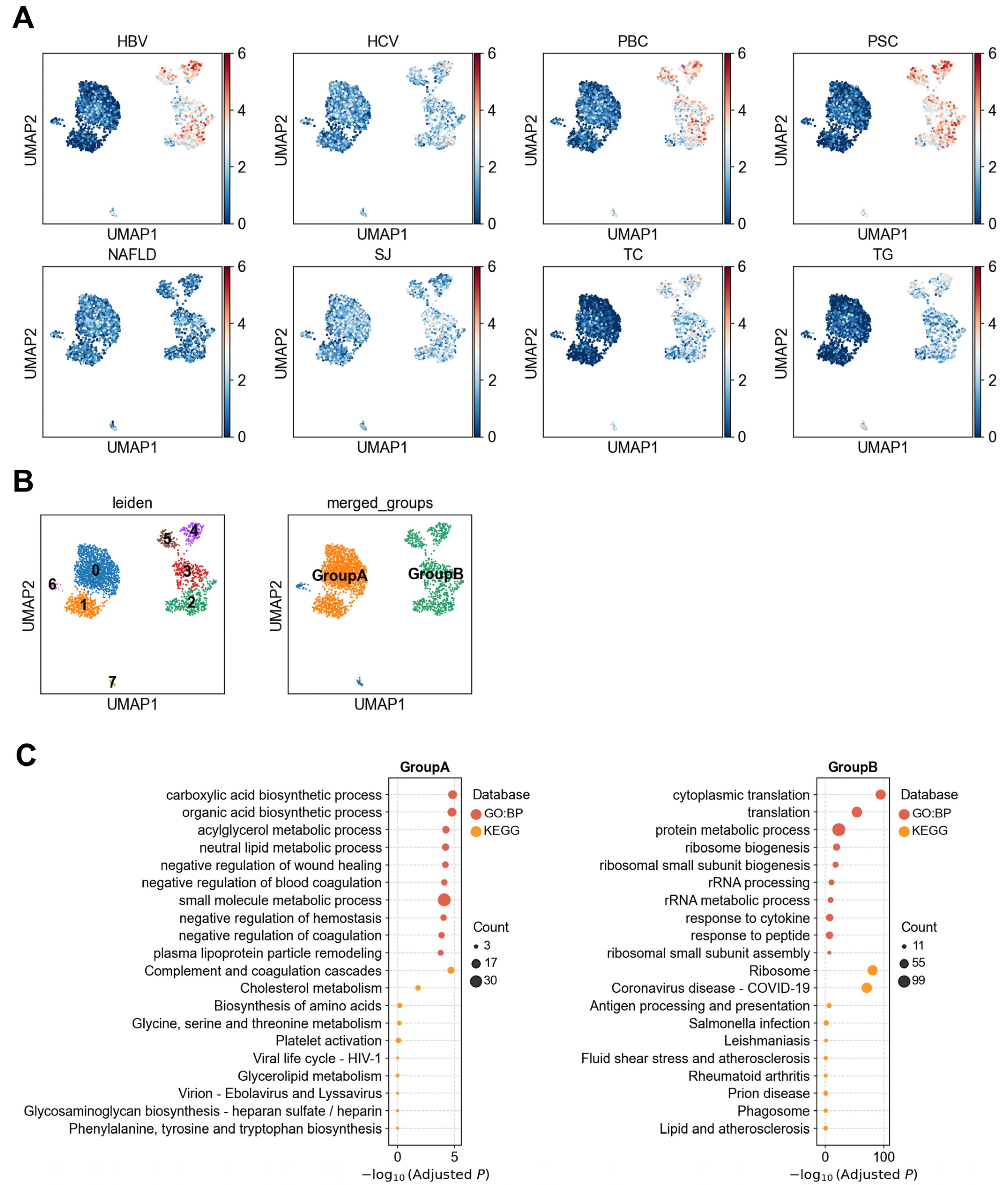
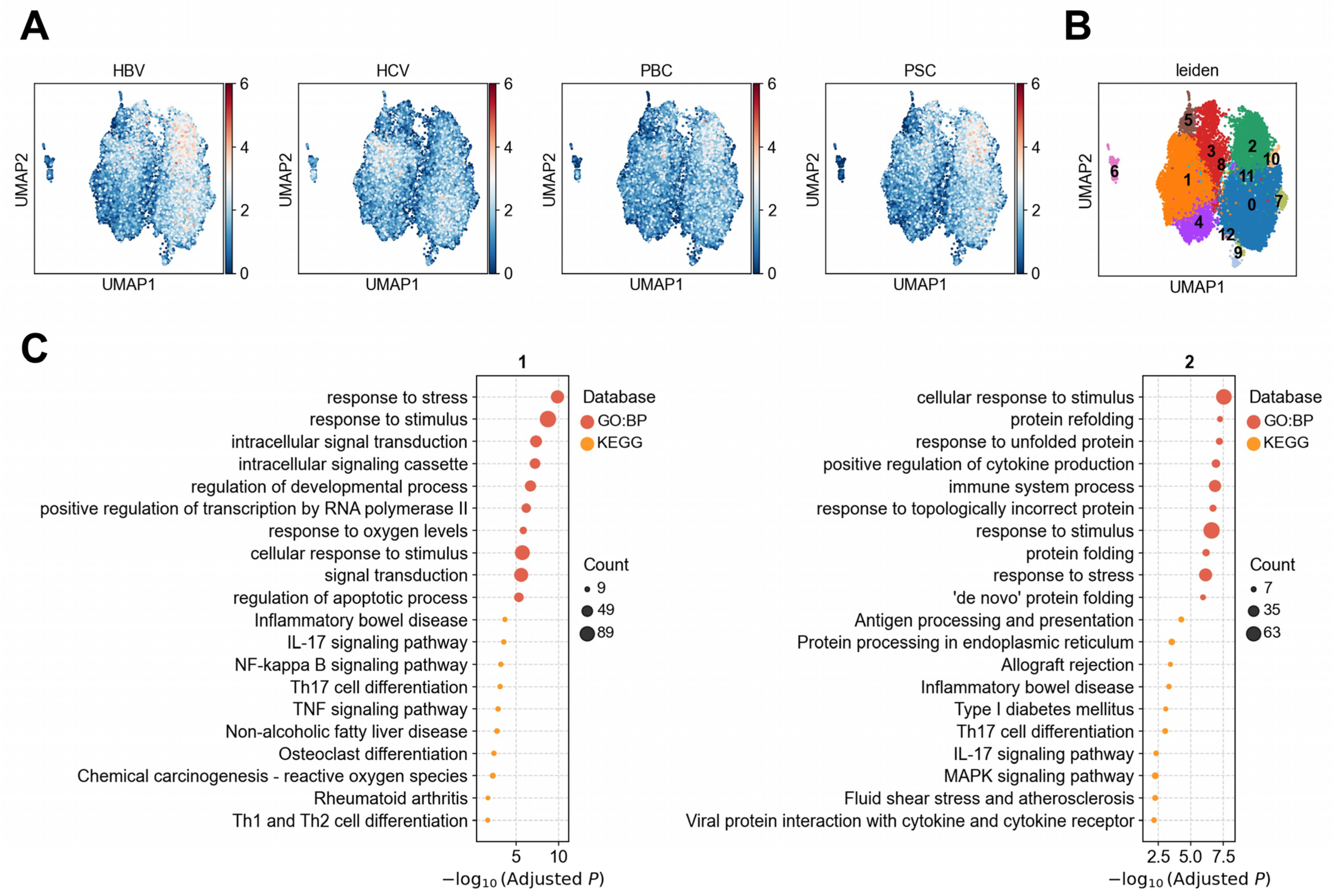
3.1.2. Heterogeneity of LSECs in Association with Liver Diseases
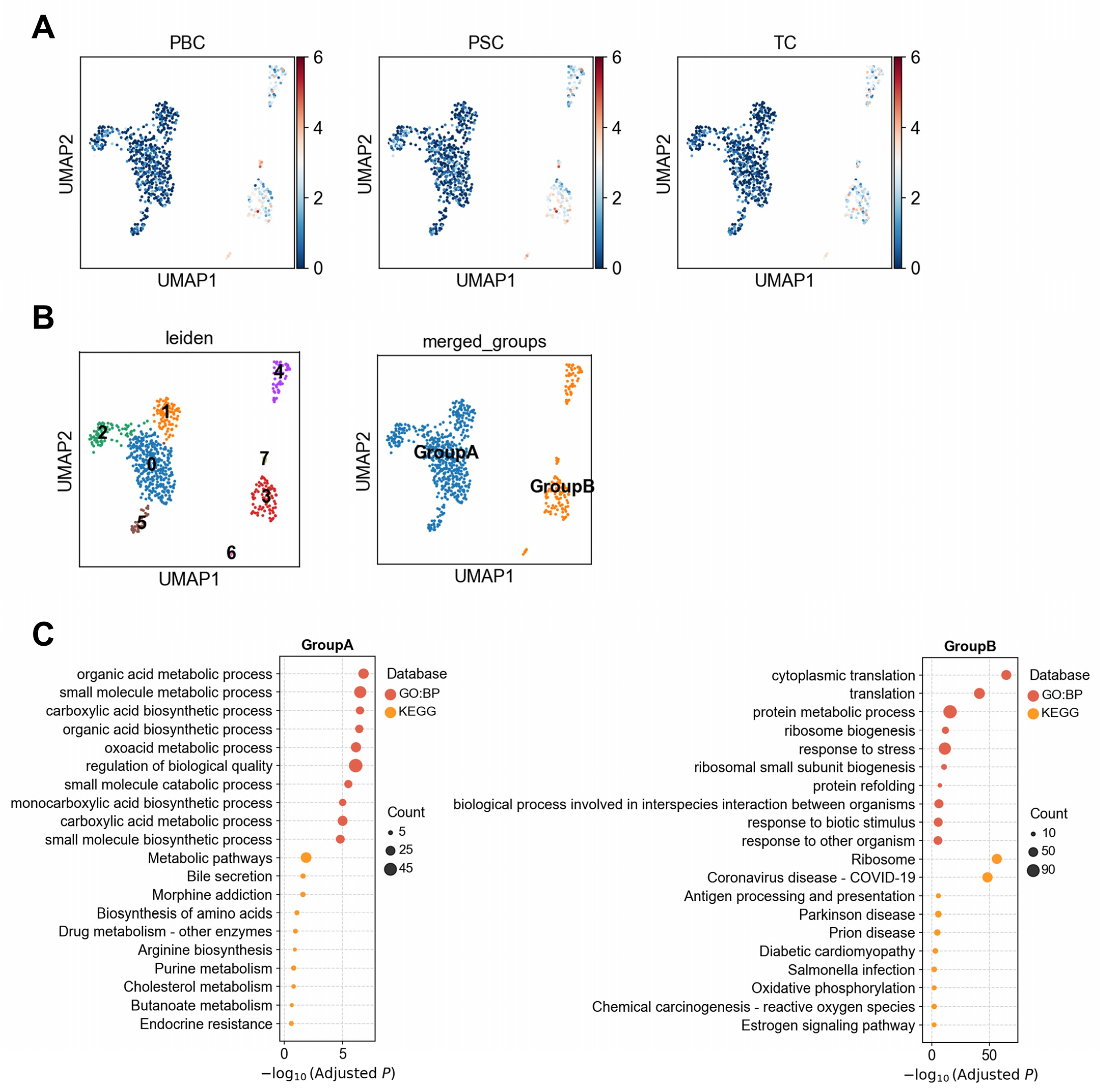
3.1.3. Heterogeneous Association Between Cholangiocytes and PBC/PSC Reveals Functionally Distinct Subpopulations
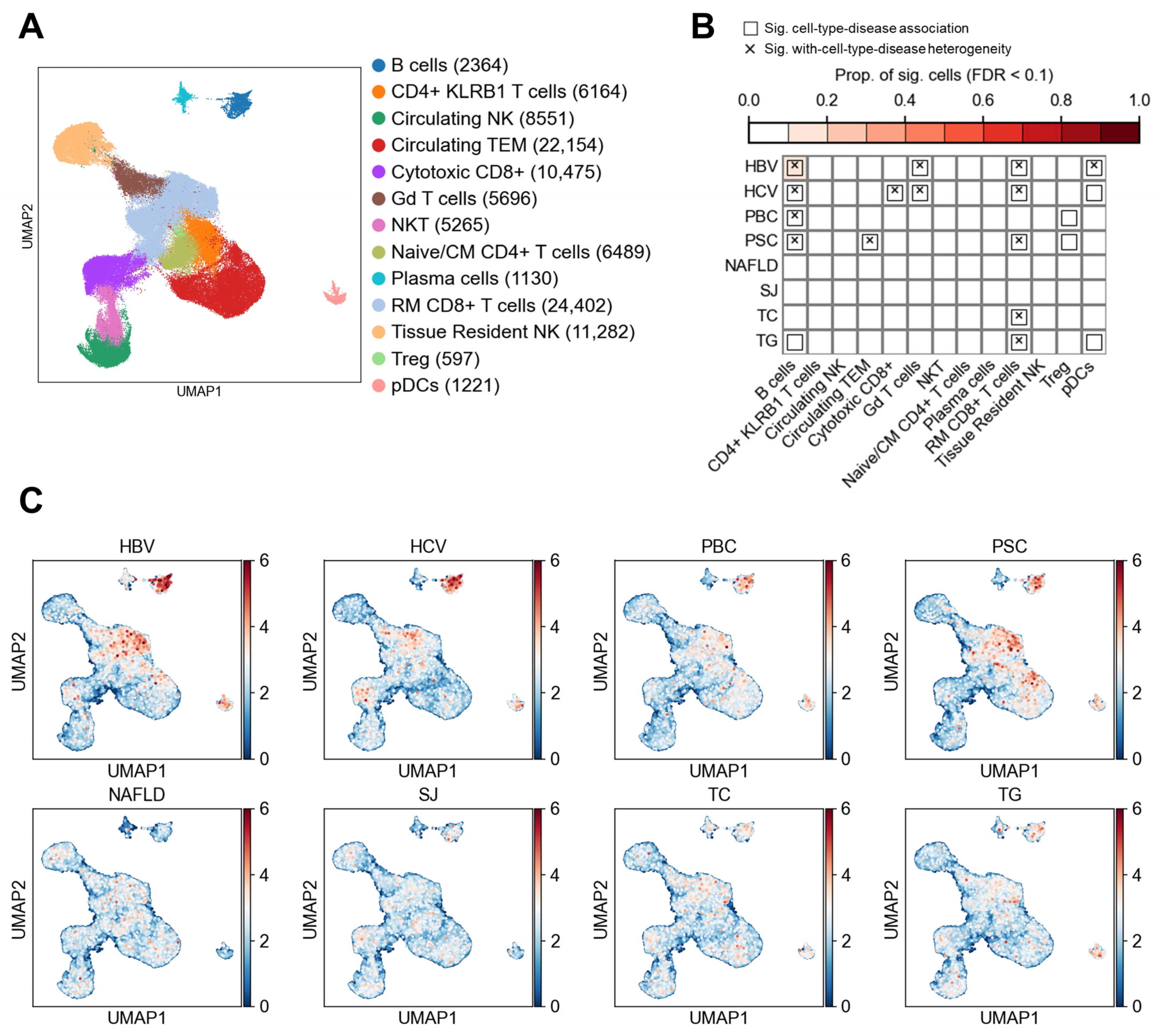
3.2. Lymphocyte Subtypes and Their Associations with Liver Diseases
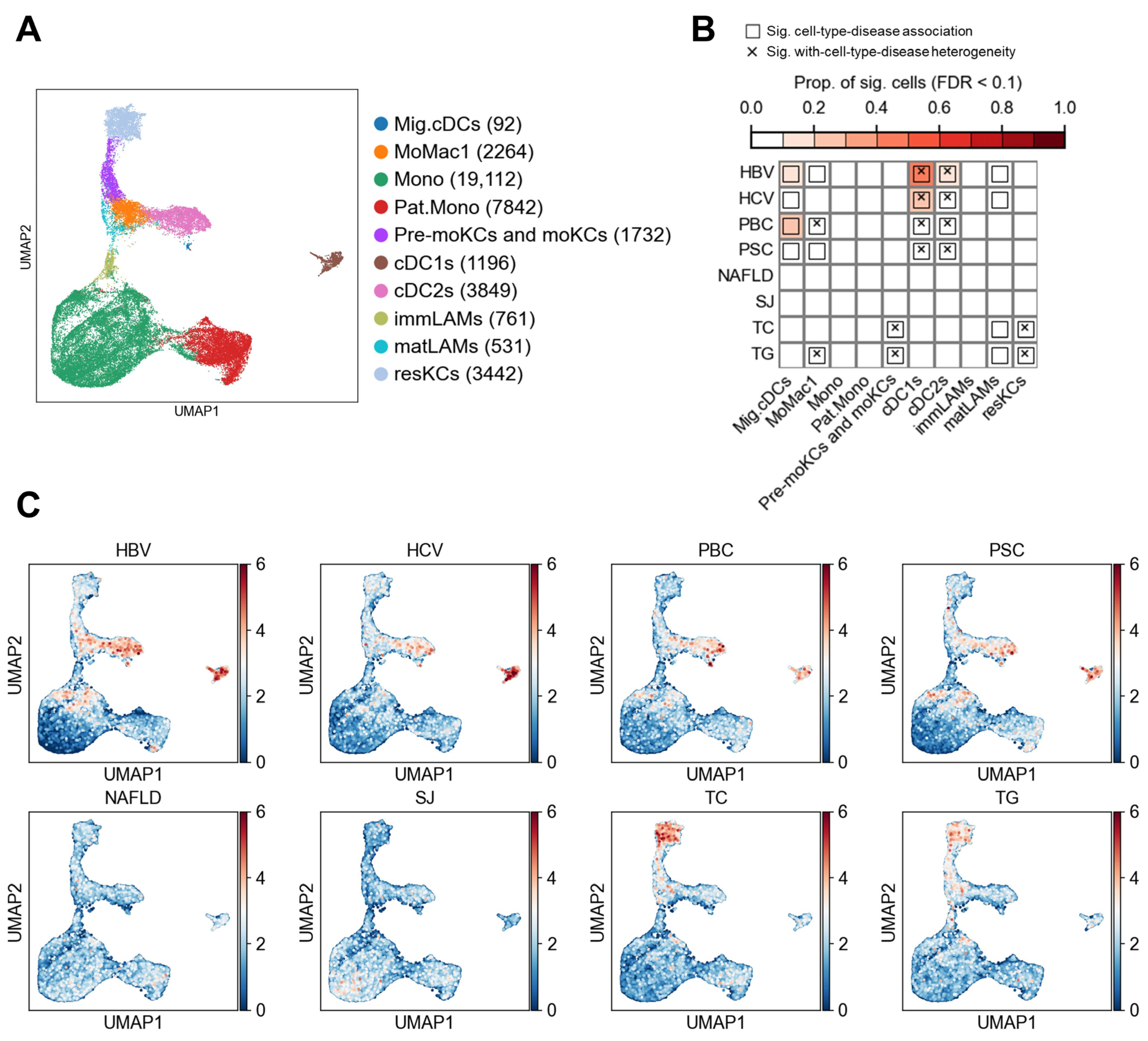
3.3. Myeloid Cell Associations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association study |
| scRNA-seq | Single-cell RNA-sequencing |
| LD | Linkage disequilibrium |
| GO | Gene Ontology |
| HBV | Hepatitis B virus infection |
| HCV | Hepatitis C virus infection |
| PBC | Primary biliary cholangitis |
| PSC | Primary sclerosing cholangitis |
| NAFLD | Non-alcoholic fatty liver disease |
| SJ | Schistosomiasis progression |
| TC | Cholesterol |
References
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, C.; Liu, Z.; Zou, G.; Li, J.; Lu, M. Host Genetic Determinants of Hepatitis B Virus Infection. Front. Genet. 2019, 10, 696. [Google Scholar] [CrossRef]
- Mosbruger, T.L.; Duggal, P.; Goedert, J.J.; Kirk, G.D.; Hoots, W.K.; Tobler, L.H.; Busch, M.; Peters, M.G.; Rosen, H.R.; Thomas, D.L.; et al. Large-scale candidate gene analysis of spontaneous clearance of hepatitis C virus. J. Infect. Dis. 2010, 201, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Almarri, M.A.; Gaffney, D.J.; Mells, G.F.; Jostins, L.; Cordell, H.J.; Ducker, S.J.; Day, D.B.; Heneghan, M.A.; Neuberger, J.M.; et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2012, 44, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, X.; Kuppa, A.; Feitosa, M.F.; Bielak, L.F.; O’Connell, J.R.; Musani, S.K.; Guo, X.; Kahali, B.; Chen, V.L.; et al. Genome-wide association meta-analysis identifies 17 loci associated with nonalcoholic fatty liver disease. Nat. Genet. 2023, 55, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Cordell, H.J.; Fryett, J.J.; Ueno, K.; Darlay, R.; Aiba, Y.; Hitomi, Y.; Kawashima, M.; Nishida, N.; Khor, S.S.; Gervais, O.; et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J. Hepatol. 2021, 75, 572–581. [Google Scholar] [CrossRef]
- Ji, S.G.; Juran, B.D.; Mucha, S.; Folseraas, T.; Jostins, L.; Melum, E.; Kumasaka, N.; Atkinson, E.J.; Schlicht, E.M.; Liu, J.Z.; et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat. Genet. 2017, 49, 269–273. [Google Scholar] [CrossRef]
- Namjou, B.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; Niu, X.; et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zheng, Z.; Fang, H.; Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 2021, 53, 1616–1621. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.P.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019, 29, 1832–1847.e8. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; Grompe, M.; Sander, M. Stem cells versus plasticity in liver and pancreas regeneration. Nat. Cell Biol. 2016, 18, 238–245. [Google Scholar] [CrossRef]
