
Profiling Genetic Variation: Divergence Patterns and Population Structure of Thailand’s Endangered Celastrus paniculatus Willd


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
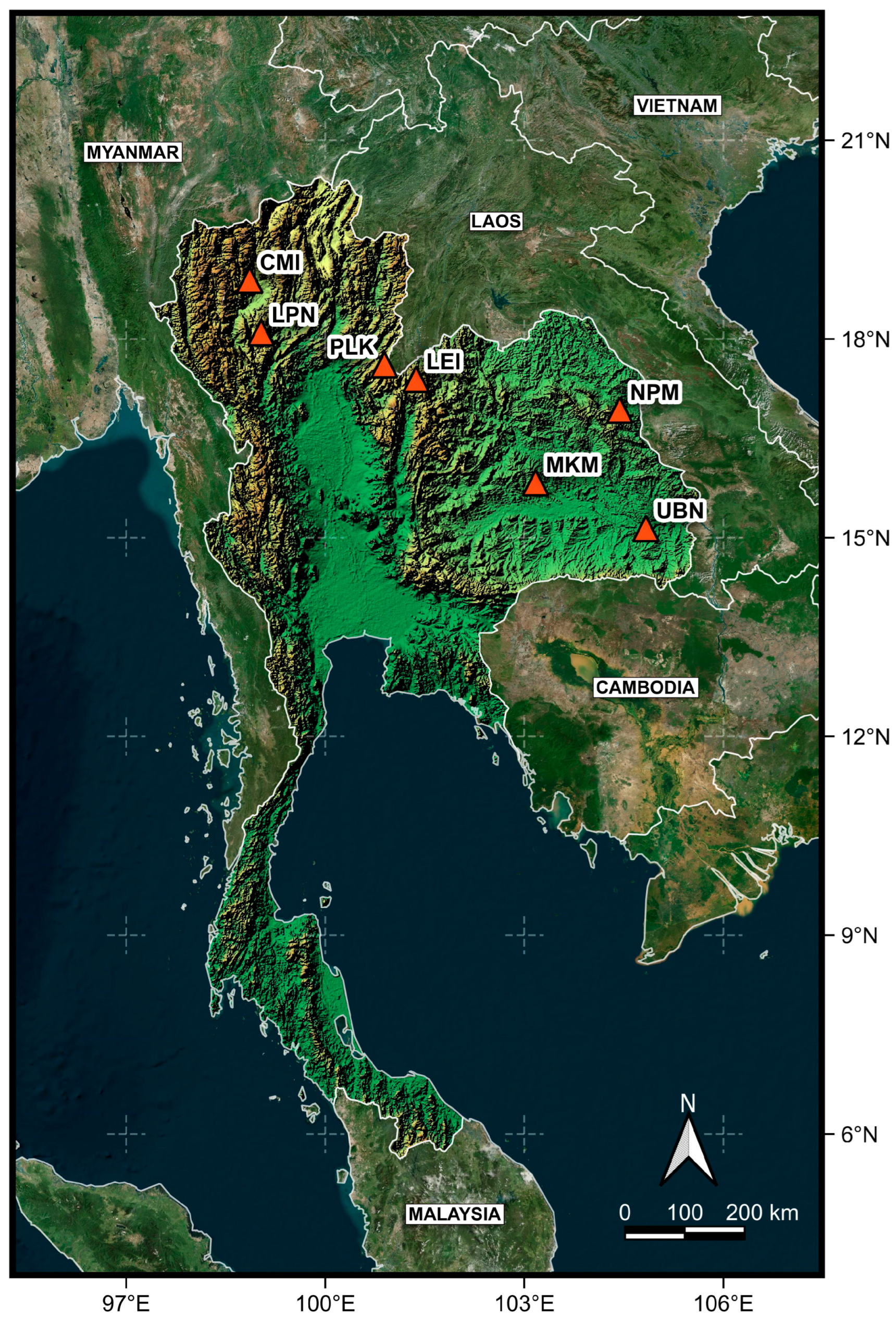
2.2. Sample Collection
2.3. Molecular Analysis
2.4. Data Analysis
3. Results and Discussion
3.1. Genetic Variation and Molecular Diversity Indices
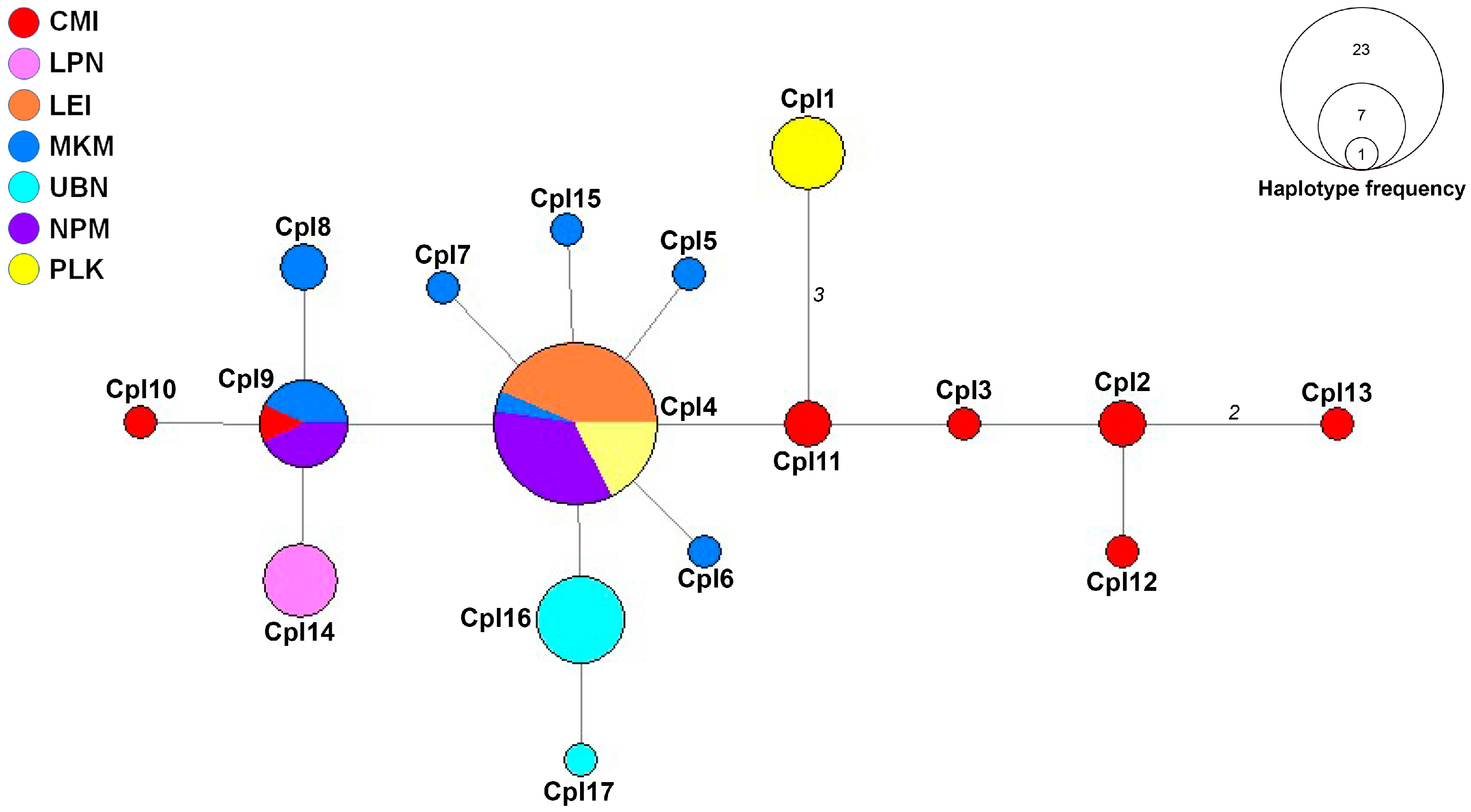
3.2. Haplotype Network Analysis
3.3. Genetic Differences and Gene Flow Analyses
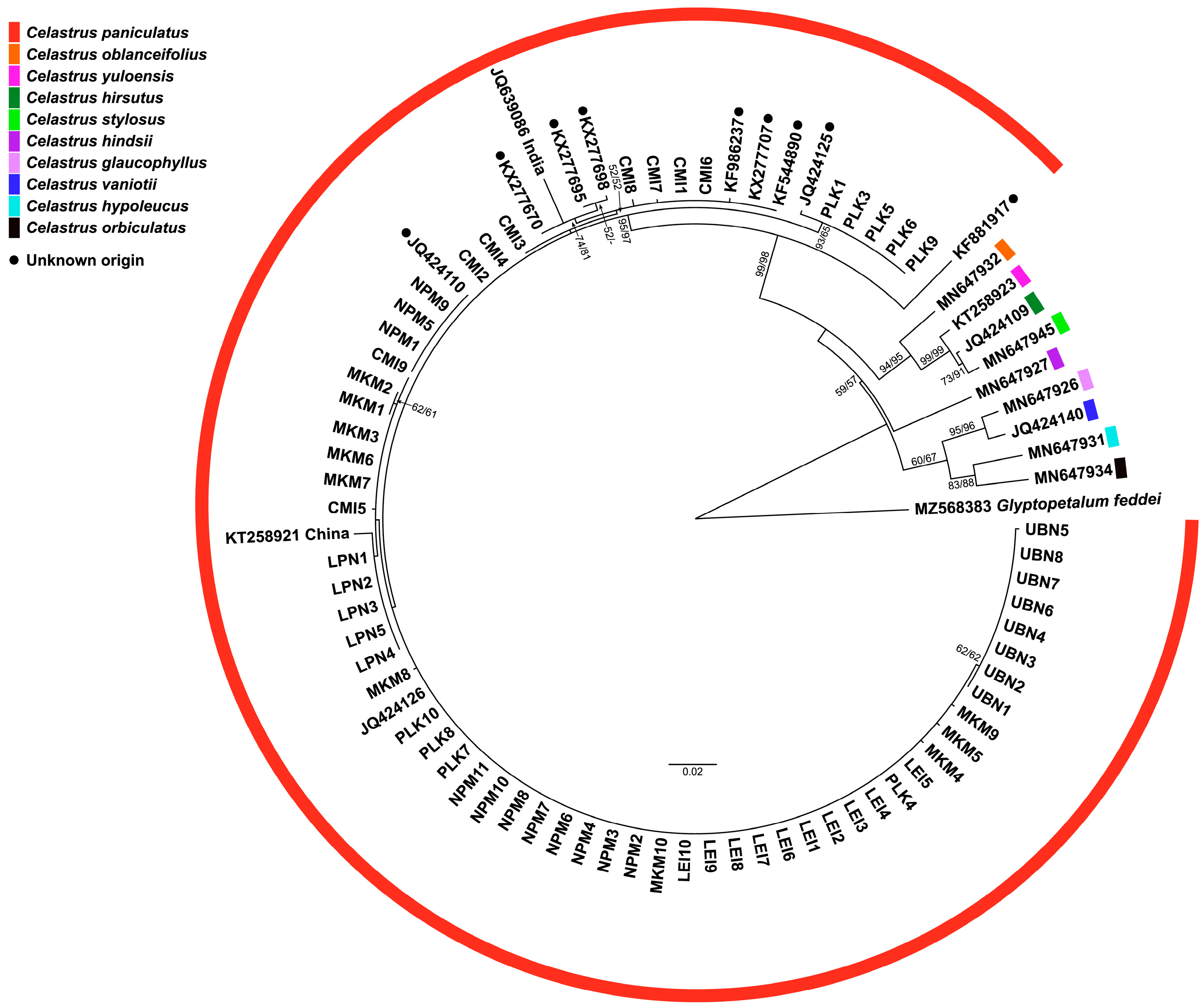
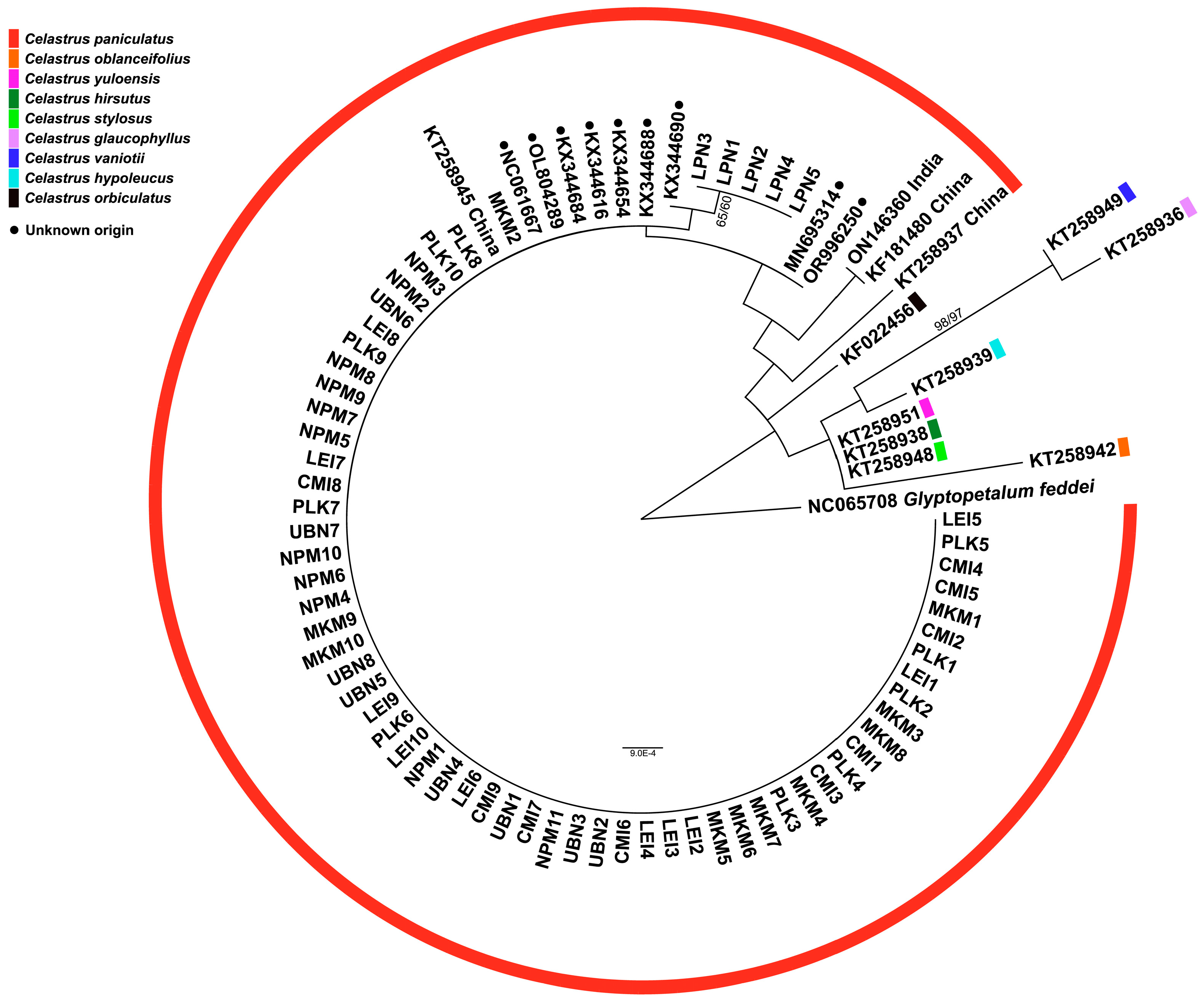
3.4. Phylogenetic Tree Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Francis, J.W.; Dandu, M.M.; Sardesai, M.M.; Dhabe, A.S. Note on Celastrus paniculatus Willd. ssp. Aggregatus K.M. Matthew ex K.T. Matthew (Celastraceae). JoTT Note 2012, 4, 3450–3453. [Google Scholar] [CrossRef]
