3.2. Characterization of Gnathostome Orthogroups Indicates That the TNF Superfamily Is More Complex than Hitherto Known
When the 385 sequences detected in the model species were aligned and analyzed as described in the
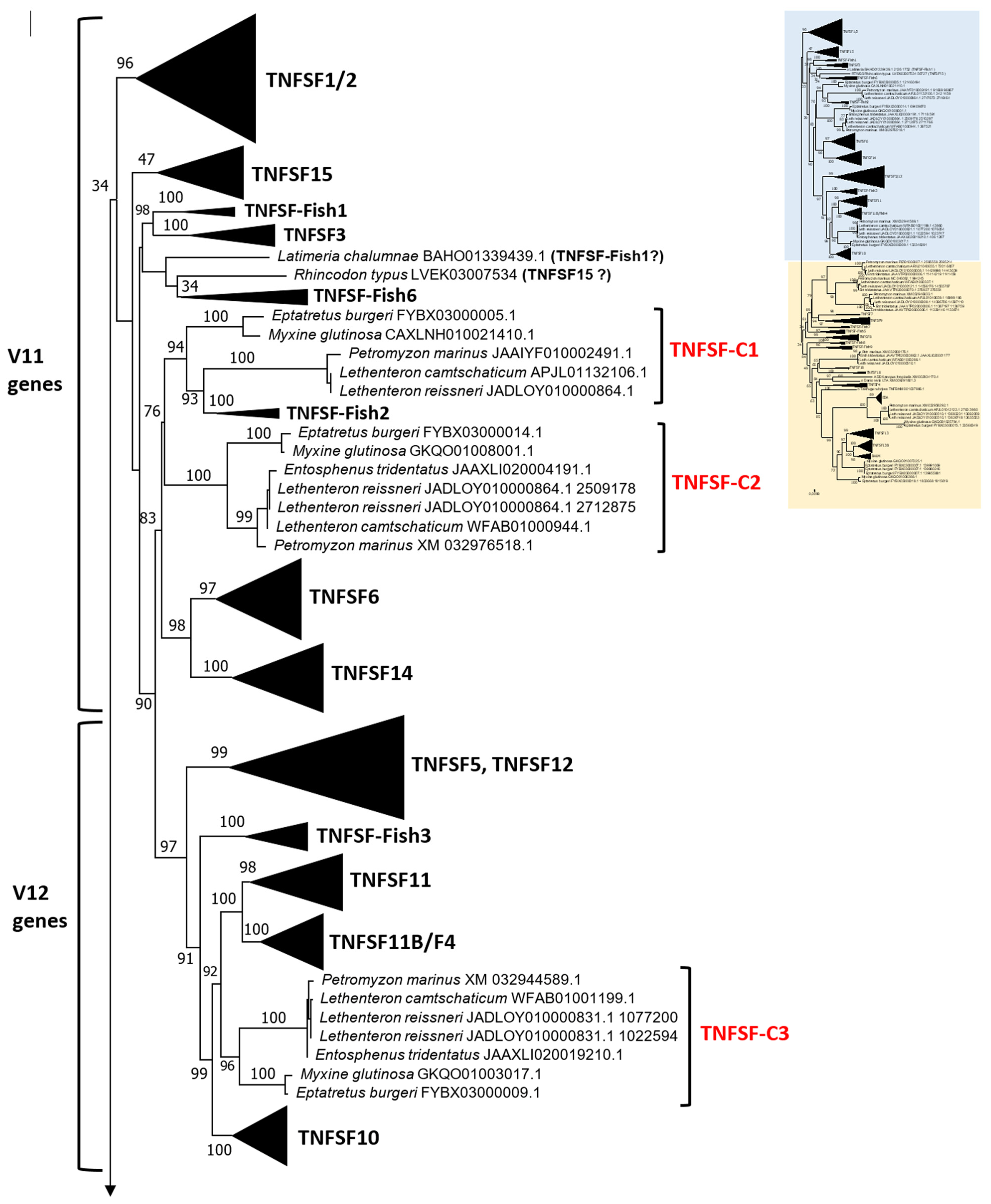
Section 2, the best tree obtained (both highest ML and lowest BIC) was the one shown in
Figure 2. The tree has been divided into two sections to facilitate visualization. The corresponding optimal alignment can be found in
Supplementary File S1, and a fully expanded version of the tree (in Newick format) is available in
Supplementary File S2. According to both their ML and BIC values, this tree, obtained using the MAFFT-NWNSI algorithm, is much better than those derived for the same sequences with ClustalX, used in my first study [
14], or MAFFT-LINSI, which was chosen in Redmond et al. [
13].