- Schaub, J.R.; Huppert, K.A.; Kurial, S.N.T.; Hsu, B.Y.; Cast, A.E.; Donnelly, B.; Karns, R.A.; Chen, F.; Rezvani, M.; Luu, H.Y.; et al. De novo formation of the biliary system by TGFβ-mediated hepatocyte transdifferentiation. Nature 2018, 557, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Yanger, K.; Zong, Y.; Maggs, L.R.; Shapira, S.N.; Maddipati, R.; Aiello, N.M.; Thung, S.N.; Wells, R.G.; Greenbaum, L.E.; Stanger, B.Z. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes. Dev. 2013, 27, 719–724. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, X.; Li, W.; Feng, R.X.; Li, L.; Yi, G.R.; Zhang, X.N.; Yin, C.; Yu, H.Y.; Zhang, J.P.; et al. Chronic Liver Injury Induces Conversion of Biliary Epithelial Cells into Hepatocytes. Cell Stem Cell 2018, 23, 114–122.e3. [Google Scholar] [CrossRef]
- Ye, X.; Wei, J.; Yue, M.; Wang, Y.; Chen, H.; Zhang, Y.; Wang, Y.; Zhang, M.; Huang, P.; Yu, R. Leveraging Single-Cell RNA-seq Data to Uncover the Association Between Cell Type and Chronic Liver Diseases. Front. Genet. 2021, 12, 637322. [Google Scholar] [CrossRef]
- Zhang, M.J.; Hou, K.; Dey, K.K.; Sakaue, S.; Jagadeesh, K.A.; Weinand, K.; Taychameekiatchai, A.; Rao, P.; Pisco, A.O.; Zou, J.; et al. Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data. Nat. Genet. 2022, 54, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thone, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef]
