
FPCAM: A Weighted Dictionary-Driven Model for Single-Cell Annotation in Pulmonary Fibrosis

 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Data Preprocessing and Quality Control
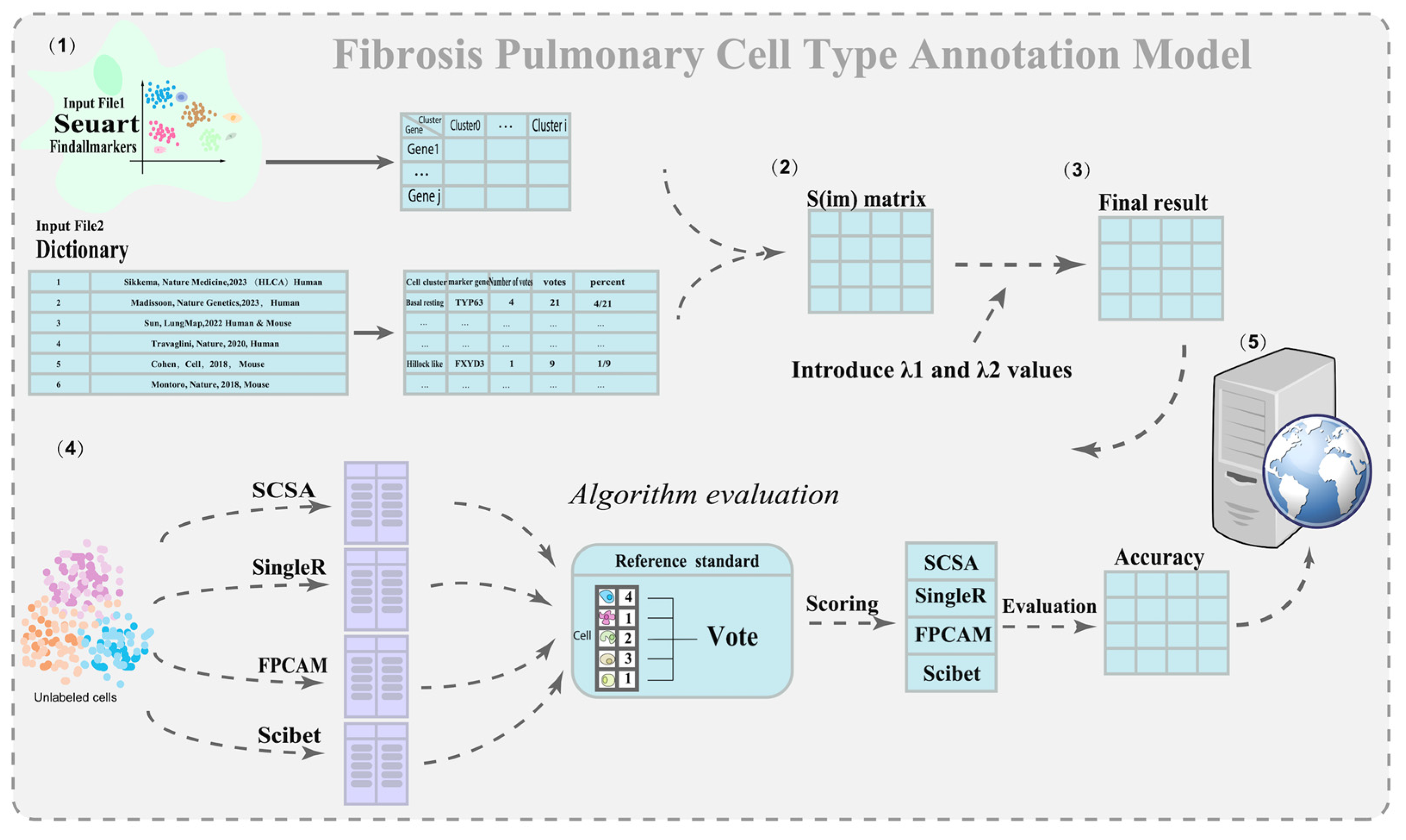
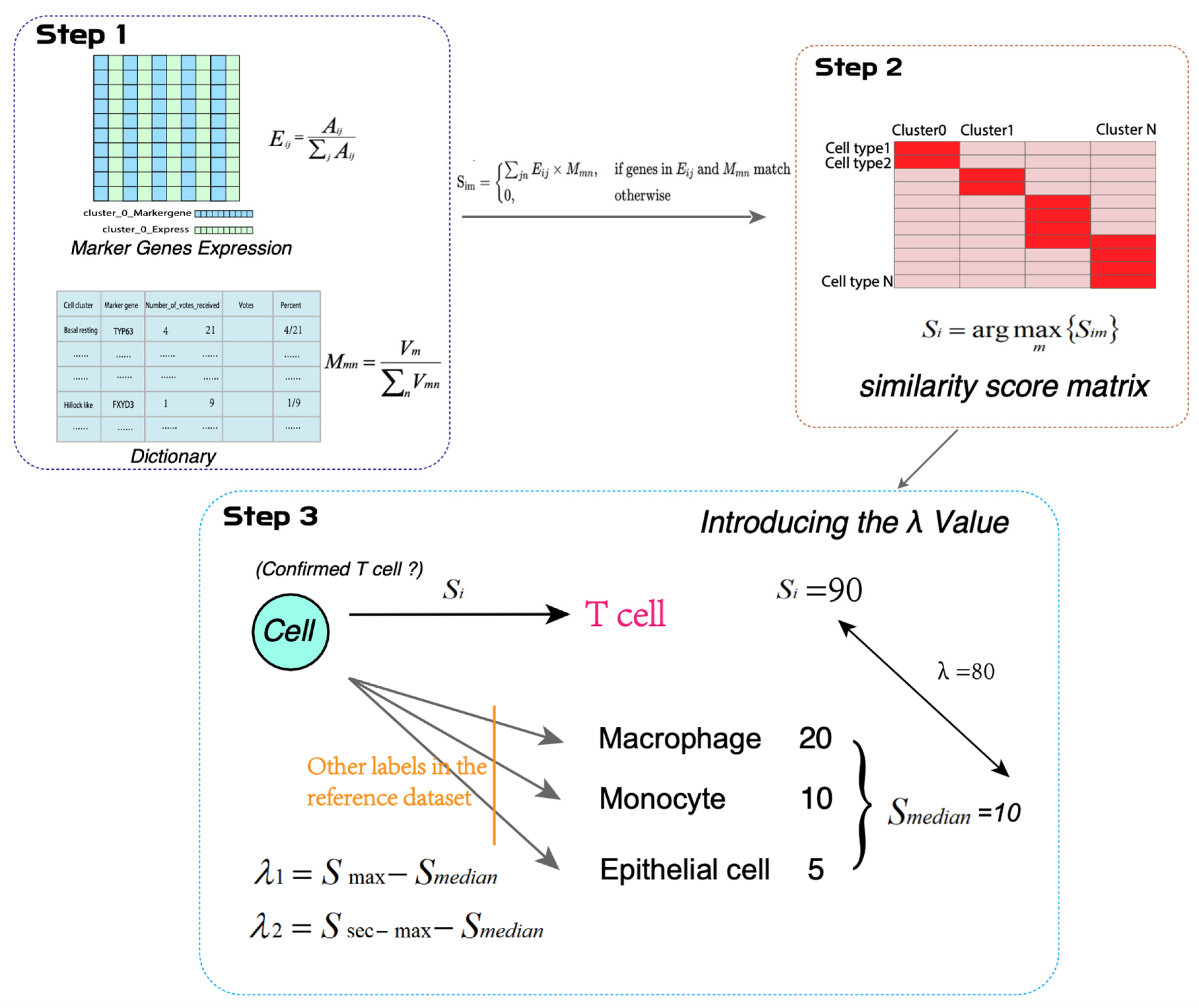
2.3. Construction of the Pulmonary Fibrosis Annotation Dictionary
2.4. Construction of the Marker Gene Expression Matrix
2.5. Construction of the Similarity Score Matrix
2.6. Model Evaluation and Comparison
2.7. Online Data Analysis Platform and Validation Dataset
3. Results
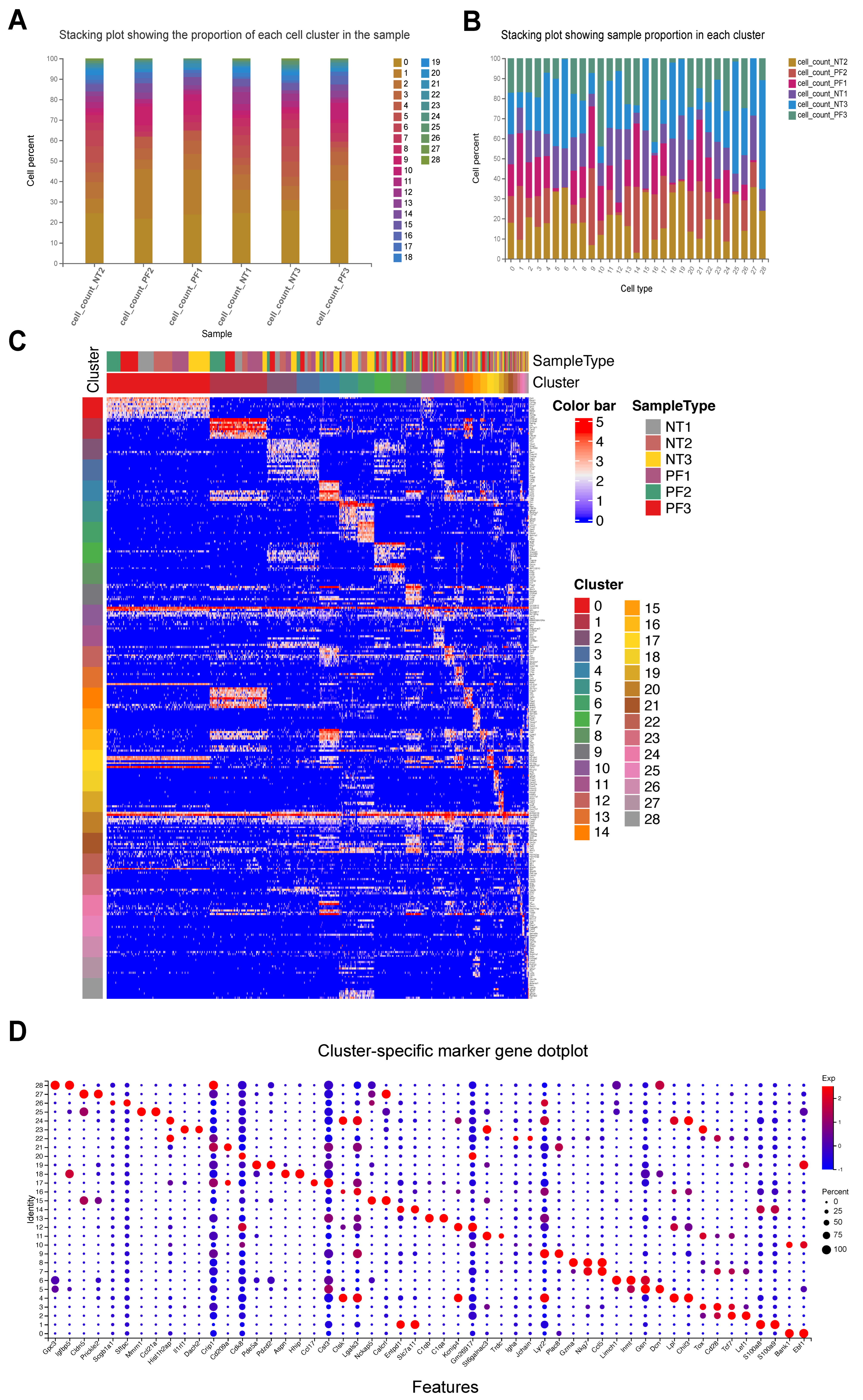
3.1. Quantitative Statistics of Sequencing Results
3.2. FPCAM Workflow and Construction of a Pulmonary Fibrosis-Specific Cell–Gene Dictionary
3.3. Similarity Score Calculation Between Known Cells and Unknown Cell Clusters
3.4. Results of Model Evaluation and Comparison
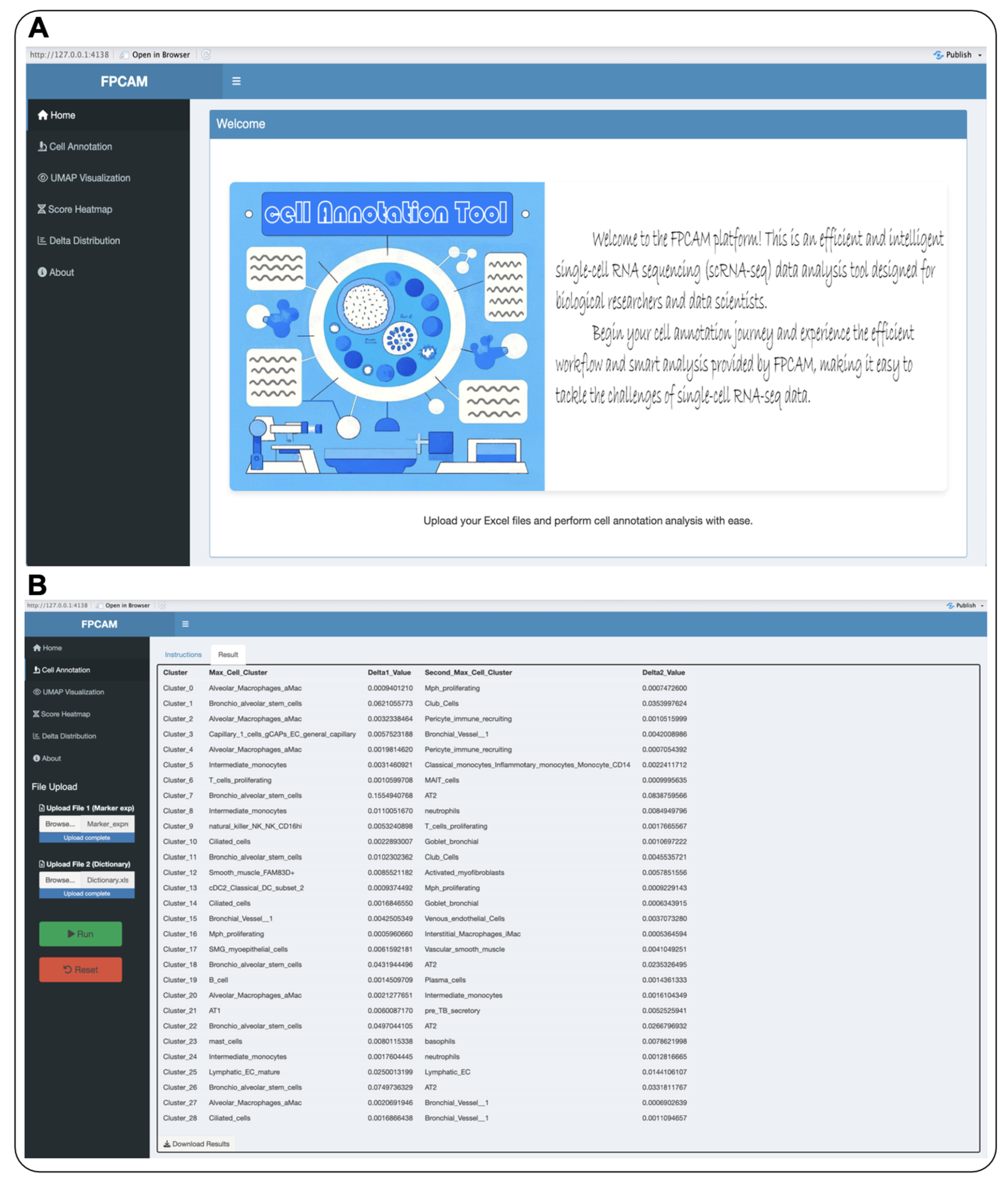
3.5. Construction of the Online Data Analysis Platform FPCAM
3.6. Visualization and Interactive Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhang, S.; Li, X.; Lin, J.; Lin, Q.; Wong, K.-C. Review of single-cell RNA-seq data clustering for cell-type identification and characterization. RNA 2023, 29, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Li, G.; Huang, T.; Geng, W.; Pei, H.; Yang, J.; Zhu, M.; Zhang, P.; Hou, R.; Tian, G.; et al. Single-cell technologies: From research to application. Innovation 2022, 3, 100342. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Huynh, T.; Cang, Z. Topological and geometric analysis of cell states in single-cell transcriptomic data. Brief. Bioinform. 2024, 25, bbae176. [Google Scholar] [CrossRef]
