
AI-Driven Transcriptome Prediction in Human Pathology: From Molecular Insights to Clinical Applications

Simple Summary
Abstract
1. Introduction
2. The Pivotal Role of Gene Expression in Disease Pathogenesis
3. The Integration of Artificial Intelligence Algorithms and Biomedicine
3.1. Multimodal Data Types
3.2. Machine Learning
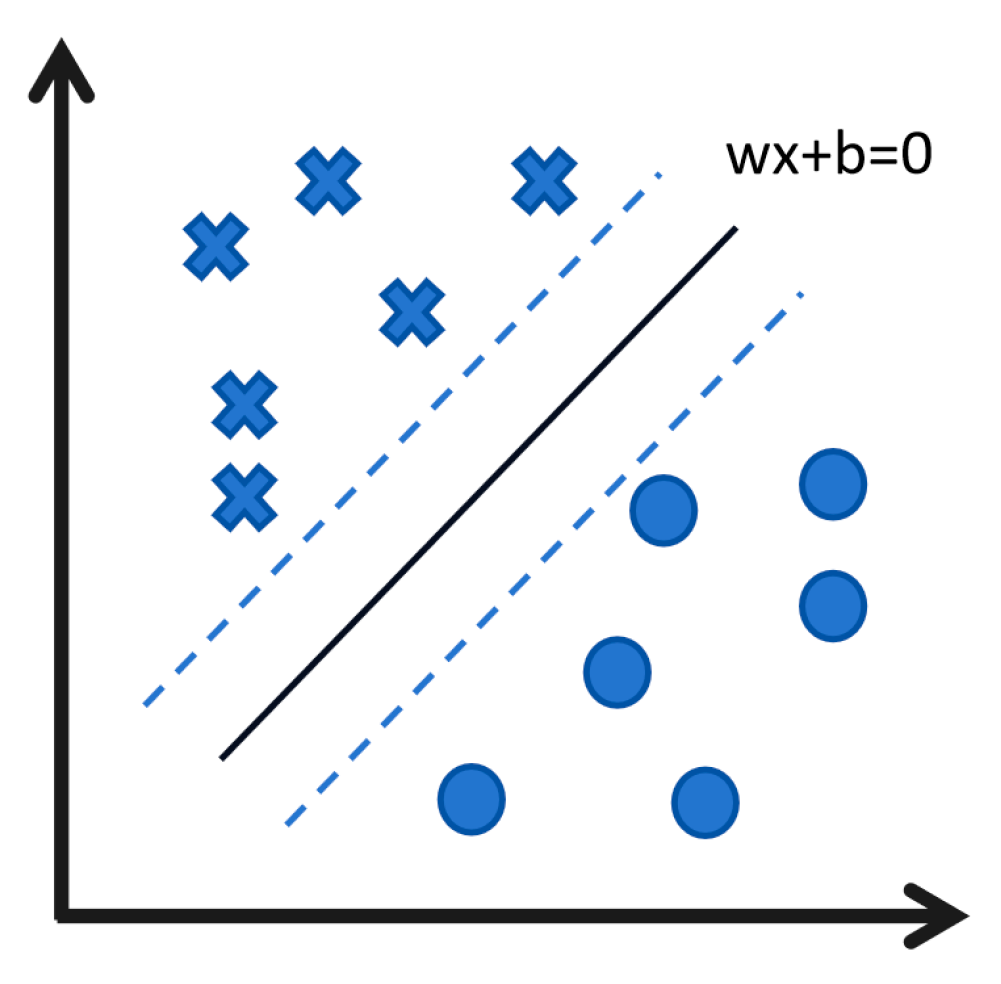
3.2.1. Support Vector Machine (SVM): Achieving Precise Classification Through Clearly Defined Boundaries
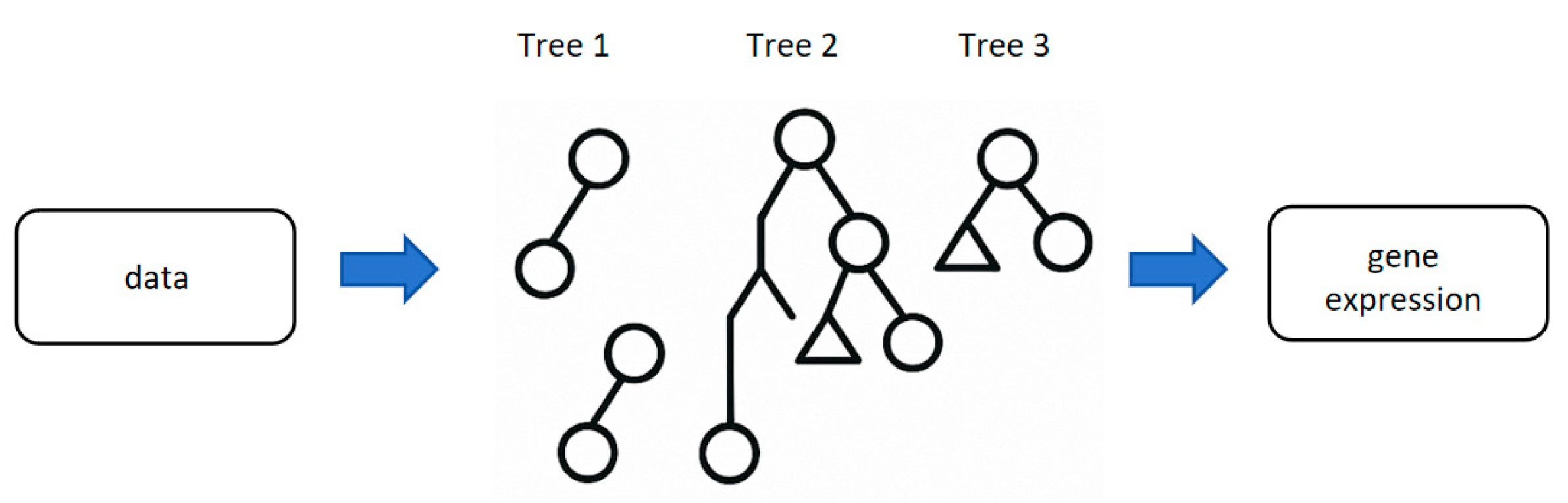
3.2.2. Random Forest (RF): The “Decision-Making Brain Trust” for Multi-Dimensional Data Integration
3.2.3. Gradient Boosting Machine (GBM): Iterative Optimization of Complex Disease Models
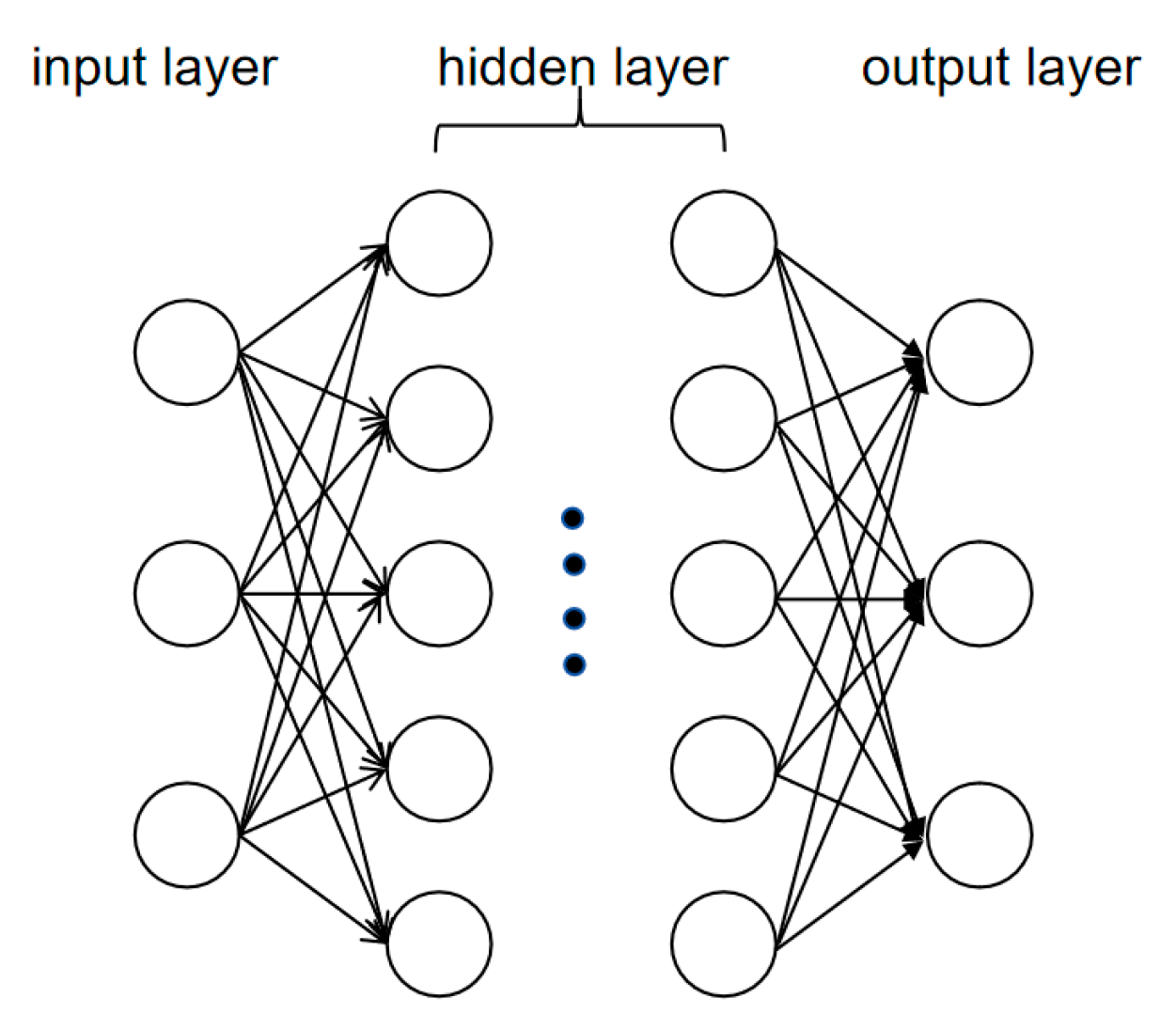
3.3. Deep Learning
3.3.1. Convolutional Neural Network (CNN): Specialized in Local Feature Extraction for Sequence and Image Analysis
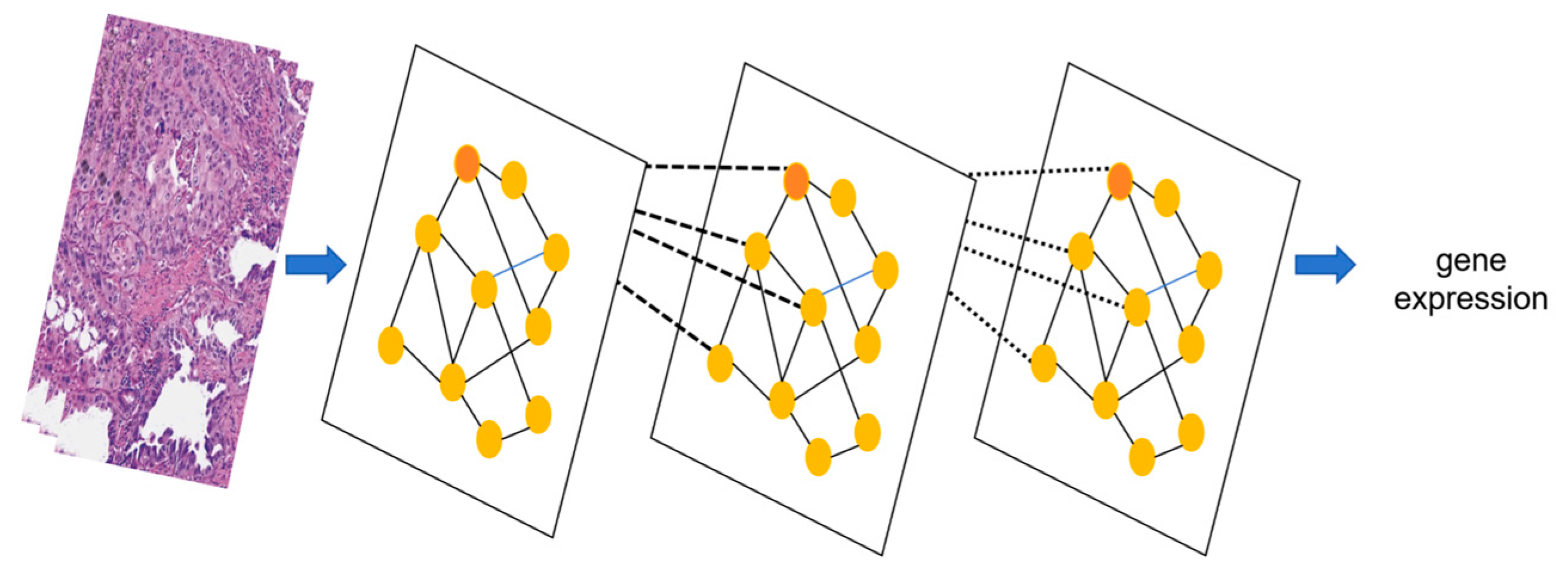
3.3.2. Graph Neural Network (GNN): Specialized in Relational Modeling for Biological Network Analysis
3.3.3. Transformer: Leveraging Self-Attention for Multimodal Integration and Long-Range Dependency Modeling
3.4. Multimodal Data Fusion
- Cross-Modal Representation Learning
- 2.
- Knowledge Graph Bridging
- 3.
- Hierarchical Attention Mechanisms
3.5. Privacy and Ethical Issues
4. Existing Models and Applications of Artificial Intelligence in Predicting Gene Expression
4.1. Disease Diagnosis and Mechanism Research
4.2. Drug Development and Synthetic Biology
4.3. Precision Medicine and Health Management
5. Challenges in AI-Driven Prediction of Gene Expression
5.1. Data-Related Challenges: Bridging the Heterogeneity–Fusion Gap
5.2. Model-Related Challenges: From Architectural Innovation to Interpretability
5.3. Ethical and Legal Challenges
6. Prospects for the Future
- Innovation in Multimodal Fusion
- 2.
- Interpretability Enhancement
- 3.
- Data Governance and Ethical Frameworks
- 4.