- Zhou, M.; Xue, C.; Wu, Z.; Wu, X.; Li, M. Genome-Wide Association Study Identifies New Risk Loci for Progression of Schistosomiasis Among the Chinese Population. Front. Cell Infect. Microbiol. 2022, 12, 871545. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Gene Ontology, C.; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef]
- Miao, G.; Yang, Y.; Yang, X.; Chen, D.; Liu, L.; Lei, X. The multifaceted potential of TPT1 as biomarker and therapeutic target. Heliyon 2024, 10, e38819. [Google Scholar] [CrossRef]
- Amson, R.; Pece, S.; Marine, J.-C.; Fiore, P.P.D.; Telerman, A. TPT1/ TCTP-regulated pathways in phenotypic reprogramming. Trends Cell Biol. 2013, 23, 37–46. [Google Scholar] [CrossRef]
- Aalkjaer, C.; Frische, S.; Leipziger, J.; Nielsen, S.; Praetorius, J. Sodium coupled bicarbonate transporters in the kidney, an update. Acta Physiol. Scand. 2004, 181, 505–512. [Google Scholar] [CrossRef]
- Wang, H.; An, J.; Jin, H.; He, S.; Liao, C.; Wang, J.; Tuo, B. Roles of Cl−/HCO3− anion exchanger 2 in the physiology and pathophysiology of the digestive system (Review). Mol. Med. Rep. 2021, 24, 491. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Willard, B.; Smith, J.D.; Stuehr, D.J.; Hazen, S.L.; Fox, P.L. Protection of Extraribosomal RPL13a by GAPDH and Dysregulation by S-Nitrosylation. Mol. Cell 2012, 47, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Peng, J.; Ma, S.; Ding, Y.; Huang, T.; Zhao, S.; Gao, L.; Liang, X.; Li, C.; Ma, C. Ribosomal protein S26 serves as a checkpoint of T-cell survival and homeostasis in a p53-dependent manner. Cell Mol. Immunol. 