
Beclin 1-Mediated Autophagy Is Potentiated by an Interaction with the Neuronal Adaptor FE65


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Transfection
2.2. Plasmids
2.3. FE65, FE65ΔCt, and GFP-LC3 Stably Transfected Cell Line
2.4. FE65 Knock out (KO) Cell Line
2.5. FE65 Knockdown
2.6. GFP-LC3 Cleavage and Puncta Formation Assay
2.7. LC3 Conversion and p62 Turnover Assays
2.8. Proximity Ligation Assay (PLA)
2.9. PI3KC3 Kinase Activity Assay
2.10. Protein–Protein Interaction Assays
2.11. Immunostaining
2.12. Statistical Analysis
3. Results
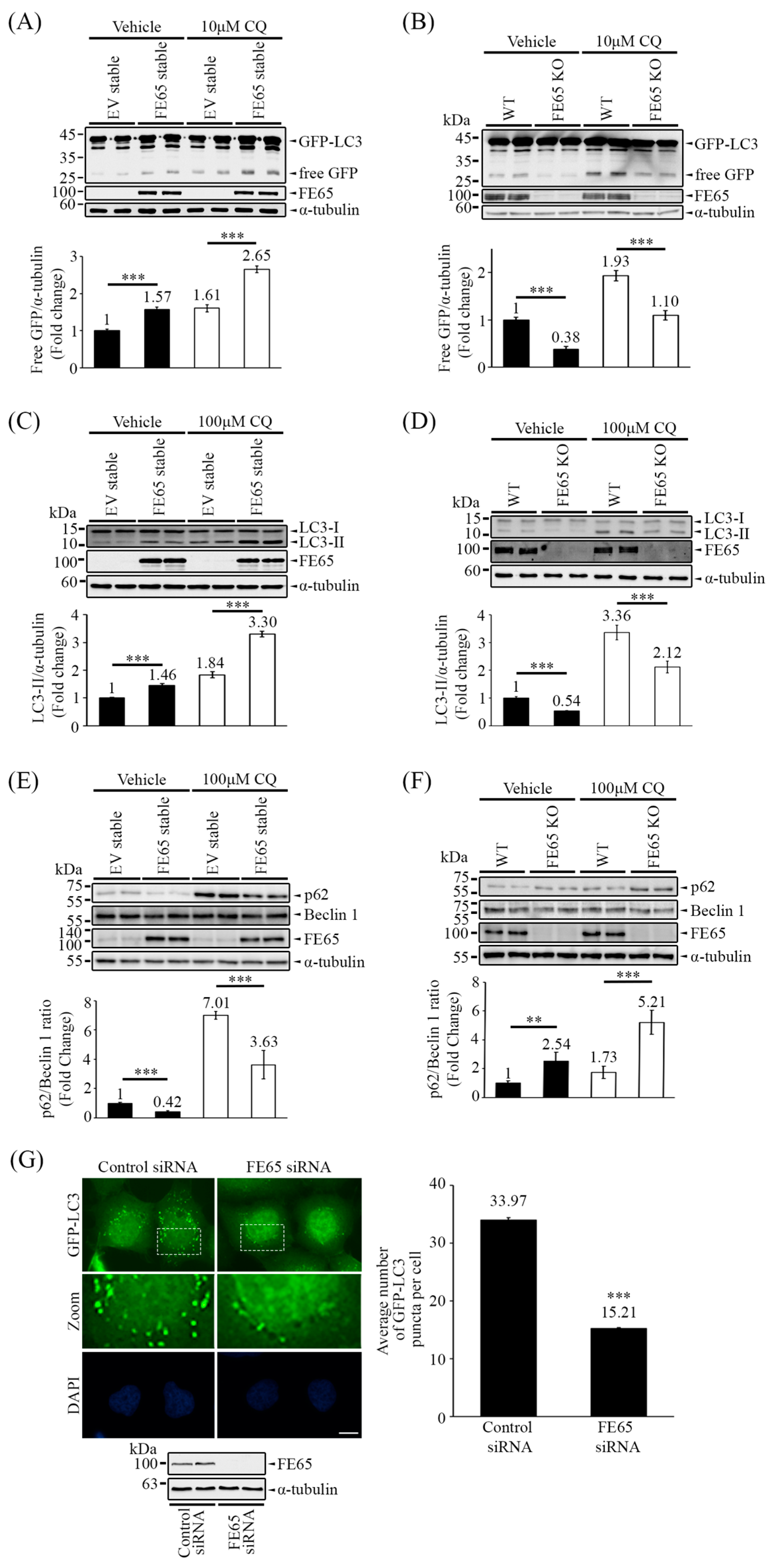
3.1. FE65 Positively Regulates Autophagy
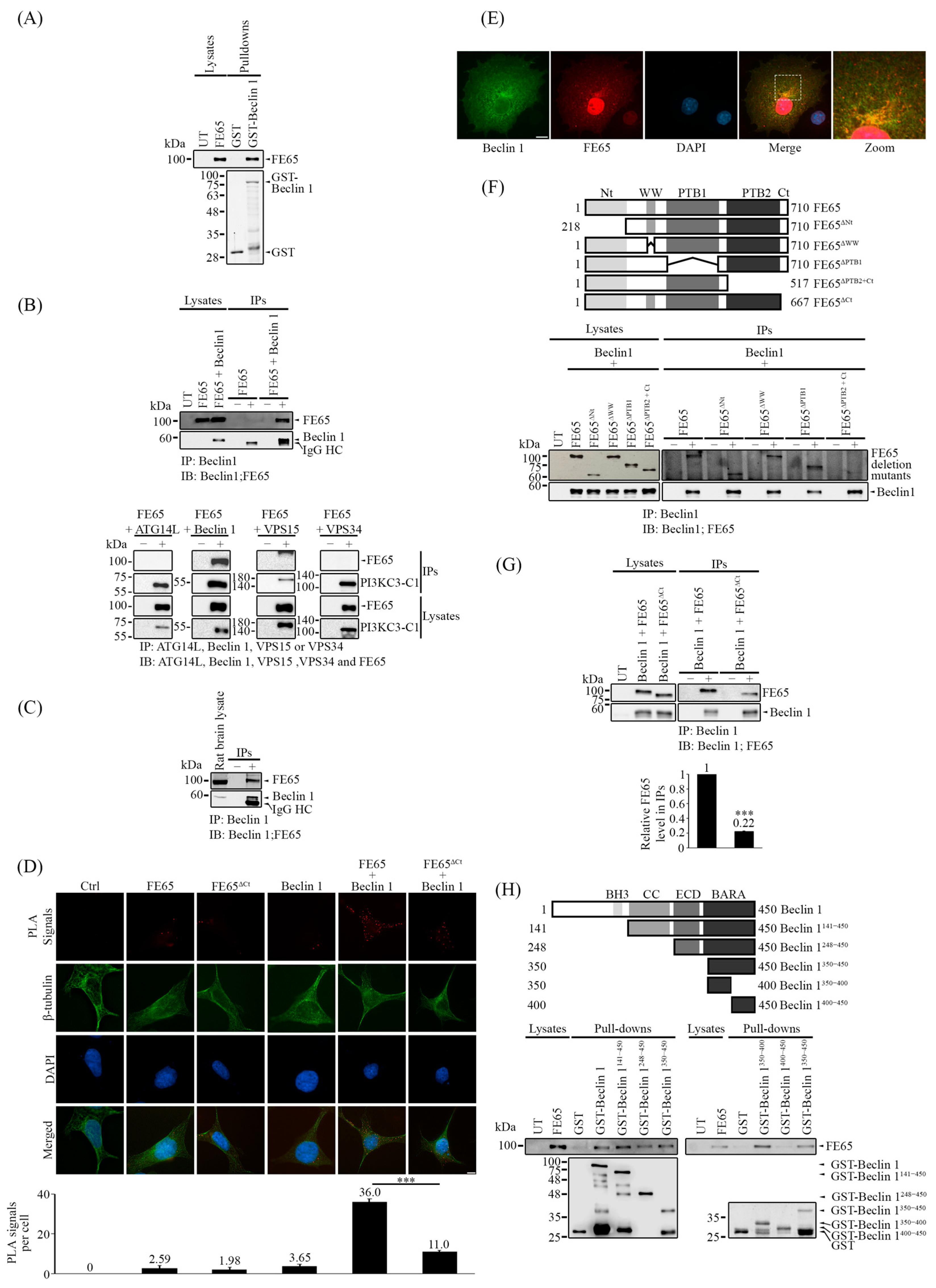
3.2. FE65 Is a Novel Interactor to Beclin 1
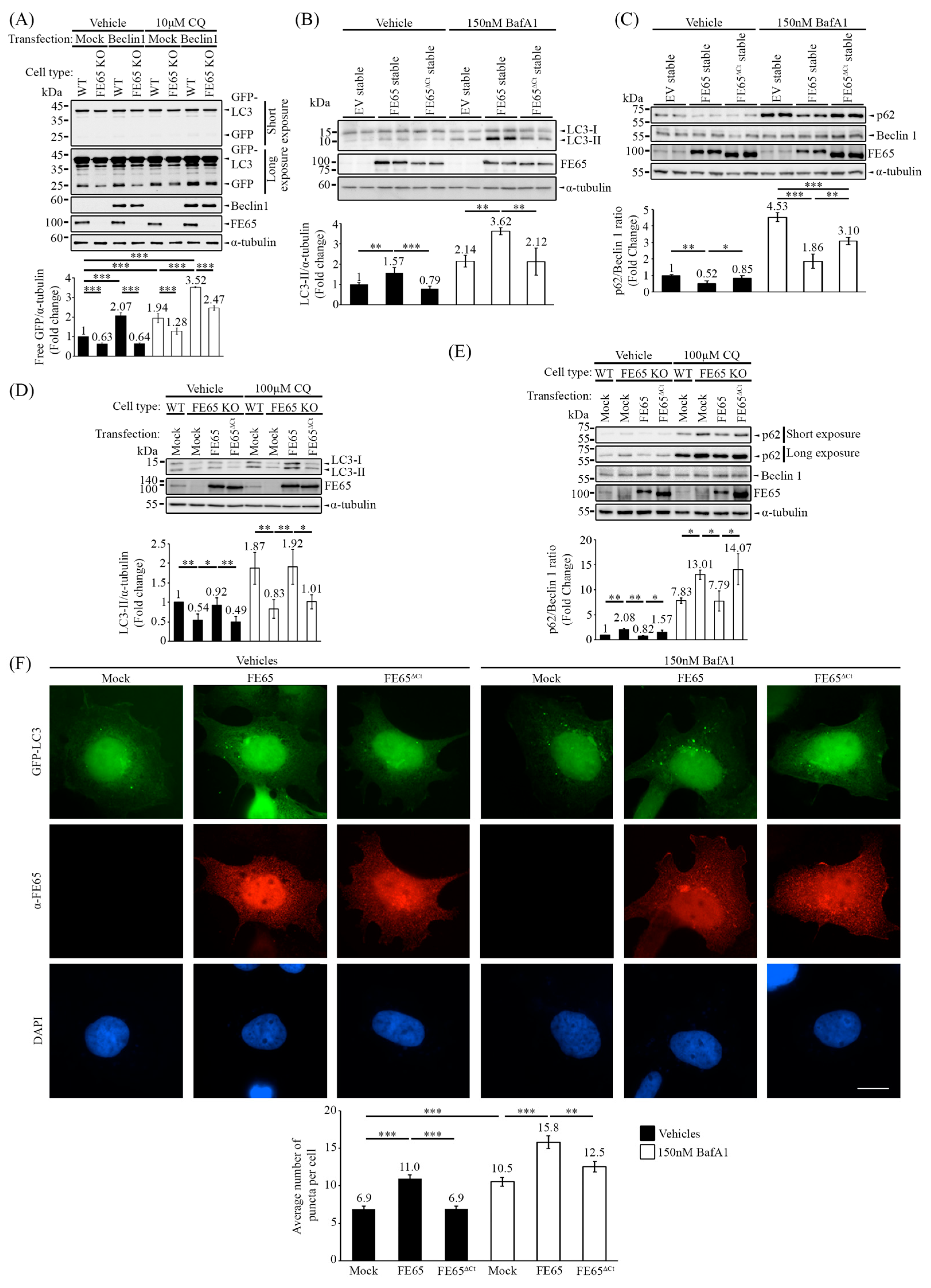
3.3. The Loss of Beclin 1 Interaction with the FE65 Mutant Has Attenuated Beclin 1-Mediated Autophagy
3.4. The Interaction of FE65-Beclin 1 Upregulates PI3KC3 Complex I Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AICD | Amyloid Precursor Protein Intracellular Domain |
| AMPK | AMP-Activated Protein Kinase |
| APP | Amyloid Precursor Protein |
| ARF6 | ADP-Ribosylation Factor 6 |
| ARNO | Arf Nucleotide Binding Site Opener |
| ATG14L | Autophagy-Related Gene 14 |
| Baf A1 | Bafilomycin A1 |
| BARA domain | β-α Autophagy-Specific Domain |
| BATS | Barkor/Atg14L Autophagosome-Targeting Sequence |
| Bcl-2 | B-Cell Leukemia/Lymphoma 2 Protein |
| c-Abl | Abelson Tyrosine Kinase |
| c-FLIP | Cellular Flice-Like Inhibitory Protein |
| CaMKII | Calcium/Calmodulin-Dependent Protein Kinase II |
| Chk1 | Checkpoint Kinase 1 |
| CQ | Chloroquine |
| EBSS | Earle’s Balanced Salt Solution |
| ECD | Evolutionarily Conserved Domain |
| ELMO1 | Engulfment and Cell Motility Protein 1 |
| ER | Endoplasmic Reticulum |
| GEF | Guanine Nucleotide Exchange Factor |
| GST | Glutathione S-Transferase |
| HEK293 | Human Embryonic Kidney 293 |
| LC3 | Microtubule-Associated Protein 1A/1B-Light Chain 3 |
| MENA | Mammalian-Enabled Protein |
| Mst1 | Mammalian Ste20-Like Kinase 1 |
| mTORC1 | Mammalian Target of Rapamycin Complex 1 |
| PE | Phosphatidylethanolamine |
| PI3KC3-C1 | Class III Phosphatidylinositol 3-Kinase Complex 1 |
| PI3P | Phosphatidylinositol 3-Phosphate |