- Clinical Translation Optimization
7. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alharbi, F.; Vakanski, A. Machine learning methods for cancer classification using gene expression data: A review. Bioengineering 2023, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, L.; Wang, Y.; Wu, P.; Hou, X.; Ouyang, J.; Fan, C.; Li, Z.; Wang, F.; Guo, C.; et al. RNA Modifications in Cancer. Br. J. Cancer 2023, 129, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Liu, Y.; Gerasimov, E.S.; Vattathil, S.M.; Wynne, M.E.; Liu, J.; Lori, A.; Faundez, V.; Bennett, D.A.; Seyfried, N.T.; et al. Shared Mechanisms across the Major Psychiatric and Neurodegenerative Diseases. Nat. Commun. 2022, 13, 4314. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 2023, 19, 509–524. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Chakrabortty, A.; Patton, D.J.; Smith, B.F.; Agarwal, P. miRNAs: Potential as Biomarkers and Therapeutic Targets for Cancer. Genes 2023, 14, 1375. [Google Scholar] [CrossRef]
- Loganathan, T.; Doss, C.G.P. Non-coding RNAs in human health and disease: Potential function as biomarkers and therapeutic targets. Funct. Integr. Genom. 2023, 23, 33. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, H.; Jiang, S.; Wang, W. Role of Tumor-Associated Neutrophils in Lung Cancer (Review). Oncol. Lett. 2022, 25, 2. [Google Scholar] [CrossRef]
- Yang, L.; Bai, B.; Chen, H.; Li, T.; Wang, M.; Zhu, X. Cinobufotalin Effect and Mechanism on Serum MMP-2, MMP-9, Beclin1, LC3-II in Advanced NSCLC Patients. Cell. Mol. Biol. 2023, 69, 58–62. [Google Scholar] [CrossRef]
- Pi, C.; Xu, C.R.; Zhang, M.F.; Peng, X.X.; Wei, X.W.; Gao, X.; Yan, H.H.; Zhou, Q. EGFR Mutations in Early-Stage and Advanced-Stage Lung Adenocarcinoma: Analysis Based on Large-Scale Data from China. Thorac. Cancer 2018, 9, 814–819. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Sharma, G.; Karmakar, S.; Banerjee, S. Multi-OMICS Approaches in Cancer Biology: New Era in Cancer Therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167120. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Wu, L.R.; Yan, Y.H.; Zhang, J.X.; Chu, T.; Kwong, L.N.; Patel, A.A.; Zhang, D.Y. Limitations and Opportunities of Technologies for the Analysis of Cell-Free DNA in Cancer Diagnostics. Nat. Biomed. Eng. 2022, 6, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Bera, K.; Schalper, K.A.; Rimm, D.L.; Velcheti, V.; Madabhushi, A. Artificial Intelligence in Digital Pathology—New Tools for Diagnosis and Precision Oncology. Nat. Rev. Clin. Oncol. 2019, 16, 703–715. [Google Scholar] [CrossRef]
- Song, A.H.; Jaume, G.; Williamson, D.F.; Lu, M.Y.; Vaidya, A.; Miller, T.R.; Mahmood, F. Artificial intelligence for digital and computational pathology. Nat. Rev. Bioeng. 2023, 1, 930–949. [Google Scholar] [CrossRef]
- Arya, N.; Saha, S. Multi-modal classification for human breast cancer prognosis prediction: Proposal of deep-learning based stacked ensemble model. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 1032–1041. [Google Scholar] [CrossRef]
- Hoang, D.T.; Dinstag, G.; Shulman, E.D.; Hermida, L.C.; Ben-Zvi, D.S.; Elis, E.; Caley, K.; Sammut, S.J.; Sinha, S.; Sinha, N.; et al. A deep-learning framework to predict cancer treatment response from histopathology images through imputed transcriptomics. Nat. Cancer 2024, 5, 1305–1317. [Google Scholar] [CrossRef]
- Lu, M.Y.; Chen, B.; Williamson, D.F.K.; Chen, R.J.; Zhao, M.; Chow, A.K.; Ikemura, K.; Kim, A.; Pouli, D.; Patel, A.; et al. A multimodal generative AI copilot for human pathology. Nature 2024, 634, 466–473. [Google Scholar] [CrossRef]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 103798. [Google Scholar] [CrossRef]
- Fu, X.; Mo, S.; Buendia, A.; Laurent, A.P.; Shao, A.; Alvarez-Torres, M.D.M.; Yu, T.; Tan, J.; Su, J.; Sagatelian, R.; et al. A foundation model of transcription across human cell types. Nature 2025, 637, 965–973. [Google Scholar] [CrossRef]
- Acosta, J.N.; Falcone, G.J.; Rajpurkar, P.; Topol, E.J. Multimodal biomedical AI. Nat. Med. 2022, 28, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Mataraso, S.J.; Espinosa, C.A.; Seong, D.; Reincke, S.M.; Berson, E.; Reiss, J.D.; Kim, Y.; Ghanem, M.; Shu, C.-H.; James, T.; et al. A Machine Learning Approach to Leveraging Electronic Health Records for Enhanced Omics Analysis. Nat. Mach. Intell. 2025, 7, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Monjo, T.; Koido, M.; Nagasawa, S.; Suzuki, Y.; Kamatani, Y. Efficient prediction of a spatial transcriptomics profile better characterizes breast cancer tissue sections without costly experimentation. Sci. Rep. 2022, 12, 4133. [Google Scholar] [CrossRef] [PubMed]
- Anil, R.; Borgeaud, S.; Alayrac, J.-B.; Yu, J.; Soricut, R.; Schalkwyk, J.; Dai, A.M.; Hauth, A.; Millican, K.; Silver, D.; et al. Gemini: A family of highly capable multimodal models. arXiv 2023, arXiv:2312.11805. [Google Scholar]
- Jia, X.; He, X.; Huang, C.; Li, J.; Dong, Z.; Liu, K. Protein translation: Biological processes and therapeutic strategies for human diseases. Signal Transduct. Target. Ther. 2024, 9, 44. [Google Scholar] [CrossRef]
- Carthew, R.W. Gene regulation and cellular metabolism: An essential partnership. Trends Genet. 2021, 37, 389–400. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Petit, D.; Fernández, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O’Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C. Aβ profiles generated by Alzheimer’s disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol. Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef]
- Kulminski, A.M.; Huang, J.; Wang, J.; He, L.; Loika, Y.; Culminskaya, I. Apolipoprotein E region molecular signatures of Alzheimer’s disease. Aging Cell 2018, 17, e12779. [Google Scholar] [CrossRef]
- Fortea, J.; Pegueroles, J.; Alcolea, D.; Belbin, O.; Dols-Icardo, O.; Vaqué-Alcázar, L.; Videla, L.; Gispert, J.D.; Suárez-Calvet, M.; Johnson, S.C. APOE4 homozygozity represents a distinct genetic form of Alzheimer’s disease. Nat. Med. 2024, 30, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Solomon, G.M.; Marshall, S.G.; Ramsey, B.W.; Rowe, S.M. Breakthrough therapies: Cystic fibrosis (CF) potentiators and correctors. Pediatr. Pulmonol. 2015, 50 (Suppl. S40), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.J.; Chale, R.S.; Abad, A.C.; Schally, A.V. Acute promyelocytic leukemia (APL): A review of the literature. Oncotarget 2020, 11, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, J.; Yu, W.; Jin, J. Current views on the genetic landscape and management of variant acute promyelocytic leukemia. Biomark. Res. 2021, 9, 33. [Google Scholar] [CrossRef]