This type of tree can be dissected at two different levels. First, it is possible to determine orthogroups, that is, groups of orthologous genes present in multiple species, characterized by strong sequence similarity, causing them to appear very close in the tree. Orthologs also often share identical genome locations across species. Such analyses were already performed in the earlier studies, but with quite different results. In my prior analysis [
14], 24 orthogroups were characterized in gnathostomes while 23 were identified in Redmond et al. [
13]. However, only 17 of these orthogroups are equivalent in both studies, i.e., they include either identical or almost identical sequences in the same groups of species. These were the following: TNFSF1/2, TNFSF5, TNFSF6, TNFSF10, TNFSF11, TNFSF12, TNFSF13, TNFSF13B, TNFSF14, TNFSF15, EDA, BALM, and TNFSF-F1 to -F5. In three other cases, TNFSF3, TNFSF8, and TNFSF18, the groups are not identical, because Redmond et al. [
13] identified putative orthologs in sharks that, apparently, I did not detect [
14]. They also found a potentially new shark-specific gene, which they named TNFSF33. Finally, two differences are due to Redmond et al. [
13] lumping together as orthologous some genes that I considered different: TNFSF4 was merged with TNFSF-New and TNFSF7 with TNFSF9. That these two associations are unsupported by the data will be discussed below. In any case, combining the results of the two studies, and assuming no additional genes remain to be discovered, a maximum of 25 orthogroups would be expected in gnathostomes: the 24 already described in my study [
14], plus the additional shark-specific gene identified by Redmond et al. [
13].
The tree based on sequence similarity shown in
Figure 2, combined with synteny data, (detailed in
Figure 3,
Figure 4,
Figure 5,
Figure 6,
Figure 7,
Figure 8,
Figure 9 and
Supplementary File S3, which includes the complete synteny results along with additional notes, useful to interpret the results), demonstrates that the gnathostome TNF superfamily is quite more complex than assumed so far. A total of twenty-eight TNFSF orthogroups can be defined in gnathostome species, namely the twenty-four already characterized in my previous work, which this new analysis fully confirms, plus four additional ones. These newly identified groups have been named, given that they are absent in tetrapods and following my previous convention, TNFSF-Fish6 to TNFSF-Fish9 (abbreviated as TNFSF-F6 to -F9). All but two of the three hundred and thirty-five genes characterized in the gnathostome model species can be included in one of these twenty-eight groups. The genes in 21 of these 28 orthogroups are together in branches with significant (≥95%) bootstrap support and also show congruent synteny. The exceptions are TNFSF5, TNFSF12, TNFSF15, TNFSF18, TNFSF-New, TNFSF-F6, and EDA. The first two, TNFSF5 and TNFSF12, fail to appear as two clearly separated branches, because the chondrichthyan
TNFSF5 genes are closer to
TNFSF12 genes than to their orthologs in osteichthyan species (see
Supplementary File S2). This was already noted in my previous analysis [
14]. However, as already concluded in that study, two different orthology groups can be defined incorporating synteny data. In many species, both in osteichthyans and chondrichthyans, the
TNFSF12 and
TNFSF13 genes are adjacent in a location that in humans corresponds to region 17p13 (
Figure 3), while all
TNFSF5 genes, again both in osteichthyans and chondrichthyans, are located in a totally different genomic region in a different chromosome. In humans, it corresponds to Xq26 (
Figure 4). The lack of separation in the tree is likely due to different rates of evolution in the
TNFSF5 genes of osteichthyans and chondrichthyans, with the latter either evolving at a slower pace, thereby retaining more similarity to
TNFSF12 genes, or, alternatively, having convergent features with them.
