


A TRIM Family-Based Strategy for TRIMCIV Target Prediction in a Pan-Cancer Context with Multi-Omics Data and Protein Docking Integration
 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
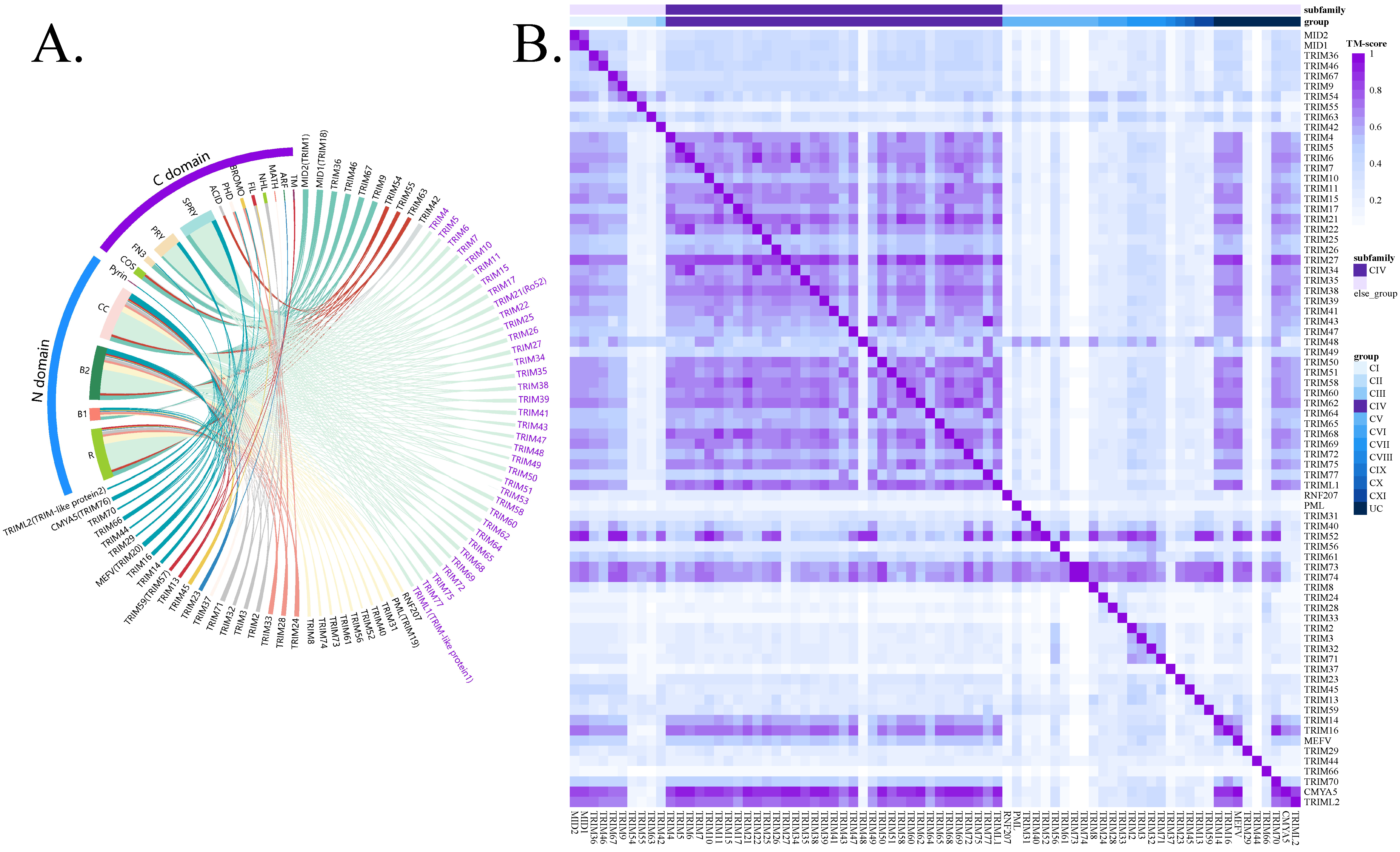
2.1. TRIM–Target Interaction Database Construction
2.2. Structural Alignment
2.3. Expression Data Collection, Processing, and Analysis
2.4. Correlation Analysis
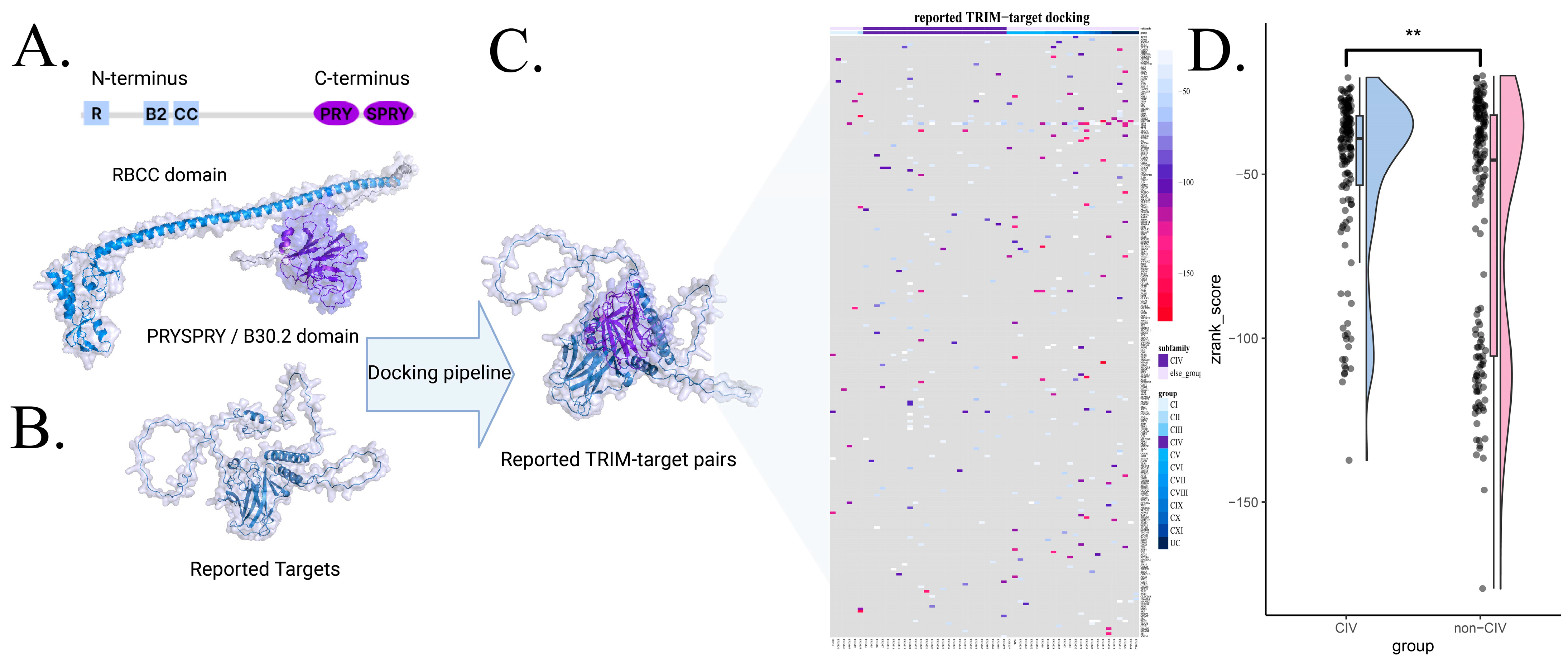
2.5. Docking Pipeline
2.6. Predictor Building
2.7. Evaluation of Model Performance
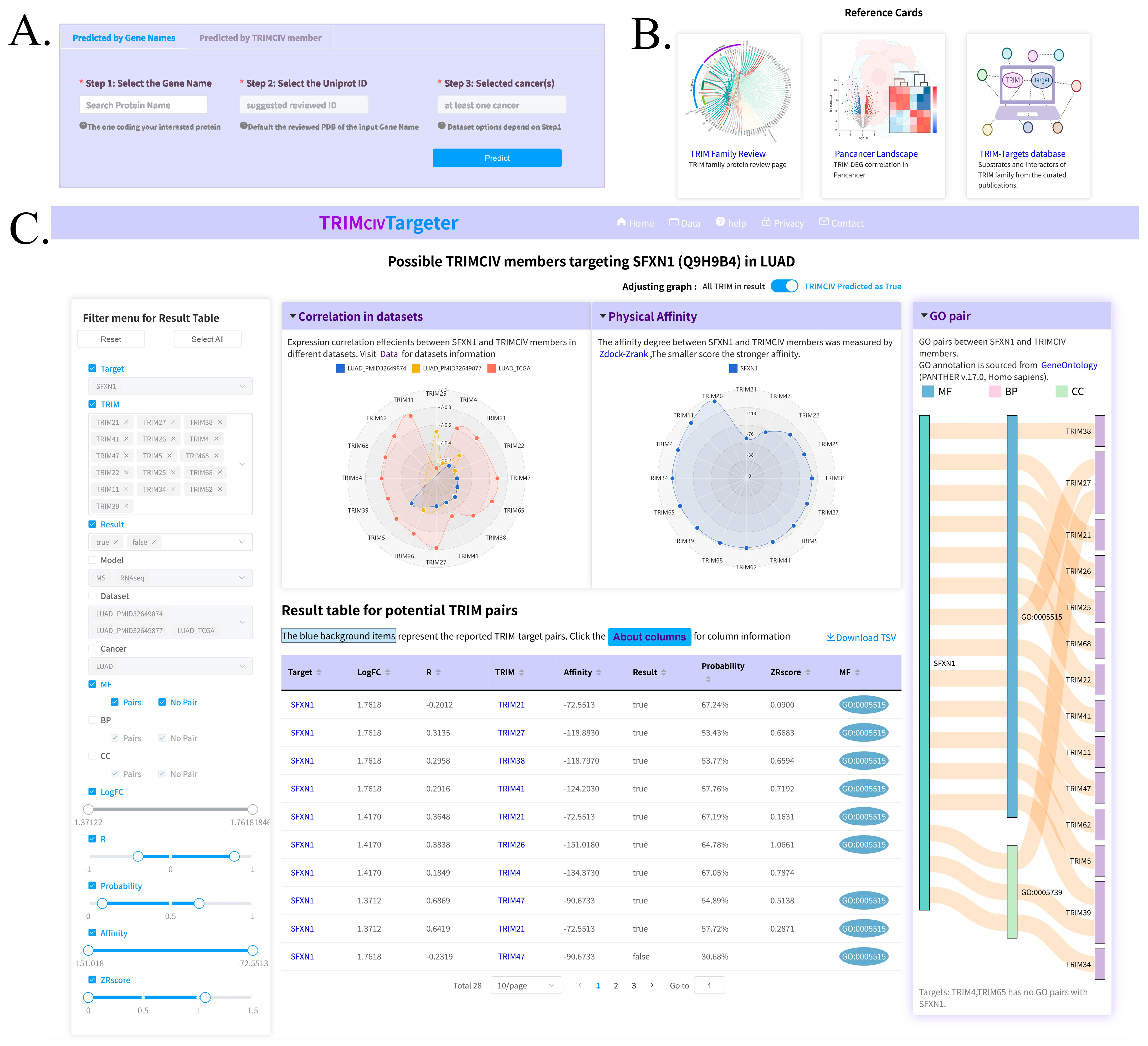
2.8. Web Interface Building
3. Results and Discussions
3.1. TRIM Family Overview and Reported TRIM–Target Pair Database Construction
3.2. Distinct Correlation of TRIMCIV with DEG
3.3. Interaction Scoring via Protein Docking
3.4. Evaluation of Prediction Model
3.5. Web Server and Utility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-T.; Hur, S. Substrate recognition by TRIM and TRIM-like proteins in innate immunity. Semin. Cell Dev. Biol. 2021, 111, 76–85. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, C.; Wen, X.; Luan, G.; Xie, L.; Guo, X. The translational values of TRIM family in pan-cancers: From functions and mechanisms to clinics. Pharmacol. Ther. 2021, 227, 107881. [Google Scholar] [CrossRef]
- Perfetto, L.; Gherardini, P.F.; Davey, N.E.; Diella, F.; Helmer-Citterich, M.; Cesareni, G. Exploring the diversity of SPRY/B30. 2-mediated interactions. Trends Biochem. Sci. 2013, 38, 38–46. [Google Scholar] [CrossRef]
- James, L.C.; Keeble, A.H.; Khan, Z.; Rhodes, D.A.; Trowsdale, J. Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc. Natl. Acad. Sci. USA 2007, 104, 6200–6205. [Google Scholar] [CrossRef]
- Gao, W.; Li, Y.; Liu, X.; Wang, S.; Mei, P.; Chen, Z.; Liu, K.; Li, S.; Xu, X.-W.; Gan, J.; et al. TRIM21 regulates pyroptotic cell death by promoting Gasdermin D oligomerization. Cell Death Differ. 2022, 29, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Singh, K.; Shinde, A.; Singh, J.; Mane, M.; Bedekar, S.; Tailor, Y.; Gohel, D.; Vasiyani, H.; Currim, F.; et al. TNF-α-induced E3 ligase, TRIM15 inhibits TNF-α-regulated NF-κB pathway by promoting turnover of K63 linked ubiquitination of TAK1. Cell. Signal. 2022, 91, 110210. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Heikel, G.; Trubitsyna, M.; Kubik, P.; Nowak, J.S.; Webb, S.; Granneman, S.; Spanos, C.; Rappsilber, J.; Castello, A.; et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 2017, 15, 105. [Google Scholar] [CrossRef]