- Ropero, P.; González, F.A.; Nieto, J.M.; Villegas, A.; Sevilla, J.; Pérez, G.; Alonso, J.M.; Recasens, V.; Abio, M.; Vagace, J.M. C>A substitution in NT 46 of the 3′ UTR region (the α complex protected region) of the alpha-1 globin gene: A non-deletional mutation or polymorphism? J. Clin. Pathol. 2020, 73, 14–16. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Vadolas, J. Controlling α-globin: A review of α-globin expression and its impact on β-thalassemia. Haematologica 2008, 93, 1868–1876. [Google Scholar] [CrossRef]
- Zerr, I.; Ladogana, A.; Mead, S.; Hermann, P.; Forloni, G.; Appleby, B.S. Creutzfeldt-Jakob disease and other prion diseases. Nat. Rev. Dis. Primers 2024, 10, 14. [Google Scholar] [CrossRef]
- Canzler, S.; Schor, J.; Busch, W.; Schubert, K.; Rolle-Kampczyk, U.E.; Seitz, H.; Kamp, H.; von Bergen, M.; Buesen, R.; Hackermüller, J. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 2020, 94, 371–388. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef]
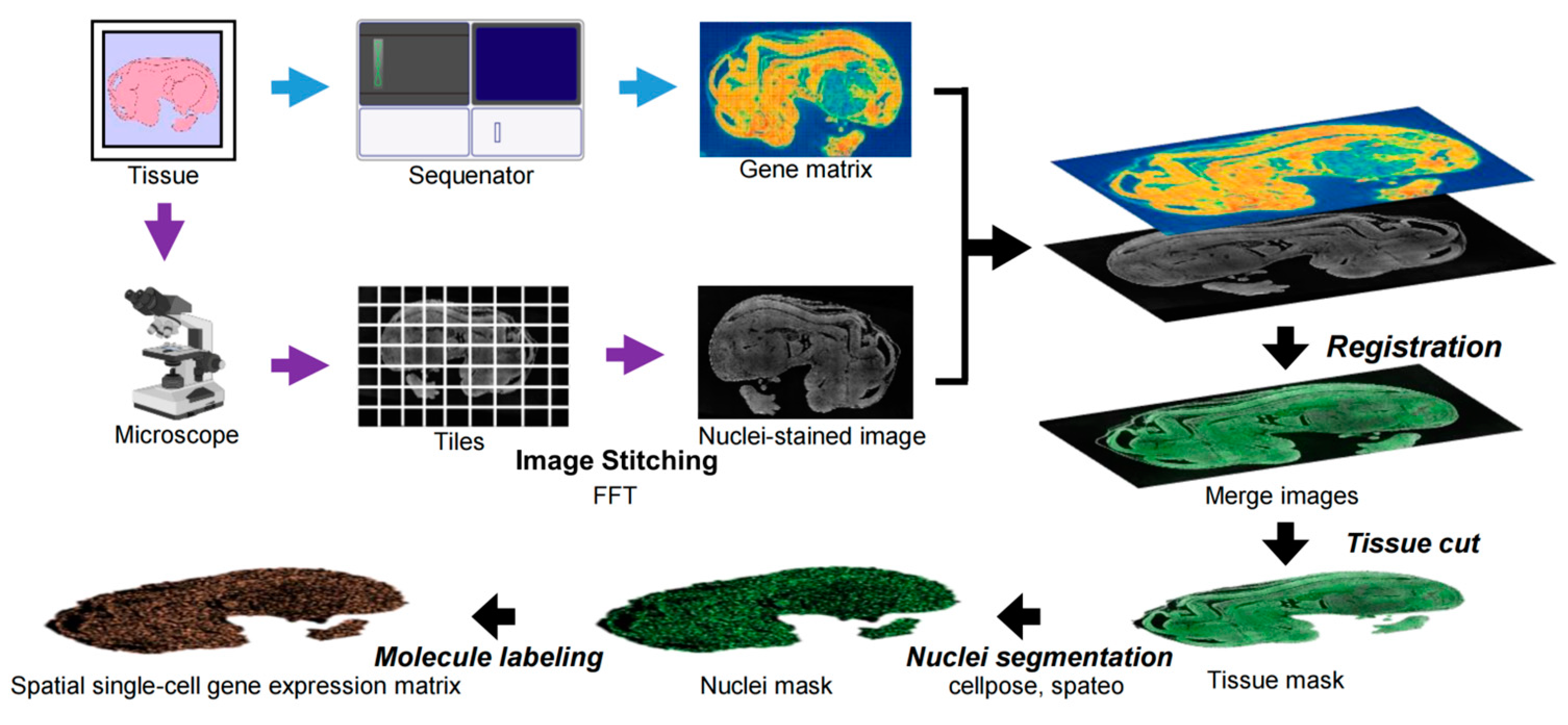
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Barkley, D.; França, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Unterauer, E.M.; Shetab Boushehri, S.; Jevdokimenko, K.; Masullo, L.A.; Ganji, M.; Sograte-Idrissi, S.; Kowalewski, R.; Strauss, S.; Reinhardt, S.C.M.; Perovic, A. Spatial proteomics in neurons at single-protein resolution. Cell 2024, 187, 1785–1800.e16. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, A.; Zhou, Y.; Chen, P.; Wang, X.; Huang, J.; Gao, J.; Wang, X.; Shu, L.; Lu, J. Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat. Commun. 2023, 14, 2692. [Google Scholar] [CrossRef]
- Paris, J.; Henderson, N.C. Liver zonation, revisited. Hepatology 2022, 76, 1219–1230. [Google Scholar] [CrossRef]
- Liu, C.; Li, R.; Li, Y.; Lin, X.; Zhao, K.; Liu, Q.; Wang, S.; Yang, X.; Shi, X.; Ma, Y. Spatiotemporal mapping of gene expression landscapes and developmental trajectories during zebrafish embryogenesis. Dev. Cell. 2022, 57, 1284–1298.e5. [Google Scholar] [CrossRef]
- Bassiouni, R.; Idowu, M.O.; Gibbs, L.D.; Robila, V.; Grizzard, P.J.; Webb, M.G.; Song, J.; Noriega, A.; Craig, D.W.; Carpten, J.D. Spatial Transcriptomic Analysis of a Diverse Patient Cohort Reveals a Conserved Architecture in Triple-Negative Breast Cancer. Cancer Res. 2023, 83, 34–48. [Google Scholar] [CrossRef]
- Moffet, J.J.D.; Fatunla, O.E.; Freytag, L.; Kriel, J.; Jones, J.J.; Roberts-Thomson, S.J.; Pavenko, A.; Scoville, D.K.; Zhang, L.; Liang, Y. Spatial architecture of high-grade glioma reveals tumor heterogeneity within distinct domains. Neurooncol. Adv. 2023, 5, vdad142. [Google Scholar]
- Stringer, C.; Wang, T.; Michaelos, M.; Pachitariu, M. Cellpose: A generalist algorithm for cellular segmentation. Nat. Methods 2021, 18, 100–106. [Google Scholar] [CrossRef]
- Qiu, X.; Zhu, D.Y.; Lu, Y.; Yao, J.; Jing, Z.; Min, K.H.; Cheng, M.; Pan, H.; Zuo, L.; King, S. Spatiotemporal modeling of molecular holograms. Cell 2024, 187, 7351–7373.e61. [Google Scholar] [CrossRef]
- Krallinger, M.; Leitner, F.; Valencia, A. Analysis of biological processes and diseases using text mining approaches. Bioinform. Methods Clin. Res. 2010, 341–382. [Google Scholar]
- Rana, A.; Lowe, A.; Lithgow, M.; Horback, K.; Janovitz, T.; Da Silva, A.; Tsai, H.; Shanmugam, V.; Bayat, A.; Shah, P. Use of Deep Learning to Develop and Analyze Computational Hematoxylin and Eosin Staining of Prostate Core Biopsy Images for Tumor Diagnosis. JAMA Netw. Open 2020, 3, e205111. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Overview of deep learning in medical imaging. Radiol. Phys. Technol. 2017, 10, 257–273. [Google Scholar] [CrossRef]
- Ardila, D.; Kiraly, A.P.; Bharadwaj, S.; Choi, B.; Reicher, J.J.; Peng, L.; Tse, D.; Etemadi, M.; Ye, W.; Corrado, G. End-to-end lung cancer screening with three-dimensional deep learning on low-dose chest computed tomography. Nat. Med. 2019, 25, 954–961. [Google Scholar] [CrossRef]
- Burges, C.J.C. A tutorial on support vector machines for pattern recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobot. 2013, 7, 21. [Google Scholar] [CrossRef]
- Khalsan, M.; Machado, L.; Al-Shamery, E.S.; Ajit, S.; Anthony, K.; Mu, M.; Opoku Agyeman, M. A survey of machine learning approaches applied to gene expression analysis for cancer prediction. IEEE Access 2022, 10, 27522–27534. [Google Scholar] [CrossRef]
- Steinwart, I.; Christmann, A. Support Vector Machines; Springer Science & Business Media: Berlin, Germany, 2008. [Google Scholar]
- Perfecto-Avalos, Y.; Garcia-Gonzalez, A.; Hernandez-Reynoso, A.; Sánchez-Ante, G.; Ortiz-Hidalgo, C.; Scott, S.-P.; Fuentes-Aguilar, R.Q.; Diaz-Dominguez, R.; León-Martínez, G.; Velasco-Vales, V.; et al. Discriminant analysis and machine learning approach for evaluating and improving the performance of immunohistochemical algorithms for COO classification of DLBCL. J. Transl. Med. 2019, 17, 198. [Google Scholar] [CrossRef]
- Santhosh Krishna, B.V.; Jijin Godwin, J.; Tharanee Shree, S.; Sreenidhi, B.; Abinaya, T. Detection of leukemia and its types using combination of support vector machine and K-nearest neighbors algorithm. In Next Generation of Internet of Things: Proceedings of ICNGIoT 2021; Springer: Singapore, 2021; pp. 435–444. [Google Scholar]
- Biau, G.; Scornet, E. A random forest guided tour. Test 2016, 25, 197–227. [Google Scholar] [CrossRef]
- Lima, D.V.C.; Terrematte, P.; Stransky, B.; Silva, A.C.; Souza, M.F.; Oliveira, R.S.; Pereira, L.M.; Costa, J.F.; Almeida, T.R.; Fernandes, H.L.; et al. An Integrated Data Analysis Using Bioinformatics and Random Forest to Predict Prognosis of Patients with Squamous Cell Carcinoma. IEEE Access 2024, 12, 59335–59345. [Google Scholar] [CrossRef]
- Khan, O.; Ajadi, J.O.; Hossain, M.P. Predicting malaria outbreak in The Gambia using machine learning techniques. PLoS ONE 2024, 19, e0299386. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wen, X.; Tang, H.; Zhou, Y.; Liang, J. Predictive tool for the risk of hypothermia during laparoscopic gynecologic tumor resection. Eur. J. Obstet. Gynecol. Reprod. Biol. 2025, 306, 147–153. [Google Scholar] [CrossRef]