For the TNFSF18 and TNFSF-New orthogroups, the sequences appear in two or even three separated branches, close to one another in the tree (
Figure 2). However, it is easy to determine that these related branches include orthologs, as the genes have the same genomic positions in closely related species (
Figure 5:
TNFSF-New genes are present only in
Danio and
Takifugu;
Figure 6:
TNFSF18 is found in some tetrapods). Finally, all
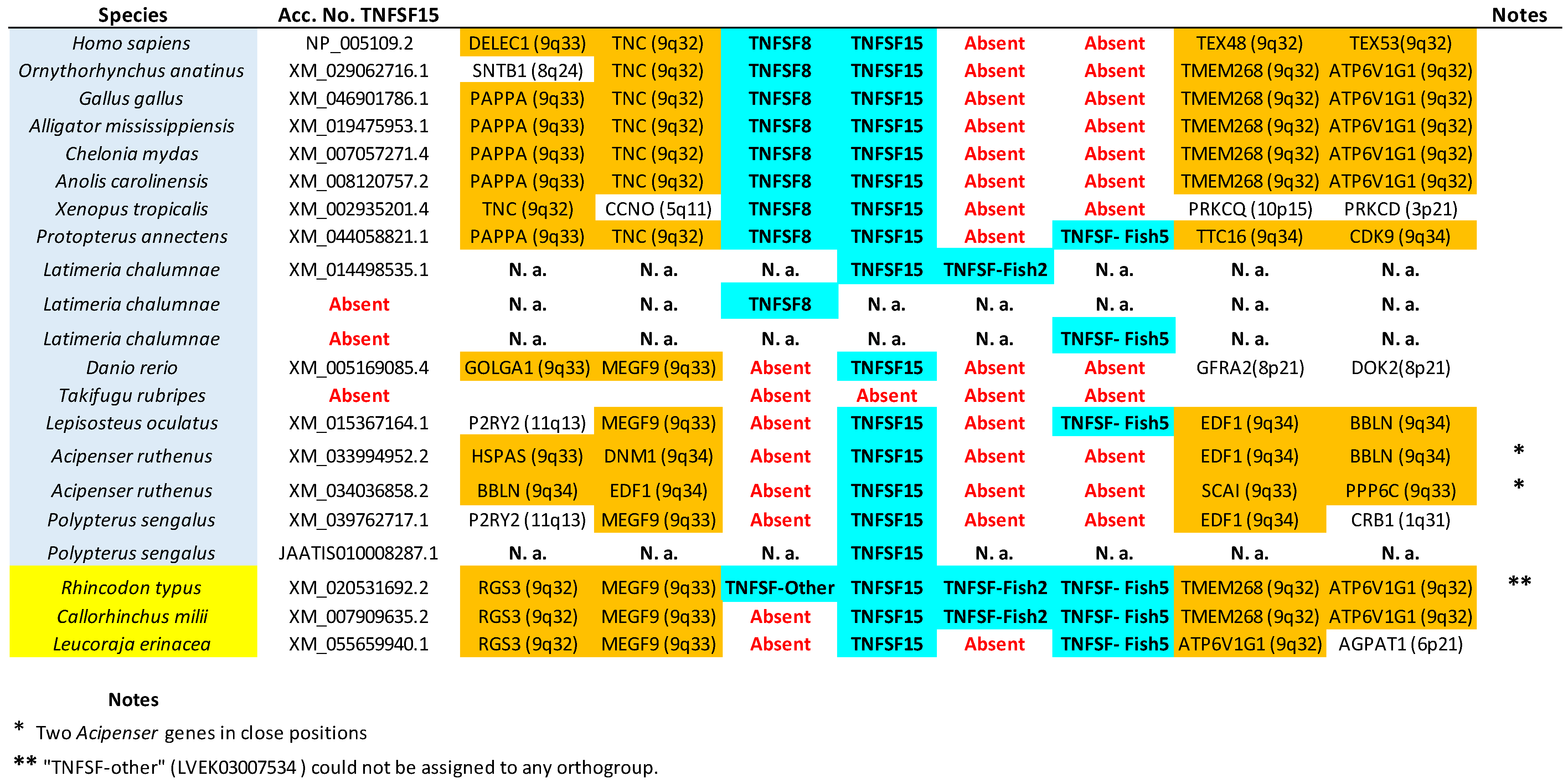
TNFSF15,
TNFSF-F6, or
EDA gene sequences indeed appear together in the tree, but within branches with non-significant (i.e., <95%) bootstrap support. For
TNFSF15, this lack of support was already noted before [
14], and the reason is that the genes of a few osteichthyan fishes evolve very rapidly, distorting the results (see details in
Supplementary File S2). In the other two cases, the most likely explanation is the presence of similar paralogous genes in other species, which may have been included within these groups in some bootstrap replicates, lowering the support values. For
TNFSF-F6, the closer sequences are precisely the two which have not been included in any orthogroup (
Latimeria BAHO01339439.1,
Rhincodon LVEK03007534.1;
Figure 2). However, according to synteny data, both are unrelated to
TNFSF-F6. They may, respectively, be rapidly evolving
TNFSF-F1 (see genomic location in
Figure 5; the gene is indicated as “TNFSF-other”) and
TNFSF15 genes (see below). Finally, for the EDA orthogroup, it is the presence of very similar genes in cyclostomes that diminishes the bootstrap values (
Figure 2). However, the branch that includes both the gnathostome
EDA genes and these cyclostome
EDA-like genes has 100% bootstrap support (
Figure 2), strongly suggesting that the cyclostome sequences may also be
EDA orthologs, a result that will be examined in detail below. In any case, despite low bootstrap support values for these three proposed orthogroups, TNFSF15, TNFSF-F6, and EDA, synteny data indicate that the genes included in them are true orthologs, as they are located in the same regions in multiple species (
Figure 6,
Figure 7 and
Figure 8).