- Sikkema, L.; Ramírez-Suástegui, C.; Strobl, D.C.; Gillett, T.E.; Zappia, L.; Madissoon, E.; Markov, N.S.; Zaragosi, L.-E.; Ji, Y.; Ansari, M.; et al. An integrated cell atlas of the lung in health and disease. Nat. Med. 2023, 29, 1563–1577. [Google Scholar] [CrossRef]
- Madissoon, E.; Oliver, A.J.; Kleshchevnikov, V.; Wilbrey-Clark, A.; Polanski, K.; Richoz, N.; Orsi, A.R.; Mamanova, L.; Bolt, L.; Elmentaite, R.; et al. A spatially resolved atlas of the human lung characterizes a gland-associated immune niche. Nat. Genet. 2023, 55, 66–77. [Google Scholar] [CrossRef]
- Sun, X.; Perl, A.K.; Li, R.; Bell, S.M.; Sajti, E.; Kalinichenko, V.V.; Kalin, T.V.; Misra, R.S.; Deshmukh, H.; Clair, G.; et al. A census of the lung: CellCards from LungMAP. Dev. Cell 2022, 57, 112–145.e2. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. Lung single-cell signaling interaction map reveals basophil role in macrophage imprinting. Cell 2018, 175, 1031–1044.e18. [Google Scholar] [CrossRef]
- Zhang, A.W.; O’Flanagan, C.; Chavez, E.A.; Lim, J.L.P.; Ceglia, N.; McPherson, A.; Wiens, M.; Walters, P.; Chan, T.; Hewitson, B.; et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 2019, 16, 1007–1015. [Google Scholar] [CrossRef]
- Zhang, Z.; Luo, D.; Zhong, X.; Choi, J.H.; Ma, Y.; Wang, S.; Mahrt, E.; Guo, W.; Stawiski, E.W.; Modrusan, Z.; et al. SCINA: A semi-supervised subtyping algorithm of single cells and bulk samples. Genes 2019, 10, 531. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. scmap: Projection of single-cell RNA-seq data across data sets. Nat. Methods 2018, 15, 359–362. [Google Scholar] [CrossRef]