2021, 18, 1844–1846. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Wu, X. Diverse Functions of γδ T Cells in the Progression of Hepatitis B Virus and Hepatitis C Virus Infection. Front. Immunol. 2020, 11, 619872. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, J.Y.; Huang, A.; Li, Y.Y.; Zhang, S.; Wei, J.; Xia, S.; Wan, Y.; Chen, W.; Zhang, Z.; et al. Decreased Vδ2 γδ T cells associated with liver damage by regulation of Th17 response in patients with chronic hepatitis B. J. Infect. Dis. 2013, 208, 1294–1304. [Google Scholar] [CrossRef]
- Tseng, C.T.; Miskovsky, E.; Houghton, M.; Klimpel, G.R. Characterization of liver T-cell receptor gammadelta T cells obtained from individuals chronically infected with hepatitis C virus (HCV): Evidence for these T cells playing a role in the liver pathology associated with HCV infections. Hepatology 2001, 33, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Agrati, C.; D’Offizi, G.; Narciso, P.; Abrignani, S.; Ippolito, G.; Colizzi, V.; Poccia, F. Vdelta1 T lymphocytes expressing a Th1 phenotype are the major gammadelta T cell subset infiltrating the liver of HCV-infected persons. Mol. Med. 2001, 7, 11–19. [Google Scholar] [CrossRef]
- Oo, Y.H.; Sakaguchi, S. Regulatory T-cell directed therapies in liver diseases. J. Hepatol. 2013, 59, 1127–1134. [Google Scholar] [CrossRef]