| PLA | Proximity Ligation Assay |
| PTB domain | Phosphotyrosine-Binding Domain |
| Rac1 | Ras-Related C3 Botulinum Toxin Substrate 1 |
| SNAREs | Soluble N-ethylmaleimide-Sensitive Factor Attachment Protein Receptors |
| Tip60 | Tat Interactive Protein 60 kDa Histone Acetyltransferase |
| TRAF6 | Tumor Necrosis Factor Receptor-Associated Factor 6 |
| UVRAG | UV-Radiation Resistance-Associated Gene Protein |
| VPS15 | Vacuolar Protein Sorting 15 |
| VPS34 | Vacuolar Protein Sorting 34 |
| WIPI-2 | WD-repeat PI3P effector protein-2 |
| WW domain | Tryptophan-Tryptophan Domain |
References
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Covarrubias-Pinto, A.; Bhaskara, R.M.; Glogger, M.; Kuncha, S.K.; Xavier, A.; Seemann, E.; Misra, M.; Hoffmann, M.E.; Bräuning, B.; et al. Ubiquitination regulates ER-phagy and remodelling of endoplasmic reticulum. Nature 2023, 618, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Z.; Zhang, S.; Zhang, T.; Liu, Y.; Zhang, L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liu, J.J.; Li, Y.; Huang, Y.; Ta, N.; Chen, Y.; Fu, H.; Ye, M.D.; Ding, Y.; Huang, W.; et al. Cryo-EM structure and biochemical analysis reveal the basis of the functional difference between human PI3KC3-C1 and -C2. Cell Res 2017, 27, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A. Current views on the source of the autophagosome membrane. Essays Biochem. 2013, 55, 29–38. [Google Scholar] [PubMed]
- Brier, L.W.; Ge, L.; Stjepanovic, G.; Thelen, A.M.; Hurley, J.H.; Schekman, R. Regulation of LC3 lipidation by the autophagy-specific class III phosphatidylinositol-3 kinase complex. Mol. Biol. Cell 2019, 30, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Lindmo, K.; Brech, A.; Finley, K.D.; Gaumer, S.; Contamine, D.; Rusten, T.E.; Stenmark, H. The PI 3-kinase regulator Vps15 is required for autophagic clearance of protein aggregates. Autophagy 2008, 4, 500–506. [Google Scholar] [CrossRef]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Ohsumi, Y. Atg14: A key player in orchestrating autophagy. Int. J. Cell Biol. 2011, 2011, 713435. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, S.; Carlson, L.A.; Stjepanovic, G.; Young, L.N.; Kim, D.J.; Grob, P.; Stanley, R.E.; Nogales, E.; Hurley, J.H. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife 2014, 3, e05115. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jia, Y.; Zhang, Y.; Ma, F.; Zhu, Y.; Hong, X.; Zhou, Q.; He, R.; Zhang, H.; Jin, J.; et al. Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 2019, 15, 1130–1149. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ni, D.; Ma, B.; Lee, J.H.; Zhang, T.; Ghozalli, I.; Pirooz, S.D.; Zhao, Z.; Bharatham, N.; Li, B.; et al. PtdIns(3)P-bound UVRAG coordinates Golgi-ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nat. Cell Biol. 2013, 15, 1206–1219. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, X.Q.; Deng, R.; Li, D.D.; Tang, J.; Chen, W.D.; Chen, J.H.; Ji, J.; Jiao, L.; Jiang, S.; et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat. Commun. 2017, 8, 1159. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gorantla, S.P.; Muller-Rudorf, A.; Muller, T.A.; Kreutmair, S.; Albers, C.; Jakob, L.; Lippert, L.J.; Yue, Z.; Engelhardt, M.; et al. Phosphorylation of BECLIN-1 by BCR-ABL suppresses autophagy in chronic myeloid leukemia. Haematologica 2020, 105, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Tomaipitinca, L.; Petrungaro, S.; D’Acunzo, P.; Facchiano, A.; Dubey, A.; Rizza, S.; Giulitti, F.; Gaudio, E.; Filippini, A.; Ziparo, E.; et al. c-FLIP regulates autophagy by interacting with Beclin-1 and influencing its stability. Cell Death Dis. 2021, 12, 686. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.N.; Cheung, H.N.; Li, W.; Lau, K.F. FE65: Roles beyond amyloid precursor protein processing. Cell. Mol. Biol. Lett. 2015, 20, 66–87. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Schrotter, A.; Loosse, C.; Pfeiffer, K.; Theiss, C.; Kauth, M.; Meyer, H.E.; Marcus, K. A ternary complex consisting of AICD, FE65, and TIP60 down-regulates Stathmin1. Biochim. Biophys. Acta 2013, 1834, 387–394. [Google Scholar] [CrossRef]