- Tan, Y.; Cahan, P. SingleCellNet: A computational tool to classify single cell RNA-seq data across platforms and across species. Cell Syst. 2019, 9, 207–213.e2. [Google Scholar] [CrossRef]
- Alquicira-Hernandez, J.; Sathe, A.; Ji, H.P.; Nguyen, Q.; Powell, J.E. scPred: Accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019, 20, 264. [Google Scholar] [CrossRef] [PubMed]
- Clarke, Z.A.; Andrews, T.S.; Atif, J.; Pouyabahar, D.; Innes, B.T.; MacParland, S.A.; Bader, G.D. Tutorial: Guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc. 2021, 16, 2749–2764. [Google Scholar] [CrossRef]
- Abdelaal, T.; Michielsen, L.; Cats, D.; Hoogduin, D.; Mei, H.; Reinders, M.J.T.; Mahfouz, A. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 2019, 20, 194. [Google Scholar] [CrossRef]
- Domínguez Conde, C.; Xu, C.; Jarvis, L.B.; Rainbow, D.B.; Wells, S.B.; Gomes, T.; Howlett, S.K.; Suchanek, O.; Polanski, K.; King, H.W.; et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 2022, 376, eabl5197. [Google Scholar] [CrossRef]
- Ma, F.; Pellegrini, M. ACTINN: Automated identification of cell types in single cell RNA sequencing. Bioinformatics 2020, 36, 533–538. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Peng, G. SCSA: A cell type annotation tool for single-cell RNA-seq data. Front. Genet. 2020, 11, 490. [Google Scholar] [CrossRef]
- Li, C.; Liu, B.; Kang, B.; Liu, Z.; Liu, Y.; Chen, C.; Ren, X.; Zhang, Z. SciBet as a portable and fast single cell type identifier. Nat. Commun. 2020, 11, 1818. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Abrams, Z.B.; Johnson, T.S.; Huang, K.; Payne, P.R.O.; Coombes, K. A protocol to evaluate RNA sequencing normalization methods. BMC Bioinform. 