- Kokubo, K.; Hirahara, K.; Kiuchi, M.; Tsuji, K.; Shimada, Y.; Sonobe, Y.; Shinmi, R.; Hishiya, T.; Iwamura, C.; Onodera, A.; et al. Thioredoxin-interacting protein is essential for memory T cell formation via the regulation of the redox metabolism. Proc. Natl. Acad. Sci. USA 2023, 120, e2218345120. [Google Scholar] [CrossRef]
- Maldonado, R.A.; von Andrian, U.H. How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 2010, 108, 111–165. [Google Scholar] [CrossRef] [PubMed]
- Kumar Sahu, A.K.; Kashyap, P.; Mishra, K.; Mishra, S.P.; Dutta, S.; Sahu, B. Hepatocytes and Their Role in Metabolism. In Drug Metabolism; Dunnington, K., Ed.; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar]
- Massalha, H.; Bahar Halpern, K.; Abu-Gazala, S.; Jana, T.; Massasa, E.E.; Moor, A.E.; Buchauer, L.; Rozenberg, M.; Pikarsky, E.; Amit, I.; et al. A single cell atlas of the human liver tumor microenvironment. Mol. Syst. Biol. 2020, 16, e9682. [Google Scholar] [CrossRef]
- Ren, A.; He, W.; Rao, J.; Ye, D.; Cheng, P.; Jian, Q.; Fu, Z.; Zhang, X.; Deng, R.; Gao, Y.; et al. Dysregulation of innate cell types in the hepatic immune microenvironment of alcoholic liver cirrhosis. Front. Immunol. 2023, 14, 1034356. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef]
- He, Q.; He, W.; Dong, H.; Guo, Y.; Yuan, G.; Shi, X.; Wang, D.; Lu, F. Role of liver sinusoidal endothelial cell in metabolic dysfunction-associated fatty liver disease. Cell Commun. Signal 2024, 22, 346. [Google Scholar] [CrossRef] [PubMed]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Jalan-Sakrikar, N.; Guicciardi, M.E.; O’Hara, S.P.; Azad, A.; LaRusso, N.F.; Gores, G.J.; Huebert, R.C. Central role for cholangiocyte pathobiology in cholestatic liver diseases. Hepatology 2024. [Google Scholar] [CrossRef]