- Jia, Y. Research on Factor Interaction Effects and Nonlinear Relationships in Quantitative Models. In Proceedings of the 2024 6th International Conference on Machine Learning, Big Data and Business Intelligence (MLBDBI), Hangzhou, China, 1–3 November 2024; IEEE: Piscataway, NJ, USA, 2024; pp. 106–109. [Google Scholar]
- Yan, C.; Wu, F.X.; Wang, J.; Duan, G. PESM: Predicting the essentiality of miRNAs based on gradient boosting machines and sequences. BMC Bioinform. 2020, 21, 111. [Google Scholar] [CrossRef]
- Bentéjac, C.; Csörgő, A.; Martínez-Muñoz, G. A comparative analysis of gradient boosting algorithms. Artif. Intell. Rev. 2021, 54, 1937–1967. [Google Scholar] [CrossRef]
- Yao, X.; Fu, X.; Zong, C. Short-term load forecasting method based on feature preference strategy and LightGBM-XGboost. IEEE Access 2022, 10, 75257–75268. [Google Scholar] [CrossRef]
- Aniesch, C.; Zschech, P.; Heinrich, K. Machine learning and deep learning. Electron. Mark 2021, 31, 685–695. [Google Scholar] [CrossRef]
- Bouvrie, J. Notes on Convolutional Neural Networks; Massachusetts Institute of Technology: Cambridge, MA, USA, 2006. [Google Scholar]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. [Google Scholar] [CrossRef]
- Han, K.; Xiao, A.; Wu, E.; Guo, J.; Xu, C.; Wang, Y. Transformer in transformer. Adv. Neural Inf. Process. Syst. 2021, 34, 15908–15919. [Google Scholar]
- de Almeida, B.P.; Reiter, F.; Pagani, M.; Stark, A. DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat. Genet. 2022, 54, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Gunavathi, C.; Sivasubramanian, K.; Keerthika, P.; Paramasivam, C. A review on convolutional neural network based deep learning methods in gene expression data for disease diagnosis. Mater. Today Proc. 2021, 45, 2282–2285. [Google Scholar]
- Wang, S.; He, M.; Qin, M.; Zhou, Y. Cross-tissue Graph Attention Networks for Semi-supervised Gene Expression Prediction. bioRxiv 2024. bioRxiv:2024.11.15.623881. [Google Scholar]
- Shahir, J.A.; Stanley, N.; Purvis, J.E. Cellograph: A semi-supervised approach to analyzing mul-ti-condition single-cell RNA-sequencing data using graph neural networks. BMC Bioinform. 2024, 25, 25. [Google Scholar] [CrossRef]
- Mai, S.; Hu, H.; Xing, S. Modality to modality translation: An adversarial representation learning and graph fusion network for multimodal fusion. AAAI Conf. Artif. Intell. 2020, 34, 164–172. [Google Scholar] [CrossRef]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Wang, M.Y. Deep graph library: Towards efficient and scalable deep learning on graphs. In Proceedings of the ICLR Workshop on Representation Learning on Graphs and Manifolds, New Orleans, LA, USA, 6–9 May 2019. [Google Scholar]
- Choi, S.R.; Lee, M. Transformer architecture and attention mechanisms in genome data analysis: A comprehensive review. Biology 2023, 12, 1033. [Google Scholar] [CrossRef]
- Poria, S.; Cambria, E.; Howard, N.; Huang, G.-B.; Hussain, A. Fusing audio, visual and textual clues for sentiment analysis from multimodal content. Neurocomputing 2016, 174, 50–59. [Google Scholar] [CrossRef]
- Barnum, G.; Talukder, S.; Yue, Y. On the benefits of early fusion in multimodal representation learning. arXiv 2020, arXiv:2011.07191. [Google Scholar]
- Wang, Y.; Li, M.; Wan, X.; Zhang, C.; Wang, Y. Multiparameter space decision voting and fusion features for facial expression recognition. Comput. Intell. Neurosci. 2020, 2020, 8886872. [Google Scholar] [CrossRef]
- Gadzicki, K.; Khamsehashari, R.; Zetzsche, C. Early vs late fusion in multimodal convolutional neural networks. In Proceedings of the 2020 IEEE 23rd International Conference on Information Fusion (FUSION), Rustenburg, South Africa, 6–9 July 2020; IEEE: Piscataway, NJ, USA, 2020; pp. 1–6. [Google Scholar]
- Chen, X.; Zhang, N.; Li, L.; Deng, S.; Tan, C.; Xu, C.; Huang, F.; Si, L.; Chen, H. Hybrid transformer with multi-level fusion for multimodal knowledge graph completion. In Proceedings of the 45th International ACM SIGIR Conference on Research and Development in Information Retrieval, Madrid, Spain, 11–15 July 2022; pp. 904–915. [Google Scholar]
- Gao, Y.; Zhou, M.; Metaxas, D.N. UTNet: A hybrid transformer architecture for medical image segmentation. In Proceedings of the Medical Image Computing and Computer Assisted Intervention–MICCAI 2021, 24th International Conference, Proceedings, Part III 24. Strasbourg, France, 27 September–1 October 2021; Springer International Publishing: Berlin/Heidelberg, Germany, 2021; pp. 61–71. [Google Scholar]
- Guarrasi, V.; Aksu, F.; Caruso, C.M.; Di Feola, F.; Rofena, A.; Ruffini, F.; Soda, P. A systematic review of intermediate fusion in multimodal deep learning for biomedical applications. Image Vis. Comput. 2025, 158, 105509. [Google Scholar] [CrossRef]
- Jiao, T.; Guo, C.; Feng, X.; Chen, Y.; Song, J. A comprehensive survey on deep learning multi-modal fusion: Methods, technologies and applications. Comput. Mater. Contin. 2024, 80, 1–35. [Google Scholar] [CrossRef]
- Shankar, S.; Thompson, L.; Fiterau, M. Progressive fusion for multimodal integration. arXiv 2022, arXiv:2209.00302. [Google Scholar]
- Sutton, R.S.; Barto, A.G. Reinforcement Learning: An Introduction; MIT Press: Cambridge, MA, USA, 1998. [Google Scholar]
- Huang, S.C.; Pareek, A.; Jensen, M.; Lungren, M.P.; Yeung, S.; Chaudhari, A.S. Self-supervised learning for medical image classification: A systematic review and implementation guidelines. NPJ Digit. Med. 2023, 6, 74. [Google Scholar] [CrossRef]
- Mnih, V.; Kavukcuoglu, K.; Silver, D.; Rusu, A.A.; Veness, J.; Bellemare, M.G.; Graves, A.; Riedmiller, M.; Fidjeland, A.K.; Ostrovski, G. Human-level control through deep reinforcement learning. Nature 2015, 518, 529–533. [Google Scholar] [CrossRef]
- Khajuria, R.; Sarwar, A. Active reinforcement learning based approach for localization of target ROI (region of interest) in cervical cell images. Multimed. Tools Appl. 2024, 84, 18467–18479. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, K.; Yuan, Q.; Peng, H.; Zheng, Y.; Hanratty, T.; Wang, S.; Han, J. Regions, periods, activities: Uncovering urban dynamics via cross-modal representation learning. In Proceedings of the 26th International Conference on World Wide Web, Perth, Australia, 3–7 April 2017; pp. 361–370. [Google Scholar]