2019, 20, 679. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Jia, L.; Yao, W.; Jiang, Y.; Li, Y.; Wang, Z.; Li, H.; Huang, F.; Li, J.; Chen, T.; Zhang, H. Development of interactive biological web applications with R/shiny. Brief. Bioinform. 2022, 23, bbab415. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Tsukui, T.; Sun, K.H.; Wetter, J.B.; Wilson-Kanamori, J.R.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.D.; Rosas, I.O.; et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 2020, 11, 1920. [Google Scholar] [CrossRef] [PubMed]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Franzén, L.; Olsson Lindvall, M.; Hühn, M.; Ptasinski, V.; Setyo, L.; Keith, B.P.; Collin, A.; Oag, S.; Volckaert, T.; Borde, A.; et al. Mapping spatially resolved transcriptomes in human and mouse pulmonary fibrosis. Nat. Genet. 2024, 56, 1725–1736. [Google Scholar] [CrossRef]
- Xie, T.; Kulur, V.; Liu, N.; Deng, N.; Wang, Y.; Rowan, S.C.; Yao, C.; Huang, G.; Liu, X.; Taghavifar, F.; et al. Mesenchymal growth hormone receptor deficiency leads to failure of alveolar progenitor cell function and severe pulmonary fibrosis. Sci. Adv. 2021, 7, 6005. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Wei, H.; Chen, B.; Wu, L.; Song, J.; Zhang, Q.; Wu, X.; Liang, G.; Chen, W.; Wang, Y.; et al. Inhalation of itraconazole mitigates bleomycin-induced lung fibrosis via regulating SPP1 and C3 signaling pathway pivotal in the interaction between phagocytic macrophages and diseased fibroblasts. J. Transl. Med. 2024, 22, 1058. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, 1972. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care. Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, 1983. [Google Scholar] [CrossRef]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI. Insight. 2016, 1, 90558. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- King, C.S.; Mannem, H.; Kukreja, J.; Aryal, S.; Tang, D.; Singer, J.P.; Bharat, A.; Behr, J.; Nathan, S.D. Lung transplantation for patients with COVID-19. Chest 2022, 161, 169–178. [Google Scholar] [CrossRef]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary fibrosis in COVID-19 survivors: Predictive factors and risk reduction strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, H.; Lu, T.; Li, T.; Wang, Y.; Fan, X.; Jiang, Y.; Cai, M.; He, P.; Liu, J. Progress in understanding the role and therapeutic targets of polarized subtypes of macrophages in pulmonary fibrosis. Cell Biochem. Biophys. 2023, 81, 673–682. [Google Scholar] [CrossRef]
- Sun, J.; Jin, T.; Niu, Z.; Guo, J.; Guo, Y.; Yang, R.; Wang, Q.; Gao, H.; Zhang, Y.; Li, T.; et al. LncRNA DACH1 protects against pulmonary fibrosis by binding to SRSF1 to suppress CTNNB1 accumulation. Acta Pharm. Sin. B 2022, 12, 3602–3617. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Tsao, D.; Bai, K.; Tsao, M.; Xing, L.; Zhang, X. scAnnotate: An automated cell-type annotation tool for single-cell RNA-sequencing data. Bioinform. Adv. 2023, 3, vbad030. [Google Scholar] [CrossRef]