- Pinto, C.; Giordano, D.M.; Maroni, L.; Marzioni, M. Role of inflammation and proinflammatory cytokines in cholangiocyte pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1270–1278. [Google Scholar] [CrossRef]
- McKinney, E.F.; Smith, K.G.C. Metabolic exhaustion in infection, cancer and autoimmunity. Nat. Immunol. 2018, 19, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Aron, J.H.; Bowlus, C.L. The immunobiology of primary sclerosing cholangitis. Semin. Immunopathol. 2009, 31, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, Z.; Yu, Y.; Wu, S.; Huang, H.; Huang, Y.; Liu, J.; Mo, K.; Tan, J.; Han, Z.; et al. Deciphering the dynamic single-cell transcriptional landscape in the ocular surface ectoderm differentiation system. Life Med. 2024, 3, lnae033. [Google Scholar] [CrossRef] [PubMed]
- Neavin, D.; Nguyen, Q.; Daniszewski, M.S.; Liang, H.H.; Chiu, H.S.; Wee, Y.K.; Senabouth, A.; Lukowski, S.W.; Crombie, D.E.; Lidgerwood, G.E.; et al. Single cell eQTL analysis identifies cell type-specific genetic control of gene expression in fibroblasts and reprogrammed induced pluripotent stem cells. Genome Biol. 2021, 22, 76. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease/Trait Name | Abbreviation | Population | Sample Size | PMID |
|---|---|---|---|---|