- Andonian, A.; Chen, S.; Hamid, R. Robust cross-modal representation learning with progressive self-distillation. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, New Orleans, LA, USA, 18–24 June 2022; pp. 16430–16441. [Google Scholar]
- Wang, T.; Fan, J.; Zhang, D.; Liu, D.; Xia, Y.; Huang, H.; Cai, W. MIRROR: Multi-Modal Pathological Self-Supervised Representation Learning via Modality Alignment and Retention. arXiv 2025, arXiv:2503.00374. [Google Scholar]
- Bourgeais, V.; Zehraoui, F.; Hanczar, B. GraphGONet: A self-explaining neural network encapsulating the Gene Ontology graph for phenotype prediction on gene expression. Bioinformatics 2022, 38, 2504–2511. [Google Scholar] [CrossRef]
- Li, M.; Liu, H.; Li, M.; Fang, S.; Kang, Q.; Zhang, J. StereoCell enables highly accurate single-cell segmentation for spatial transcriptomics. bioRxiv 2023. [Google Scholar] [CrossRef]
- Li, H.; Han, Z.; Sun, Y.; Wang, F.; Hu, P.; Gao, Y.; Bai, X.; Peng, S.; Ren, C.; Xu, X. CGMega: Explainable graph neural network framework with attention mechanisms for cancer gene module dissection. Nat. Commun. 2024, 15, 5997. [Google Scholar] [CrossRef]
- Pizurica, M.; Zheng, Y.; Carrillo-Perez, F.; Noor, H.; Yao, W.; Wohlfart, C.; Vladimirova, A.; Marchal, K.; Gevaert, O. Digital profiling of gene expression from histology images with linearized attention. Nat. Commun. 2024, 15, 9886. [Google Scholar] [CrossRef] [PubMed]
- Jin, I.; Nam, H. HiDRA: Hierarchical network for drug response prediction with attention. J. Chem. Inf. Model. 2021, 61, 3858–3867. [Google Scholar] [CrossRef] [PubMed]
- Saeed, D.; Xing, H.; AlBadani, B.; Feng, L.; Al-Sabri, R.; Abdullah, M.; Rehman, A. MGATAF: Multi-channel graph attention network with adaptive fusion for cancer-drug response prediction. BMC Bioinform. 2025, 26, 19. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Liang, Y.; Li, L.; Liao, S.; Zou, Y.; Yang, C.; Ouyang, D. Inferring circRNA-drug sensitivity associations via dual hierarchical attention networks and multiple kernel fusion. BMC Genom. 2023, 24, 796. [Google Scholar] [CrossRef]
- Lu, M.Y.; Chen, R.J.; Kong, D.; Lipková, J.; Singh, R.; Williamson, D.F.K.; Chen, T.Y.; Mahmood, F. Federated learning for computational pathology on gigapixel whole slide images. Med. Image Anal. 2022, 76, 102298. [Google Scholar] [CrossRef]
- Saldanha, O.L.; Quirke, P.; West, N.P.; James, J.A.; Loughrey, M.B.; Grabsch, H.I.; Salto-Tellez, M.; Alwers, E.; Cifci, D.; Ghaffari Laleh, N. Swarm learning for decentralized artificial intelligence in cancer histopathology. Nat. Med. 2022, 28, 1232–1239. [Google Scholar] [CrossRef]
- Beguier, C.; du Terrail, J.O.; Meah, I.; Andreux, M.; Tramel, E.W. Differentially private federated learning for cancer prediction. arXiv 2021, arXiv:2101.02997. [Google Scholar]
- Brancato, V.; Esposito, G.; Coppola, L.; Cavaliere, C.; Mirabelli, P.; Scapicchio, C.; Borgheresi, R.; Neri, E.; Salvatore, M.; Aiello, M. Standardizing digital biobanks: Integrating imaging, genomic, and clinical data for precision medicine. J. Transl. Med. 2024, 22, 136. [Google Scholar] [CrossRef]
- Bohannon, J. Genealogy databases enable naming of anonymous DNA donors. Science 2013, 339, 262. [Google Scholar] [CrossRef]
- Custers, B.; Dechesne, F.; Sears, A.M.; Tani, T.; van der Hof, S. A comparison of data protection legislation and policies across the EU. Comput. Law Secur. Rev. 2018, 34, 234–243. [Google Scholar] [CrossRef]
- Xu, J.; Glicksberg, B.S.; Su, C.; Walker, P.; Bian, J.; Wang, F. Federated learning for healthcare informatics. J. Healthc. Inform. Res. 2021, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gichoya, J.W.; Banerjee, I.; Bhimireddy, A.R.; Burns, J.L.; Celi, L.A.; Chen, L.C.; Correa, R.; Dullerud, N.; Ghassemi, M.; Huang, S.C. AI recognition of patient race in medical imaging: A modelling study. Lancet Digit. Health 2022, 4, e406–e414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Lemoine, B.; Mitchell, M. Mitigating unwanted biases with adversarial learning. In Proceedings of the 2018 AAAI/ACM Conference on AI, Ethics, and Society, New Orleans, LA, USA, 2–7 February 2018; pp. 335–340. [Google Scholar]
- Veale, M.; Matus, K.; Gorwa, R. AI and global governance: Modalities, rationales, tensions. Annu. Rev. Law Soc. Sci. 2023, 19, 255–275. [Google Scholar] [CrossRef]
- Clayton, E.W.; Evans, B.J.; Hazel, J.W.; Rothstein, M.A. The law of genetic privacy: Applications, implications, and limitations. J. Law Biosci. 2019, 6, 1–36. [Google Scholar] [CrossRef]
- Li, J.; Dada, A.; Puladi, B.; Kleesiek, J.; Egger, J. ChatGPT in healthcare: A taxonomy and systematic review. Comput. Methods Programs Biomed. 2024, 245, 108013. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Y.; Wang, Q.; Zhang, C.; Li, M.; Wang, G.; Song, Q. Gene expression prediction from histology images via hypergraph neural networks. Brief. Bioinform. 2024, 25, bbae500. [Google Scholar] [CrossRef]
- Nguyen, E.; Poli, M.; Faizi, M.; Thomas, A.; Birch-Sykes, C.; Wornow, M.; Patel, A.; Rabideau, C.; Massaroli, S.; Bengio, Y. Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution. Adv. Neural Inf. Process. Syst. 2023, 36, 43177–43201. [Google Scholar]
- Baokar, P.; Ioannidis, N. Evaluating the Use of Sequence-to-Expression Predictors for Personalized Expression Prediction; University of California: Berkeley, CA, USA, 2022. [Google Scholar]
- Andersson, A.; Larsson, L.; Stenbeck, L.; Salmén, F.; Ehinger, A.; Wu, S.Z.; Al-Eryani, G.; Roden, D.; Swarbrick, A.; Borg, Å. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun. 2021, 12, 6012. [Google Scholar] [CrossRef]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; ‘t Hoen, P.A.C.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef]
- Zhou, Y.; Geng, P.; Zhang, S.; Xiao, F.; Cai, G.; Chen, L.; Lu, Q. Multimodal functional deep learning for multiomics data. Brief. Bioinform. 2024, 25, bbae448. [Google Scholar] [CrossRef]