- Shao, X.; Liao, J.; Lu, X.; Xue, R.; Ai, N.; Fan, X. scCATCH: Automatic annotation on cell types of clusters from single-cell RNA sequencing data. iScience 2020, 23, 100882. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, P.; Alamo, I.; Nepomuceno-Chamorro, I.A.; Dopazo, J.; Loucera, C. SigPrimedNet: A Signaling-Informed Neural Network for scRNA-seq Annotation of Known and Unknown Cell Types. Biology 2023, 12, 579. [Google Scholar] [CrossRef]
- Xu, C.; Jackson, S.A. complex and complex biological data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Cell Identities | Citations | PMID | References |
|---|---|---|---|---|
| Human | 61 | 380 | PMID: 37291214 | Sikkema et al., Nature Medicine, 2023 [5] |
| Human | 80 | 123 | PMID: 36543915 | Madissoon et al., Nature Genetics, 2023 [6] |
| Human and Mouse | 27 | 121 | PMID: 34936882 | Sun et al., Developmental Cell, 2022 [7] |
| Human | 58 | 1416 | PMID: 33208946 | Travaglini et al., Nature, 2020 [8] |
| Mouse | 22 | 387 | PMID: 30318149 | Cohen et al., Cell, 2018 [9] |
| Mouse | 7 | 1206 | PMID: 30069044 | Montoro et al., Nature, 2018 [30] |
| Cell Cluster | Marker Gene | Number of Votes Received | Votes | Percent |
|---|---|---|---|---|
| Basal resting | TP63 | 4 | 21 | 4/21 |
| Basal resting | KRT5 | 4 | 21 | 4/21 |
| Basal resting | KRT15 | 3 | 21 | 1/7 |
| Basal resting | FXYD3 | 2 | 21 | 2/21 |
| Basal resting | KRT14 | 2 | 21 | 2/21 |
| Basal resting | KRT17 | 2 | 21 | 2/21 |
| Basal resting | EPCAM | 1 | 21 | 1/21 |
| Basal resting | ELF3 | 1 | 21 | 1/21 |
| Basal resting | IGFBP2 | 1 | 21 | 1/21 |
| Basal resting | SERPINF1 | 1 | 21 | 1/21 |
| Cells/Clusters | Cluster 0 | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 |
|---|---|---|---|---|---|---|
| AT1 | 0 | 0 | 0 | 5.89 × 10−5 | 1.80 × 10−5 | 0 |