| Hepatitis B virus infection | HBV | East Asian | 171,822 | 34594039 |
| Hepatitis C virus infection | HCV | East Asian | 176,698 | 34594039 |
| Primary biliary cholangitis | PBC | European | 24,510 | 34033851 |
| Primary sclerosing cholangitis | PSC | European | 14,890 | 27992413 |
| Non-alcoholic fatty liver disease | NAFLD | European | 9677 | 31311600 |
| Schistosoma japonicum hyaluronan levels | SJ | East Asian | 637 | 35493725 |
| Cholesterol | TC | European | 340,162 | 25826379 |
| Triglycerides | TG | European | 340,162 | 25826379 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; Liu, M.; Xue, C. Dissecting the Cellular Heterogeneity Underlying Liver Diseases Through the Integration of GWASs and Single-Cell RNA Sequencing. Biology 2025, 14, 777. https://doi.org/10.3390/biology14070777
Zhou M, Liu M, Xue C. Dissecting the Cellular Heterogeneity Underlying Liver Diseases Through the Integration of GWASs and Single-Cell RNA Sequencing. Biology. 2025; 14(7):777. https://doi.org/10.3390/biology14070777
Chicago/Turabian StyleZhou, Miao, Meng Liu, and Chao Xue. 2025. "Dissecting the Cellular Heterogeneity Underlying Liver Diseases Through the Integration of GWASs and Single-Cell RNA Sequencing" Biology 14, no. 7: 777. https://doi.org/10.3390/biology14070777
APA StyleZhou, M., Liu, M., & Xue, C. (2025). Dissecting the Cellular Heterogeneity Underlying Liver Diseases Through the Integration of GWASs and Single-Cell RNA Sequencing. Biology, 14(7), 777. https://doi.org/10.3390/biology14070777