- Zhai, Y.; Chan, W.W.R.; Li, W.; Lau, K.F. ARNO is recruited by the neuronal adaptor FE65 to potentiate ARF6-mediated neurite outgrowth. Open Biol. 2022, 12, 220071. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tam, K.M.V.; Chan, W.W.R.; Koon, A.C.; Ngo, J.C.K.; Chan, H.Y.E.; Lau, K.F. Neuronal adaptor FE65 stimulates Rac1-mediated neurite outgrowth by recruiting and activating ELMO1. J. Biol. Chem. 2018, 293, 7674–7688. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Concannon, C.G.; Ward, M.W.; Walsh, C.M.; Tirniceriu, A.L.; Tribl, F.; Kogel, D.; Prehn, J.H.; Egensperger, R. Modulation of gene expression and cytoskeletal dynamics by the amyloid precursor protein intracellular domain (AICD). Mol. Biol. Cell 2007, 18, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.W.; Concannon, C.G.; Whyte, J.; Walsh, C.M.; Corley, B.; Prehn, J.H. The amyloid precursor protein intracellular domain(AICD) disrupts actin dynamics and mitochondrial bioenergetics. J. Neurochem. 2010, 113, 275–284. [Google Scholar] [CrossRef]
- Moreau, K.; Ravikumar, B.; Puri, C.; Rubinsztein, D.C. Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D. J. Cell Biol. 2012, 196, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Perkinton, M.S.; Standen, C.L.; Lau, K.F.; Kesavapany, S.; Byers, H.L.; Ward, M.; McLoughlin, D.M.; Miller, C.C. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. J. Biol. Chem. 2004, 279, 22084–22091. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.; Dulcey, A.E.; Campos, F.; de la Fuente, C.; Acuna, M.; Castro, J.; Pinto, C.; Yanez, M.J.; Cortez, C.; McGrath, D.W.; et al. c-Abl Activation Linked to Autophagy-Lysosomal Dysfunction Contributes to Neurological Impairment in Niemann-Pick Type A Disease. Front. Cell Dev. Biol. 2022, 10, 844297. [Google Scholar] [CrossRef] [PubMed]
- Ermekova, K.S.; Zambrano, N.; Linn, H.; Minopoli, G.; Gertler, F.; Russo, T.; Sudol, M. The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J. Biol. Chem. 1997, 272, 32869–32877. [Google Scholar] [CrossRef] [PubMed]
- Sabo, S.L.; Ikin, A.F.; Buxbaum, J.D.; Greengard, P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 2001, 153, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Wang, M.; Su, J.; Dong, X.; Yang, Y.; Wang, H.; Li, Q. The mammalian actin elongation factor ENAH/MENA contributes to autophagosome formation via its actin regulatory function. Autophagy 2024, 20, 1798–1814. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.W.R.; Li, W.; Chang, R.C.C.; Lau, K.F. ARF6-Rac1 signaling-mediated neurite outgrowth is potentiated by the neuronal adaptor FE65 through orchestrating ARF6 and ELMO1. FASEB J. 2020, 34, 16397–16413. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Chau, D.D.; Li, W.; Chan, W.W.R.; Sun, J.K.; Zhai, Y.; Chow, H.M.; Lau, K.F. Insulin stimulates atypical protein kinase C-mediated phosphorylation of the neuronal adaptor FE65 to potentiate neurite outgrowth by activating ARF6-Rac1 signaling. FASEB J. 2022, 36, e22594. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Musiwaro, P.; Smith, M.; Manifava, M.; Walker, S.A.; Ktistakis, N.T. Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy 2013, 9, 1407–1417. [Google Scholar] [CrossRef]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. 2009, 452, 13–23. [Google Scholar] [PubMed]
- Kara, N.Z.; Toker, L.; Agam, G.; Anderson, G.W.; Belmaker, R.H.; Einat, H. Trehalose induced antidepressant-like effects and autophagy enhancement in mice. Psychopharmacology 2013, 229, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Schnell, H.M.; Ruoff, K.; Goldberg, A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J. Cell Biol. 2018, 217, 1757–1776. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Proikas-Cezanne, T.; Ruckerbauer, S.; Stierhof, Y.D.; Berg, C.; Nordheim, A. Human WIPI-1 puncta-formation: A novel assay to assess mammalian autophagy. FEBS Lett. 2007, 581, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Irving, N.G.; Brown, S.D.; Miller, C.C.; McLoughlin, D.M. Mapping of the human and murine X11-like genes (APBA2 and apba2), the murine Fe65 gene (Apbb1), and the human Fe65-like gene (APBB2): Genes encoding phosphotyrosine-binding domain proteins that interact with the Alzheimer’s disease amyloid precursor protein. Mamm. Genome 1998, 9, 473–475. [Google Scholar] [PubMed]
- McLoughlin, D.M.; Miller, C.C. The FE65 proteins and Alzheimer’s disease. J. Neurosci. Res. 2008, 86, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Kesavapany, S.; Banner, S.J.; Lau, K.F.; Shaw, C.E.; Miller, C.C.; Cooper, J.D.; McLoughlin, D.M. Expression of the Fe65 adapter protein in adult and developing mouse brain. Neuroscience 2002, 115, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e438. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hu, Q.; Hearn, M.G.; Shimizu, K.; Ware, C.B.; Liggitt, D.H.; Jin, L.W.; Cool, B.H.; Storm, D.R.; Martin, G.M. Isoform-specific knockout of FE65 leads to impaired learning and memory. J. Neurosci. Res. 2004, 75, 12–24. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Oh, S.J.; Lee, M.S. Role of Autophagy in the Pathogenesis of Diabetes and Therapeutic Potential of Autophagy Modulators in the Treatment of Diabetes and Metabolic Syndrome. J. Korean Med. Sci. 2022, 37, e276. [Google Scholar] [CrossRef]
- Jiang, B.; Zhou, X.; Yang, T.; Wang, L.; Feng, L.; Wang, Z.; Xu, J.; Jing, W.; Wang, T.; Su, H.; et al. The role of autophagy in cardiovascular disease: Cross-interference of signaling pathways and underlying therapeutic targets. Front. Cardiovasc. Med. 2023, 10, 1088575. [Google Scholar] [CrossRef]
- Keller, C.W.; Adamopoulos, I.E.; Lunemann, J.D. Autophagy pathways in autoimmune diseases. J. Autoimmun. 2023, 136, 103030. [Google Scholar] [CrossRef]
- Park, K.; Lee, M.S. Current Status of Autophagy Enhancers in Metabolic Disorders and Other Diseases. Front. Cell Dev. Biol. 2022, 10, 811701. [Google Scholar] [CrossRef]
- Lim, H.; Lim, Y.M.; Kim, K.H.; Jeon, Y.E.; Park, K.; Kim, J.; Hwang, H.Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef]
- Rice, S.; Elia, A.; Jawad, Z.; Pellatt, L.; Mason, H.D. Metformin inhibits follicle-stimulating hormone (FSH) action in human granulosa cells: Relevance to polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E1491–E1500. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kondo, K.; Motoki, K.; Homma, H.; Okazawa, H. Fasting activates macroautophagy in neurons of Alzheimer’s disease mouse model but is insufficient to degrade amyloid-beta. Sci. Rep. 2015, 5, 12115. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F.; Wang, H.M.; Li, Q.Y.; Zhang, Y.; Pan, J.; Qiang, Q.; Xin, X.Y.; Tang, H.D.; Ding, J.Q.; Chen, S.D. Starvation triggers Abeta42 generation from human umbilical vascular endothelial cells. FEBS Lett. 2010, 584, 3101–3106. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Zhu, W.; Plowey, E.D. BECN1/Beclin 1 sorts cell-surface APP/amyloid beta precursor protein for lysosomal degradation. Autophagy 2016, 12, 2404–2419. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Xu, N.; O’Prey, J.; Lao, L.Y.; Joshi, S.; Long, J.S.; O’Prey, M.; Croft, D.R.; Beaumatin, F.; Baudot, A.D.; et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Fang, Y.; Yan, L.; Xu, L.; Zhang, S.; Cao, Y.; Xu, L.; Zhang, X.; Xie, J.; Jiang, G.; et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci. Rep. 2017, 7, 45385. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Teles, F.; Minopoli, G.; Russo, T.; Rosenfeld, M.G.; Suh, Y. An epigenomic role of Fe65 in the cellular response to DNA damage. Mutat. Res. 2015, 776, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Minopoli, G.; Stante, M.; Napolitano, F.; Telese, F.; Aloia, L.; De Felice, M.; Di Lauro, R.; Pacelli, R.; Brunetti, A.; Zambrano, N.; et al. Essential roles for Fe65, Alzheimer amyloid precursor-binding protein, in the cellular response to DNA damage. J. Biol. Chem. 2007, 282, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Revol, R.S.; Koistinen, N.A.; Menon, P.K.; Chicote-Gonzalez, A.; Iverfeldt, K.; Strom, A.L. Alpha-secretase dependent nuclear localization of the amyloid-beta precursor protein-binding protein Fe65 promotes DNA repair. Mol. Cell. Neurosci. 2023, 127, 103903. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, W.W.R.; Chow, J.; Chau, D.D.-L.; Zhai, Y.; Lau, K.-F. Beclin 1-Mediated Autophagy Is Potentiated by an Interaction with the Neuronal Adaptor FE65. Biology 2025, 14, 97. https://doi.org/10.3390/biology14010097
Chan WWR, Chow J, Chau DD-L, Zhai Y, Lau K-F. Beclin 1-Mediated Autophagy Is Potentiated by an Interaction with the Neuronal Adaptor FE65. Biology. 2025; 14(1):97. https://doi.org/10.3390/biology14010097
Chicago/Turabian StyleChan, Wai Wa Ray, Jessica Chow, Dennis Dik-Long Chau, Yuqi Zhai, and Kwok-Fai Lau. 2025. "Beclin 1-Mediated Autophagy Is Potentiated by an Interaction with the Neuronal Adaptor FE65" Biology 14, no. 1: 97. https://doi.org/10.3390/biology14010097
APA StyleChan, W. W. R., Chow, J., Chau, D. D.-L., Zhai, Y., & Lau, K.-F. (2025). Beclin 1-Mediated Autophagy Is Potentiated by an Interaction with the Neuronal Adaptor FE65. Biology, 14(1), 97. https://doi.org/10.3390/biology14010097