| AT2 | 0 | 0 | 0 | 3.34 × 10−5 | 2.04 × 10−5 | 0 |
| AT2_proliferating | 0 | 0 | 0 | 5.56 × 10−5 | 3.40 × 10−5 | 0 |
| Activated_myofibroblasts | 0 | 0 | 1.34 × 10−5 | 0 | 0 | 0.002383 |
| Airway_smooth_muscle | 0 | 0 | 0 | 0 | 4.63 × 10−5 | 7.79 × 10−6 |
| Alveolar_Macrophages_aMac | 0 | 0.000543 | 0.00017 | 0.000272 | 0.000532 | 6.92 × 10−6 |
| Alveolar_fibroblasts_1_Lipofibroblasts | 0 | 0 | 0 | 0 | 0 | 0.002304 |
| Alveolar_fibroblasts_2_Matrix_fibroblast_2_Adventitial_fibroblasts | 0 | 3.15 × 10−5 | 0 | 0 | 0 | 0.002601 |
| Arterial_endothelial_Cells | 0 | 1.28 × 10−5 | 0 | 0 | 0 | 1.91 × 10−5 |
| B_cell | 0.00119 | 0.000356 | 0.00016 | 0.000256 | 0 | 0 |
| Model | Reference File | Unannotated | Cell Subtype | Accuracy |
|---|---|---|---|---|
| SCSA | CellMarker | No | No | 0.897 |
| SCSA | PanglaoDB | No | No | 0.897 |
| SingleR | ImmGendate | No | No | 0.724 |
| SingleR | MouseRNAseqData | Yes | Yes | 0.483 |
| SciBet | Tabula Muris [20 mouse organs] | No | Yes | 0.759 |
| SciBet | Tabula Muris [Lung Smart seq2] | No | No | 0.517 |
| FPCAM | FP Dictionary | No | Yes | 0.897 |
| Dataset | Samples | Citations | PMID | References |
|---|---|---|---|---|
| GSE132771 | 24 | 551 | PMID: 32317643 | Tsukui, Tatsuya et al. Nature Communications. 2020 [31] |
| GSE141259 | 60 | 556 | PMID: 32678092 | Strunz, Maximilian et al. Nature Communications. 2020 [32] |
| GSE267904 | 24 | 21 | PMID: 38951642 | Franzén, Lovisa et al. Nature Genetics. 2024 [33] |
| GSE104154 | 6 | 15 | PMID: 34108218 | Xie, Ting et al. Science Advances. 2021 [34] |
| GSE267861 | 2 | 0 | PMID: 39587675 | Pan, Caizhe et al. Journal of Translational Medicine. 2024 [35] |