- Ashuach, T.; Gabitto, M.I.; Koodli, R.V.; Saldi, G.A.; Jordan, M.I.; Yosef, N. MultiVI: Deep generative model for the integration of multimodal data. Nat. Methods 2023, 20, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, B.; Huang, S.; Hickey, J.W.; Lin, K.Z.; Snyder, M.; Greenleaf, W.J.; Nolan, G.P.; Zhang, N.R.; Ma, Z. Integration of spatial and single-cell data across modalities with weakly linked features. Nat. Biotechnol. 2024, 42, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, P.; Chen, Z.; Zhang, J. A survey on deep learning for multimodal data fusion. Neural Comput. 2020, 32, 829–864. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Liu, Y.; Xu, H.; Gao, K. Noise imitation based adversarial training for robust multimodal sentiment analysis. IEEE Trans. Multimedia 2023, 26, 529–539. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Acharya, D.; Mukhopadhyay, A. A comprehensive review of machine learning techniques for multi-omics data integration: Challenges and applications in precision oncology. Brief. Funct. Genom. 2024, 23, 549–560. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Waqas, A.; Tripathi, A.; Ramachandran, R.P.; Stewart, P.A.; Rasool, G. Multimodal data integration for oncology in the era of deep neural networks: A review. Front. Artif. Intell. 2024, 7, 1408843. [Google Scholar] [CrossRef]
- Waqas, A.; Tripathi, A.; Ramachandran, R.P.; Stewart, P.A.; Rasool, G. Multimodal data fusion for cancer biomarker discovery with deep learning. Nat. Mach. Intell. 2023, 5, 351–362. [Google Scholar]
- Hasan, B.M.S.; Abdulazeez, A.M. A review of principal component analysis algorithm for dimensionality reduction. J. Soft Comput. Data Min. 2021, 2, 20–30. [Google Scholar]
- Doersch, C. Tutorial on variational autoencoders. arXiv 2016, arXiv:1606.05908. [Google Scholar]
- Liu, J.; Cen, X.; Yi, C.; Wang, F.A.; Ding, J.; Cheng, J.; Wu, Q.; Gai, B.; Zhou, Y.; He, R. Challenges in AI-driven Biomedical Multimodal Data Fusion and Analysis. Genom. Proteom. Bioinform. 2025, 14, qzaf011. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Xiong, J.; Li, Y.; Ding, W. Deep learning based multimodal biomedical data fusion: An overview and comparative review. Inf. Fusion 2024, 112, 102536. [Google Scholar] [CrossRef]
- Jan, R.; Hussain, A.; Assad, A.; Khurshid, S.; Macha, M.A. Challenges with multi-omics data integration. In Multi-Omics Technology in Human Health and Diseases; Academic Press: Cambridge, MA, USA, 2025; pp. 223–242. [Google Scholar]
- Kumar, S.; Rani, S.; Sharma, S.; Min, H. Multimodality Fusion Aspects of Medical Diagnosis: A Comprehensive Review. Bioengineering 2024, 11, 1233. [Google Scholar] [CrossRef]
- Hemker, K.; Simidjievski, N.; Jamnik, M. HEALNet--Hybrid Multi-Modal Fusion for Heterogeneous Biomedical Data. arXiv 2023, arXiv:2311.09115. [Google Scholar]
- Tang, Q.; Liang, J.; Zhu, F. A comparative review on multi-modal sensors fusion based on deep learning. Signal Process. 2023, 213, 109165. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, F.; Zhao, C.; Xu, Y.; Luo, L.; Chen, H. Multimodal data integration for precision oncology: Challenges and future directions. arXiv 2024, arXiv:2406.19611. [Google Scholar]
- Cui, C.; Yang, H.; Wang, Y.; Zhao, S.; Asad, Z.; Coburn, L.A.; Wilson, K.T.; Landman, B.A.; Huo, Y. Deep multimodal fusion of image and non-image data in disease diagnosis and prognosis: A review. Prog. Biomed. Eng. 2023, 5, 022001. [Google Scholar] [CrossRef]
- He, H.; Chen, G.; Tang, Z.; Chen, C.Y.-C. Dual modality feature fused neural network integrating binding site information for drug target affinity prediction. NPJ Digit. Med. 2025, 8, 67. [Google Scholar] [CrossRef]
- Kavipriya, G.; Manjula, D. Drug–Target Interaction Prediction Model Using Optimal Recurrent Neural Network. Intell. Autom. Soft Comput. 2023, 35, 1677. [Google Scholar] [CrossRef]
- Verma, S.; Magazzù, G.; Eftekhari, N.; Lou, T.; Gilhespy, A.; Occhipinti, A.; Angione, C. Cross-attention enables deep learning on limited omics-imaging-clinical data of 130 lung cancer patients. Cell Rep. Methods 2024, 4, 100817. [Google Scholar] [CrossRef] [PubMed]
- Homer, N.; Szelinger, S.; Redman, M.; Duggan, D.; Tembe, W.; Muehling, J.; Pearson, J.V.; Stephan, D.A.; Nelson, S.F.; Craig, D.W. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genet. 2008, 4, e1000167. [Google Scholar] [CrossRef] [PubMed]
- Fung, B.C.M.; Wang, K.; Chen, R.; Yu, P.S. Privacy-preserving data publishing: A survey of recent developments. ACM Comput. Surv. Csur 2010, 42, 1–53. [Google Scholar] [CrossRef]
- Zeng, Y.; Wei, Z.; Yu, W.; Yin, R.; Yuan, Y.; Li, B.; Tang, Z.; Lu, Y.; Yang, Y. Spatial transcriptomics prediction from histology jointly through transformer and graph neural networks. Brief. Bioinform. 2022, 23, bbac297. [Google Scholar] [CrossRef]
- Avsec, Ž.; Agarwal, V.; Visentin, D.; Ledsam, J.R.; Grabska-Barwinska, A.; Taylor, K.R.; Assael, Y.; Jumper, J.; Kohli, P.; Kelley, D.R. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 2021, 18, 1196–1203. [Google Scholar] [CrossRef]
- Klil-Drori, S.; Bodenstein, K.C.; Sun, S.; Kojok, L.; Gruber, J.; Ghantous, Y.; Cummings, J.; Nasreddine, Z. Montreal Cognitive Assessment (MoCA) XpressO: Validation of a digital self-administered cognitive prescreening tool. J. Am. Geriatr. Soc. 2024, 72, 2516–2522. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Pang, M.; Su, K.; Li, M. Leveraging information in spatial transcriptomics to predict super-resolution gene expression from histology images in tumors. bioRxiv 2021. bioRxiv:2021.11.28.470212.. [Google Scholar]
- Mutso, M.; Nikonov, A.; Pihlak, A.; Žusinaite, E.; Viru, L.; Selyutina, A.; Reintamm, T.; Kelve, M.; Saarma, M.; Karelson, M. RNA interference-guided targeting of hepatitis C virus replication with antisense locked nucleic acid-based oligonucleotides containing 8-oxo-dG modifications. PLoS ONE 2015, 10, e0128686. [Google Scholar] [CrossRef]
- Wang, C.; Chan, A.S.; Fu, X.; Ghazanfar, S.; Kim, J.; Patrick, E.; Yang, J.Y.H.; Yang, Y.; Wang, Y.; Wang, L. Benchmarking the translational potential of spatial gene expression prediction from histology. Nat. Commun. 2025, 16, 1544. [Google Scholar] [CrossRef]
- Xiao, X.; Kong, Y.; Li, R.; Wang, Y.; Wang, X.; Zhang, J.; Yang, W.; Huang, J.; Han, X.; Zhang, M. Transformer with convolution and graph-node co-embedding: An accurate and interpretable vision backbone for predicting gene expressions from local histopathological image. Med. Image Anal. 2024, 91, 103040. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Liu, J.; Chen, L.; Zhao, T.; Wang, Y. THItoGene: A deep learning method for predicting spatial transcriptomics from histological images. Brief. Bioinform. 2024, 25, bbad464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Schroeder, A.; Yan, H.; Yang, H.; Hu, J.; Lee, M.Y.Y.; Cho, K.S.; Susztak, K.; Xu, G.X.; Feldman, M.D. Inferring super-resolution tissue architecture by integrating spatial transcriptomics with histology. Nat. Biotechnol. 2024, 42, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.M.; Millar, E.K.A.; Meijering, E. Breast cancer histopathology image-based gene expression prediction using spatial transcriptomics data and deep learning. Sci. Rep. 2023, 13, 13604. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type | Omics Data | Natural Language Data | Medical Imaging Data | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biological Dimension | Molecular level: gene variation, epigenetics, RNA expression, protein interactions, metabolism | Clinical and knowledge-based: phenotypes, treatments, mechanisms, clinical notes | Structural and functional: organ morphology, tissue context, physiology, pathology | |||||||||