- Mohanty, A.; Mohanty, M.; Panda, P.C.; Nayak, S.; Mohanty, S. Phytochemistry, ethnopharmacology and conservation through biotechnological approaches: A critical review on the endangered medicinal plant Celastrus paniculatus. Proc. Natl. Acad. Sci. India. Sect. B Biol. Sci. 2025; in press. [Google Scholar] [CrossRef]
- Mohan, G.K.; Sachin, Y.S.; Manohar, V.P.; Dipak, L.R.; Sanjay, J.S. Pharmacognostical investigation and physicochemical analysis of Celastrus paniculatus Willd. leaves. Asian Pac. J. Trop. Biomed. 2012, 2, 1232–1236. [Google Scholar] [CrossRef]
- Kulkarni, Y.A.; Agarwal, S.; Garud, M.S. Effect of Jyotishmati (Celastrus paniculatus) seeds in animal models of pain and inflammation. J. Ayurveda Integr. Med. 2015, 6, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.N.; Kaur, H.; Shrivastava, B.; Arora, S.C. A review from historical to current-Celastrus paniculatus. Int. J. Pharm. Pharmaceut. Sci. 2020, 12, 15–20. [Google Scholar] [CrossRef]
- Faldu, K.G.; Patel, S.S.; Shah, J.S. Celastrus paniculatus oil ameliorates NF-KB mediated neuroinflammation and synaptic plasticity in the scopolamine-induced cognitive impairment rat model. Metab. Brain Dis. 2023, 38, 1405–1419. [Google Scholar] [CrossRef]
- Bambaradeniya, C.; Karunarathna, D.M.S.S.; Kekulandala, B. The 2007 Red List of Threatened Fauna and Flora of Sri Lanka. In The World Conservation Union (IUCN) and Ministry of Environment and Natural Resources, Colombo, Sri Lanka; Bambaradeniya, C., Ed.; IUCN Sri Lanka and the Ministry of Environment and Natural Resources: Colombo, Sri Lanka, 2007; p. 148. [Google Scholar]
- Gopi, D.K.; Mattummal, R.; Narayana, S.K.K.; Parameswaran, S. IUCN red listed medicinal plants of siddha. J. Res. Sid. Med. 2018, 1, 15–22. [Google Scholar]