- Huang, N.; Sun, X.; Li, P.; Liu, X.; Zhang, X.; Chen, Q.; Xin, H. TRIM family contribute to tumorigenesis, cancer development, and drug resistance. Exp. Hematol. Oncol. 2022, 11, 75. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Hashemifar, S.; Neyshabur, B.; Khan, A.A.; Xu, J. Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics 2018, 34, i802–i810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- De Bodt, S.; Proost, S.; Vandepoele, K.; Rouzé, P.; Van de Peer, Y. Predicting protein-protein interactions in Arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genom. 2009, 10, 288. [Google Scholar] [CrossRef]
- Li, Y.; Xie, P.; Lu, L.; Wang, J.; Diao, L.; Liu, Z.; Guo, F.; He, Y.; Liu, Y.; Huang, Q.; et al. An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat. Commun. 2017, 8, 347. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Huang, F.; Li, J.; Shen, W.; Chen, L.; Feng, K.; Huang, T.; Cai, Y.-D. Identification of Protein–Protein Interaction Associated Functions Based on Gene Ontology. Protein J. 2024, 43, 477–486. [Google Scholar] [CrossRef]
- Lei, C.; Ruan, J. A novel link prediction algorithm for reconstructing protein–protein interaction networks by topological similarity. Bioinformatics 2013, 29, 355–364. [Google Scholar] [CrossRef]
- Meyer, M.J.; Beltrán, J.F.; Liang, S.; Fragoza, R.; Rumack, A.; Liang, J.; Wei, X.; Yu, H. Interactome INSIDER: A structural interactome browser for genomic studies. Nat. Methods 2018, 15, 107–114. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef]
- Wass, M.N.; Fuentes, G.; Pons, C.; Pazos, F.; Valencia, A. Towards the prediction of protein interaction partners using physical docking. Mol. Syst. Biol. 2011, 7, 469. [Google Scholar] [CrossRef] [PubMed]