| GSE135893 | 34 | 873 | PMID: 32832598 | Habermann, Arun C et al. Science Advances. 2020 [36] |
| GSE122960 | 17 | 1177 | PMID: 30554520 | Reyfman, Paul A et al. American Journal of Respiratory and Critical Care Medicine. 2019 [37] |
| GSE136831 | 78 | 1108 | PMID: 32832599 | Adams, Taylor S et al. Science Advances. 2020 [38] |
| GSE128033 | 18 | 619 | PMID: 31221805 | Morse, Christina et al. The European Respiratory Journal. 2019 [39] |
| GSE86618 | 540 | 8 | PMID: 27942595 | Xu, Yan et al. JCI Insight. 2016 [40] |
| GSE94555 | 6 | 8 | PMID: 27942595 | Xu, Yan et al. JCI Insight. 2016 [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Shi, Y.; Huang, H.; Xiao, N.; Liu, C.; Zhao, H.; Xing, Y.; Cai, L. FPCAM: A Weighted Dictionary-Driven Model for Single-Cell Annotation in Pulmonary Fibrosis. Biology 2025, 14, 479. https://doi.org/10.3390/biology14050479
Liu G, Shi Y, Huang H, Xiao N, Liu C, Zhao H, Xing Y, Cai L. FPCAM: A Weighted Dictionary-Driven Model for Single-Cell Annotation in Pulmonary Fibrosis. Biology. 2025; 14(5):479. https://doi.org/10.3390/biology14050479
Chicago/Turabian StyleLiu, Guojun, Yan Shi, Hongxu Huang, Ningkun Xiao, Chuncheng Liu, Hongyu Zhao, Yongqiang Xing, and Lu Cai. 2025. "FPCAM: A Weighted Dictionary-Driven Model for Single-Cell Annotation in Pulmonary Fibrosis" Biology 14, no. 5: 479. https://doi.org/10.3390/biology14050479
APA StyleLiu, G., Shi, Y., Huang, H., Xiao, N., Liu, C., Zhao, H., Xing, Y., & Cai, L. (2025). FPCAM: A Weighted Dictionary-Driven Model for Single-Cell Annotation in Pulmonary Fibrosis. Biology, 14(5), 479. https://doi.org/10.3390/biology14050479