- Chen, W.; Hou, L.; Zhang, Z.; Pang, X.; Li, Y. Genetic diversity, population structure, and linkage disequilibrium of a core collection of Ziziphusjujuba assessed with genome-wide SNPs developed by genotyping-by-sequencing and SSR markers. Front. Plant Sci. 2017, 8, 575. [Google Scholar]
- Bidyananda, N.; Jamir, I.; Nowakowska, K.; Varte, V.; Vendrame, W.A.; Devi, R.S.; Nongdam, P. Plant genetic diversity studies: Insights from DNA marker analyses. Int. J. Plant Biol. 2024, 15, 607–640. [Google Scholar] [CrossRef]
- Mu, X.Y.; Zhao, L.C.; Zhang, Z.X. Molecular analysis of Chinese Celastrus and Tripterygium and implications in medicinal and pharmacological studies. PLoS ONE 2017, 12, e0169973. [Google Scholar] [CrossRef]
- Xia, M.Z.; Li, Y.; Zhang, F.Q.; Yu, J.Y.; Khan, G.; Chi, X.F.; Xu, H.; Chen, S.L. Reassessment of the phylogeny and systematics of Chinese Parnassia (Celastraceae): A thorough investigation using whole plastomes and nuclear ribosomal DNA. Front. Plant Sci. 2022, 13, 855944. [Google Scholar] [CrossRef]
- Prompen, B.; Saijuntha, W.; Pilap, W.; Thanonkeo, S. Genetic diversity and population structure of Siamese rosewood (Dalbergia cochinchinensis Pierre) in Thailand using matK and internal transcribed spacer markers. Forests 2025, 16, 332. [Google Scholar] [CrossRef]
- CBOL Plant Working Group. A DNA barcode for land plants. Proc. Nat. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Sivanov, I.A.; Welzen, P.C.V.; Duyfjes, B.E.E.; Harwood, B.; Chayamarit, K.; Wilde, W.J.; Atkins, S. Flora of Thailand; The Forest Herbarium, Royal Forest Department: Bangkok, Thailand, 2010; p. 10.