- Durham, J.; Zhang, J.; Humphreys, I.R.; Pei, J.; Cong, Q. Recent advances in predicting and modeling protein–protein interactions. Trends Biochem. Sci. 2023, 48, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Zhang, X.; Liu, Y.; Peng, H.; Zheng, B.; Yin, Y.; Zeng, X. Machine learning on protein–protein interaction prediction: Models, challenges and trends. Brief. Bioinform. 2023, 24, bbad076. [Google Scholar] [CrossRef]
- Blohm, P.; Frishman, G.; Smialowski, P.; Goebels, F.; Wachinger, B.; Ruepp, A.; Frishman, D. Negatome 2.0: A database of non-interacting proteins derived by literature mining, manual annotation and protein structure analysis. Nucleic Acids Res. 2014, 42, D396–D400. [Google Scholar] [CrossRef]
- Barman, R.K.; Jana, T.; Das, S.; Saha, S. Prediction of intra-species protein-protein interactions in enteropathogens facilitating systems biology study. PLoS ONE 2015, 10, e0145648. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, G.; Guo, M.; Wang, J. Predicting protein-protein interactions using high-quality non-interacting pairs. BMC Bioinform. 2018, 19, 105–124. [Google Scholar] [CrossRef]
- Khunlertgit, N.; Yoon, B.-J. Incorporating topological information for predicting robust cancer subnetwork markers in human protein-protein interaction network. BMC Bioinform. 2016, 17, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-P.; Du, P.-F.; Shen, Z.-A.; Liu, H.-Y.; Luo, T. DPPN-SVM: Computational identification of mis-localized proteins in cancers by integrating differential gene expressions with dynamic protein-protein interaction networks. Front. Genet. 2020, 11, 600454. [Google Scholar] [CrossRef]
- Busch, J.D.; Fielden, L.F.; Pfanner, N.; Wiedemann, N. Mitochondrial protein transport: Versatility of translocases and mechanisms. Mol. Cell 2023, 83, 890–910. [Google Scholar] [CrossRef]
- Li, H.-m.; Chiu, C.-C. Protein transport into chloroplasts. Annu. Rev. Plant Biol. 2010, 61, 157–180. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, M.; Qian, Y. protein2vec: Predicting protein-protein interactions based on LSTM. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 1257–1266. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, H.; Chen, D.Z. TANGO: A GO-term embedding based method for protein semantic similarity prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 20, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Ieremie, I.; Ewing, R.M.; Niranjan, M. TransformerGO: Predicting protein–protein interactions by modelling the attention between sets of gene ontology terms. Bioinformatics 2022, 38, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- van Noort, V.; Snel, B.; Huynen, M.A. Predicting gene function by conserved co-expression. TRENDS Genet. 2003, 19, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015, 25, 1121–1136. [Google Scholar] [CrossRef]
- Li, Y.; Bao, L.; Zheng, H.; Geng, M.; Chen, T.; Dai, X.; Xiao, H.; Yang, L.; Mao, C.; Qiu, Y.; et al. E3 ubiquitin ligase TRIM21 targets TIF1γ to regulate β-catenin signaling in glioblastoma. Theranostics 2023, 13, 4919. [Google Scholar] [CrossRef]
- Xiao, Y.; Wu, J.; Lin, Z.; Zhao, X. A deep learning-based multi-model ensemble method for cancer prediction. Comput. Methods Programs Biomed. 2018, 153, 1–9. [Google Scholar] [CrossRef]
- Liang, J.-y.; Wang, D.-s.; Lin, H.-c.; Chen, X.-x.; Yang, H.; Zheng, Y.; Li, Y.-h. A novel ferroptosis-related gene signature for overall survival prediction in patients with hepatocellular carcinoma. Int. J. Biol. Sci. 2020, 16, 2430. [Google Scholar] [CrossRef]
- Zhou, Z.; Ji, Z.; Wang, Y.; Li, J.; Cao, H.; Zhu, H.H.; Gao, W.-Q. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 2014, 147, 1043–1054. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Q.; Zhao, G.; Zhang, J.; Yuan, H.; Feng, T.; Ou, D.; Gu, R.; Li, S.; Li, K. Loss of TRIM31 promotes breast cancer progression through regulating K48-and K63-linked ubiquitination of p53. Cell Death Dis. 2021, 12, 945. [Google Scholar] [CrossRef]
- Meng, J.; Yao, Z.; He, Y.; Zhang, R.; Zhang, Y.; Yao, X.; Yang, H.; Chen, L.; Zhang, Z.; Zhang, H.; et al. ARRDC4 regulates enterovirus 71-induced innate immune response by promoting K63 polyubiquitination of MDA5 through TRIM65. Cell Death Dis. 2017, 8, e2866. [Google Scholar] [CrossRef]