| Specific Type | Genomics | Epigenomics | Transcriptomics | Proteomics | Metabolomics | Scientific research literature | Medical records | Clinical reports | CT | MRI | PET | H&E |
| Data Resource | NCBI GeneBank | MethBank | GEO | Human Protein Atlas (HPA) | MetaboLights | PubMed | ClinicalTrials.gov | MIMIC-III | LIDC-IDRI | TCIA | Human Connectome Project (HCP) | TCGA |
| UCSC Genome Browser | ENCODE (Encyclopedia of DNA Elements) | HCA (Human Cell Atlas) | PRIDE (Proteomics Identifications Database) | HMDB (Human Metabolome Database) | Google Scholar | / | MIMIC-IV | TCIA (The Cancer Imaging Archive) | / | / | CDSA (Cancer Slide Digital Archive) | |
| Ensembl Genome Browser | CNGB | GTEx | / | / | / | / | PhysioNet | / | / | / | GTEx Histology | |
| DDBJ | / | SpatialDB | / | / | / | / | / | / | / | / | / | |
| CNGB | / | TCGA | / | / | / | / | / | / | / | / | / | |
| Algorithms | Specific Technology | Advantages in Predicting Gene Expression | Current Applications in Clinical Pathology | Training Requirements for Medical Staff |
|---|---|---|---|---|
| Machine Learning Algorithms | SVM [56] | Proficient in handling high-dimensional small-sample data | Diagnosing cancer subtypes via gene expression analysis, aiding personalized treatment | Basic ML and data pre-processing, short-term online training suffices |
| RF [57] | Strong ability to resist overfitting | Predicting cancer prognosis using clinical and genetic data | Feature selection and model evaluation knowledge, medium-level workshop training needed | |
| GBM [58] | Suitable for weak features in gene expression progressive learning | Predicting cancer treatment response | Gradient descent and parameter tuning, systematic courses and practice required | |
| Deep Learning Algorithms | GNN [60] | Expert in relational modeling and analyzing biological networks | Predicting gene activity in rare tissues, understanding tumor microenvironments | Graph theory and GNN principles, specialized courses and hands-on training |
| CNN [61] | Expert in extracting local sequence features | Analyzing histopathology for cancer subtype prediction and gene methylation for early detection | Image processing and CNN principles, medium-to-extensive case-based training | |
| Transformer [62] | Self-attention-based multimodal integration and long-range dependency modeling | Predicting gene expression across unseen cell types, integrating multi-omics data for diagnosis | Deep learning basics, Transformer architecture, long-term specialized training |
| Model Name | Model Type | Application | Data Type | Advantages | Limitations |
|---|---|---|---|---|---|
| GET [20] | Transformer | Cross-cell-type gene prediction | Gene expression, chromatin accessibility, sequence data | Zero-shot prediction, multimodal integration | Data privacy and interpretability challenges |
| HGGEP [118] | GEM + HAM | Disease mechanism research, pathological image–gene correlation | Histological images | Visual association of tissue morphology and molecular mechanisms | Requires high-quality images, small-sample limitation |
| DeepSTARR [75] | CNN | Enhancer activity prediction | DNA sequences, STARR-seq data | Direct DNA sequence-to-enhancer activity prediction | Reliance on STARR-seq experimental data |
| HyenaDNA [119] | Hyena Operator-based Model | Long-range genomic sequence modeling, regulatory signal capture | DNA sequences, RNA secondary structure data | Single-nucleotide resolution, distal regulatory capture | High computational power requirement |
| DeepPT [17] | Transformer | Cancer treatment response prediction, transcriptomics simulation | H&E histopathology images, transcriptomics data | Non-invasive gene expression prediction from pathology images | Requires large, matched image–transcriptomics datasets |
| GemGAT [77] | Graph Attention Network (GAT) | Rare tissue gene expression prediction | Gene expression, spatial co-expression networks | Cross-tissue generalization, small-sample robustness | High data quality dependency, model stability issues |
| Gemini Ultra [24] | Multimodal Fusion Model | Multimodal cancer classification | Gene expression data, H&E pathology images | Cross-modal association for diagnostic reliability | High edge deployment cost, limited rare cancer subtype validation |
| Basenji2 [120] | CNN | Local regulatory sequence analysis | Genome sequences, chromatin states | Efficient promoter-proximal feature processing | Ignores distal regulatory elements |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Xu, H.; Yu, S.; Hu, W.; Zhang, Z.; Wang, X.; Yuan, Y.; Wang, M.; Chen, L.; Lin, X.; et al. AI-Driven Transcriptome Prediction in Human Pathology: From Molecular Insights to Clinical Applications. Biology 2025, 14, 651. https://doi.org/10.3390/biology14060651
Chen X, Xu H, Yu S, Hu W, Zhang Z, Wang X, Yuan Y, Wang M, Chen L, Lin X, et al. AI-Driven Transcriptome Prediction in Human Pathology: From Molecular Insights to Clinical Applications. Biology. 2025; 14(6):651. https://doi.org/10.3390/biology14060651
Chicago/Turabian StyleChen, Xiaoya, Huinan Xu, Shengjie Yu, Wan Hu, Zhongjin Zhang, Xue Wang, Yue Yuan, Mingyue Wang, Liang Chen, Xiumei Lin, and et al. 2025. "AI-Driven Transcriptome Prediction in Human Pathology: From Molecular Insights to Clinical Applications" Biology 14, no. 6: 651. https://doi.org/10.3390/biology14060651
APA StyleChen, X., Xu, H., Yu, S., Hu, W., Zhang, Z., Wang, X., Yuan, Y., Wang, M., Chen, L., Lin, X., Hu, Y., & Cai, P. (2025). AI-Driven Transcriptome Prediction in Human Pathology: From Molecular Insights to Clinical Applications. Biology, 14(6), 651. https://doi.org/10.3390/biology14060651