- Wonnapinij, P.; Sriboonlert, A. Molecular phylogenetics of species of Bulbophyllum sect. Trias (Orchidaceae; Epidendroideae; Malaxidae) based on nrITS and plastid rbcL and matK. Phytotaxa 2015, 226, 1–7. [Google Scholar] [CrossRef]
- Hasebe, M.; Omori, T.; Nakazawa, M.; Sano, T.; Kato, M.; Iwatsuki, K. rbcL gene sequences provide evidence for the evolutionary lineages of leptosporangiate ferns. Proc. Nat. Acad. Sci. USA 1994, 91, 5730–5734. [Google Scholar] [CrossRef]
- Asahina, H.; Shinozaki, J.; Masuda, K.; Morimitsu, Y.; Satake, M. Identification of medicinal Dendrobium species by phylogenetic analyses using matK and rbcL sequences. J. Nat. Med. 2010, 64, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/ NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 18, 3022–3027. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution in Mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Amom, T.; Nongdam, P. The use of molecular marker methods in plants: A review. Int. J. Curr. Res. Rev. 2017, 9, 1–7. [Google Scholar]
- Ramesh, P.; Mallikarjuna, G.; Sameena, S.; Kumar, A.; Gurulakshmi, K.; Reddy, B.V.; Reddy, P.C.O.; Sekhar, A.C. Advancements in molecular marker technologies and their applications in diversity studies. J. Biosci. 2020, 45, 1–15. [Google Scholar] [CrossRef]
- Raju, N.L.; Prasad, M.N.V. Genetic diversity analysis of Celastrus paniculatus Willd.-a nearly threatened, cognitive and intelligence enhancer by RAPD markers. Func. Plant Sci. Biotechnol. 2007, 1, 195–199. [Google Scholar]
- Mu, X.Y.; Zhao, L.C.; Zhang, Z.X. Phylogeny of Celastrus L. (Celastraceae) inferred from two nuclear and three plastid markers. J. Plant Res. 2012, 125, 619–630. [Google Scholar] [CrossRef]
- Bal, P.; Panda, P.C. Molecular characterization and phylogenetic relationships of Dalbergia species of eastern India based on RAPD and ISSR analysis. Int. J. Innov. Sci. Res. Technol. 2018, 3, 417–422. [Google Scholar]
- Hartvig, I.; So, T.; Changtragoon, S.; Tran, H.T.; Bouamanivong, S.; Theilade, I.; Kjaer, E.D.; Nielsen, L.R. Population genetic structure of the endemic rosewoods Dalbergia cochinchinensis and D. oliveri at a regional scale reflects the Indochinese landscape and life-history traits. Ecol. Evol. 2018, 8, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Cheng, Y.; Li, K.; Li, H. Integrating phylogeographic analysis and geospatial methods to infer historical dispersal routes and glacial refugia of Liriodendron chinense. Forests 2019, 10, 565. [Google Scholar] [CrossRef]
- Hartvig, I.; Czako, M.; Kjaer, E.D.; Nielsen, L.R.; Theilade, I. The use of DNA barcoding in identification and conservation of rosewood (Dalbergia spp.). PLoS ONE 2015, 10, e0138231. [Google Scholar] [CrossRef]