- Chen, D.; Liu, X.; Xia, T.; Tekcham, D.S.; Wang, W.; Chen, H.; Li, T.; Lu, C.; Ning, Z.; Liu, X.; et al. A multidimensional characterization of E3 ubiquitin ligase and substrate interaction network. IScience 2019, 16, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- O’Meara, M.J.; Leaver-Fay, A.; Tyka, M.D.; Stein, A.; Houlihan, K.; DiMaio, F.; Bradley, P.; Kortemme, T.; Baker, D.; Snoeyink, J.; et al. Combined covalent-electrostatic model of hydrogen bonding improves structure prediction with Rosetta. J. Chem. Theory Comput. 2015, 11, 609–622. [Google Scholar] [CrossRef]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins Struct. Funct. Bioinform. 2007, 67, 1078–1086. [Google Scholar] [CrossRef]
- Wiehe, K.; Pierce, B.; Tong, W.W.; Hwang, H.; Mintseris, J.; Weng, Z. The performance of ZDOCK and ZRANK in rounds 6–11 of CAPRI. Proteins Struct. Funct. Bioinform. 2007, 69, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Vreven, T.; Pierce, B.G.; Hung, J.H.; Weng, Z. Performance of ZDOCK and ZRANK in CAPRI rounds 13–19. Proteins Struct. Funct. Bioinform. 2010, 78, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Vreven, T.; Pierce, B.G.; Hwang, H.; Weng, Z. Performance of ZDOCK in CAPRI rounds 20–26. Proteins Struct. Funct. Bioinform. 2013, 81, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Yang, L.; Shami, A. On hyperparameter optimization of machine learning algorithms: Theory and practice. Neurocomputing 2020, 415, 295–316. [Google Scholar] [CrossRef]
- Bradshaw, T.J.; Huemann, Z.; Hu, J.; Rahmim, A. A guide to cross-validation for artificial intelligence in medical imaging. Radiol. Artif. Intell. 2023, 5, e220232. [Google Scholar] [CrossRef]
- Huang, Y.; Gao, X.; He, Q.Y.; Liu, W. A interacting model: How TRIM21 orchestrates with proteins in intracellular immunity. Small Methods 2024, 8, 2301142. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Yugandhar, K.; Jemimah, S. Protein–protein interactions: Scoring schemes and binding affinity. Curr. Opin. Struct. Biol. 2017, 44, 31–38. [Google Scholar] [CrossRef]
- Li, Y.-H.; Tong, K.-L.; Lu, J.-L.; Lin, J.-B.; Li, Z.-Y.; Sang, Y.; Ghodbane, A.; Gao, X.-J.; Tam, M.-S.; Hu, C.-D.; et al. PRMT5-TRIM21 interaction regulates the senescence of osteosarcoma cells by targeting the TXNIP/p21 axis. Aging 2020, 12, 2507. [Google Scholar] [CrossRef]
- Yang, Z.; Li, J.; Li, J.; Zheng, H.; Li, H.; Lai, Q.; Chen, Y.; Qin, L.; Zuo, Y.; Guo, L.; et al. Engagement of the G3BP2-TRIM25 Interaction by Nucleocapsid Protein Suppresses the Type I Interferon Response in SARS-CoV-2-Infected Cells. Vaccines 2022, 10, 2042. [Google Scholar] [CrossRef]