- Yan, K.; Lu, X.; Li, W.; Sun, C.; Zhou, X.; Wang, Y. Chloroplast genome diversity and molecular evolution in hypericaceae: New insights from three Hypericum species. Int. J. Mol. Sci. 2025, 26, 323. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Chen, J.; Ran, Z.; Huang, L.; Li, Z. Comparative analysis of complete chloroplast genomes and phylogenetic relationships of 21 sect Camellia (Camellia L.) plants. Genes 2025, 16, 49. [Google Scholar]
- Hussain, H.; Nisar, M. Assessment of plant genetic variations using molecular markers: A review. J. Appl. Biol. Biotechnol. 2020, 8, 99–109. [Google Scholar]
- Svenning, J.C.; Eiserhardt, W.L.; Normand, S.; Ordonez, A.; Sandel, B. The influence of paleoclimate on present-day patterns in biodiversity and ecosystems. Annu. Rev. Ecol. Syst. 2015, 46, 551–572. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | N | District | Province | Region | Altitude (m) | Latitude | Longitude |
|---|---|---|---|---|---|---|---|
| CMI | 9 | Mae Rim | Chiang Mai | North | 776 | 18.889889 | 98.860825 |
| PLK | 9 | Chat Trakan | Phitsanulok | North | 1066 | 17.608864 | 100.902396 |
| LPN | 5 | Thung Hua Chang | Lamphun | North | 840 | 18.097083 | 99.033806 |
| LEI | 10 | Phu Ruea | Loei | Northeast | 653 | 17.391944 | 101.370556 |
| MKM | 10 | Na Chuek | Maha Sarakham | Northeast | 167 | 15.836415 | 103.159919 |
| NPM | 11 | Na Kae | Nakhon Phanom | Northeast | 140 | 16.926970 | 104.438570 |
| UBN | 8 | Warinchamrab | Ubon Ratchathani | Northeast | 120 | 15.139028 | 104.832361 |
| Total | 62 |
| Haplotypes | Nucleotide Variable Positions of ITS Region | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 4 | 5 | 5 | 5 | ||||||
| 1 | 3 | 6 | 7 | 2 | 2 | 2 | 7 | 8 | 9 | 9 | 6 | 2 | 0 | 2 | 9 | ||
| 1 | 6 | 4 | 1 | 0 | 2 | 4 | 8 | 2 | 6 | 4 | 6 | 3 | 0 | 3 | 0 | 4 | |
| CpI1 | A | C | C | C | C | C | A | A | C | A | C | G | G | T | T | G | C |
| CpI2 | . | . | T | . | . | . | . | . | . | G | T | . | . | C | G | . | . |
| CpI3 | . | . | T | . | . | . | . | . | . | G | T | . | . | . | G | . | . |
| CpI4 | . | T | . | . | . | . | . | . | . | G | T | . | . | . | G | . | . |
| CpI5 | . | T | . | G | . | . | . | . | . | G | T | . | . | . | G | . | . |
| CpI6 | . | T | . | . | G | . | . | . | . | G | T | . | . | . | G | . | . |
| CpI7 | C | T | . | . | . | . | . | . | . | G | T | . | . | . | G | . | . |
| CpI8 | . | T | . | . | . | A | T | . | . | G | T | . | . | . | G | . | . |
| CpI9 | . | T | . | . | . | A | . | . | . | G | T | . | . | . | G | . | . |
| CpI10 | . | T | . | . | . | A | . | . | . | G | T | . | . | C | G | . | . |
| CpI11 | . | . | . | . | . | . | . | . | . | G | T | . | . | . | G | . | . |
| CpI12 | . | . | T | . | . | . | . | . | . | G | T | A | . | C | G | . | . |
| CpI13 | . | . | T | . | . | . | . | . | . | G | T | . | C | C | G | C | . |
| CpI14 | . | T | T | . | . | A | . | . | . | G | T | . | . | . | G | . | . |
| CpI15 | . | T | . | . | . | . | . | C | . | G | T | . | . | . | G | . | . |
| CpI16 | . | T | . | . | . | . | . | . | A | G | T | . | . | . | G | . | . |
| CpI17 | . | T | . | . | . | . | . | . | A | G | T | . | . | . | G | . | T |
| Populations | Molecular Diversity Indices | |||||
|---|---|---|---|---|---|---|
| n | S | H | Uh | Hd ± SD | Nd ± SD | |
| PLK | 9 | 4 | 2 | 1 | 0.556 ± 0.090 | 0.0034 ± 0.0006 |
| CMI | 9 | 7 | 7 | 6 | 0.944 ± 0.070 | 0.0039 ± 0.0007 |
| LEI | 10 | 0 | 1 | 0 | 0.000 ± 0.000 | 0.0000 ± 0.0000 |
| MKM | 10 | 6 | 7 | 5 | 0.911 ± 0.077 | 0.0026 ± 0.0004 |
| LPN | 5 | 0 | 1 | 1 | 0.000 ± 0.000 | 0.0000 ± 0.0000 |
| NPM | 11 | 1 | 2 | 0 | 0.436 ± 0.133 | 0.0007 ± 0.0002 |
| UBN | 8 | 1 | 2 | 2 | 0.250 ± 0.180 | 0.0004 ± 0.0003 |
| Total | 62 | 17 | 17 | 15 | 0.832 ± 0.039 | 0.0032 ± 0.0004 |
| Codes * | CMI | LPN | LEI | MKM | UBN | NPM | PLK |
|---|---|---|---|---|---|---|---|
| CMI | – | 0.0044 | 0.0037 | 0.0051 | 0.0055 | 0.0040 | 0.0058 |
| LPN | 0.0044 | – | 0.0031 | 0.0032 | 0.0048 | 0.0026 | 0.0065 |
| LEI | 0.0038 | 0.0031 | – | 0.0017 | 0.0017 | 0.0004 | 0.0034 |
| MKM | 0.0051 | 0.0032 | 0.0017 | – | 0.0034 | 0.0017 | 0.0051 |
| UBN | 0.0055 | 0.0048 | 0.0017 | 0.0034 | – | 0.0021 | 0.0051 |
| NPM | 0.0040 | 0.0027 | 0.0004 | 0.0017 | 0.0021 | – | 0.0038 |
| PLK | 0.0059 | 0.0065 | 0.0034 | 0.0051 | 0.0052 | 0.0038 | – |
| Codes * | PLK | CMI | LEI | MKM | LPN | NPM | UBN |
|---|---|---|---|---|---|---|---|
| PLK | - | 0.4225 | 0.2326 | 0.3591 | 0.1233 | 0.2596 | 0.1552 |
| CMI | 0.3718 | - | 0.2544 | 0.4415 | 0.2862 | 0.3069 | 0.1721 |
| LEI | 0.5181 | 0.4956 | - | 0.8750 | 0.0000 | 1.0945 | 0.0273 |
| MKM | 0.4104 | 0.3615 | 0.2222 | - | 0.2570 | 8.0364 | 0.2187 |
| LPN | 0.6697 | 0.4662 | 1.0000 | 0.4931 | - | 0.0525 | 0.0133 |
| NPM | 0.4906 | 0.4489 | 0.1859 | 0.0302 | 0.8264 | - | 0.0855 |
| UBN | 0.6169 | 0.5923 | 0.9017 | 0.5333 | 0.9495 | 0.7451 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaenkham, K.; Pilap, W.; Saijuntha, W.; Thanonkeo, S. Profiling Genetic Variation: Divergence Patterns and Population Structure of Thailand’s Endangered Celastrus paniculatus Willd. Biology 2025, 14, 725. https://doi.org/10.3390/biology14060725
Kaenkham K, Pilap W, Saijuntha W, Thanonkeo S. Profiling Genetic Variation: Divergence Patterns and Population Structure of Thailand’s Endangered Celastrus paniculatus Willd. Biology. 2025; 14(6):725. https://doi.org/10.3390/biology14060725
Chicago/Turabian StyleKaenkham, Kornchanok, Warayutt Pilap, Weerachai Saijuntha, and Sudarat Thanonkeo. 2025. "Profiling Genetic Variation: Divergence Patterns and Population Structure of Thailand’s Endangered Celastrus paniculatus Willd" Biology 14, no. 6: 725. https://doi.org/10.3390/biology14060725
APA StyleKaenkham, K., Pilap, W., Saijuntha, W., & Thanonkeo, S. (2025). Profiling Genetic Variation: Divergence Patterns and Population Structure of Thailand’s Endangered Celastrus paniculatus Willd. Biology, 14(6), 725. https://doi.org/10.3390/biology14060725