- Tang, H.; Li, X.; Jiang, L.; Liu, Z.; Chen, L.; Chen, J.; Deng, M.; Zhou, F.; Zheng, X.; Liu, Z. RITA1 drives the growth of bladder cancer cells by recruiting TRIM25 to facilitate the proteasomal degradation of RBPJ. Cancer Sci. 2022, 113, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhou, Z.; Tian, Y.; Liu, Z.; Cheng, K.O.; Chen, X.; Hu, W.; Wong, Y.M.; Li, X.; Zhang, H.; et al. Opposing roles of E3 ligases TRIM23 and TRIM21 in regulation of ion channel ANO1 protein levels. J. Biol. Chem. 2021, 296, 100738. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | PRE | SPE | ACC | REC | F1 | MCC | AUC | AUPR |
|---|---|---|---|---|---|---|---|---|
| MS-based | 0.687 ± 0.051 | 0.682 ± 0.130 | 0.681 ± 0.046 | 0.682 ± 0.130 | 0.677 ± 0.068 | 0.371 ± 0.082 | 0.767 ± 0.046 | 0.758 ± 0.029 |
| RNAseq-based | 0.645 ± 0.020 | 0.763 ± 0.058 | 0.671 ± 0.022 | 0.763 ± 0.058 | 0.698 ± 0.026 | 0.350 ± 0.049 | 0.736 ± 0.032 | 0.690 ± 0.051 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Xuan, J.; Liang, J.; Liu, X.; Luo, Y.; Gao, X.; Liu, W. A TRIM Family-Based Strategy for TRIMCIV Target Prediction in a Pan-Cancer Context with Multi-Omics Data and Protein Docking Integration. Biology 2025, 14, 742. https://doi.org/10.3390/biology14070742
Huang Y, Xuan J, Liang J, Liu X, Luo Y, Gao X, Liu W. A TRIM Family-Based Strategy for TRIMCIV Target Prediction in a Pan-Cancer Context with Multi-Omics Data and Protein Docking Integration. Biology. 2025; 14(7):742. https://doi.org/10.3390/biology14070742
Chicago/Turabian StyleHuang, Yisha, Jiajia Xuan, Jiayan Liang, Xixi Liu, Yonglei Luo, Xuejuan Gao, and Wanting Liu. 2025. "A TRIM Family-Based Strategy for TRIMCIV Target Prediction in a Pan-Cancer Context with Multi-Omics Data and Protein Docking Integration" Biology 14, no. 7: 742. https://doi.org/10.3390/biology14070742
APA StyleHuang, Y., Xuan, J., Liang, J., Liu, X., Luo, Y., Gao, X., & Liu, W. (2025). A TRIM Family-Based Strategy for TRIMCIV Target Prediction in a Pan-Cancer Context with Multi-Omics Data and Protein Docking Integration. Biology, 14(7), 742. https://doi.org/10.3390/biology14070742