Let us now consider in detail the orthogroups that are defined for the first time here, TNFSF-F6 to TNFSF-F9. As already indicated, the sequences of the genes included in each of these groups are very similar, so they appear together in the tree. Synteny provides additional useful information. For example, it indicates that
TNFSF-F6 is most likely an ancient
TNFSF-F1 tandem duplicate, present in both chondrichthyan and osteichthyan fishes (
Figure 5). The chondrichthyan sequences actually correspond to those called “TNFSF33” by Redmond et al. [
13], which, however, failed to detect the osteichthyan ones.
TNFSF-F7 and
TNFSF-F8 genes are both found adjacent to
TNFSF4 and
TNFSF6 (
Figure 6). Despite this, it is very unlikely that they correspond to the same gene, given how different their sequences are (see positions in
Figure 2) and the fact that they appear only in a few closely related species:
TNFSF-F7 in
Protopterus and
Latimeria and
TNFSF-F8 in chondrichthyans. The simplest explanation for the origin of these two genes is that they are independently emerging, slow-evolving duplicates of
TNFSF4. The logic to reach that conclusion starts by considering that these three orthogroups, TNFSF4, TNFSF-F7, and TNFSF-F8, appear relatively close in the tree (
Figure 2). Moreover, the region where they reside and the one where
TNFSF7 and
TNFSF9 genes are present (shown in
Figure 9) have a common origin, emerging as two separated regions due to WGD2 (as first indicated by Collette et al. [
6]). Therefore, the fact that
TNFSF4 genes are more distant in the tree from
TNFSF7 and
TNFSF9 than
TNFSF-F7 and
TNFSF-F8 can be explained by
TNFSF4 evolving at a faster rate than the latter two genes. Finally, the last new gene,
TNFSF-F9, is found only in osteichthyans and is located in tandem with
TNFSF9 and
TNFSF7, appearing quite close to those genes in the tree (
Figure 9). The simplest hypothesis is that both
TNFSF7 (present only in mammals) and
TNFSF-F9 are duplicates of
TNFSF9, which is much older, present in both chondrichthyans and osteichthyans (
Figure 9).
As already indicated, putative shark
TNFSF3,
TNFSF8, and
TNFSF18 genes were described in Redmond et al. [
13]. The presence of
TNFSF3 in sharks is confirmed here (
Figure 5;
Supplementary File S2). On the other hand, the other two associations were not supported by our data. The putative
TNFSF8 gene of
Rhincodon typus described by Redmond et al. [
13] was indeed detected. This corresponds to one of the two sequences already mentioned that could not be included in any orthogroup (LVEK03007534.1). However, as mentioned above, that gene is related to the TNFSF15 orthogroup. It appears relatively close to
TNFSF15 and radically distant from
TNFSF8 in our phylogenetic tree (
Figure 2) and is located in tandem with
TNFSF15 in the
Rhincodon genome (
Figure 7). Thus, it can be interpreted as a shark-specific, relatively recent
TNFSF15 duplicate. Finally, their putative shark
TNFSF18 genes in fact correspond to those included here in the newly defined orthogroup TNFSF-F8. Significantly, neither in the trees obtained by Redmond et al. [
13] nor in this study was a statistically significant association of
TNFSF18 and
TNFSF-F8 sequences detected. We can therefore conclude that putting together in a single orthogroup the
TNFSF18 and
TNFSF-F8 genes, as Redmond et al. [
13] did, lacks support. Bona fide
TNFSF18 genes are found only in some tetrapod species.
The existence of the 24 gnathostome TNFSF orthogroups described in my first study [
14] has been confirmed here. On the other hand, the addition of more species, not unexpectedly, has allowed for the characterization of four additional orthology groups. In contrast, the orthogroups called “GITRL (TNFSF18)”, “OX40L (TNFSF4)”, “4-1BBL/CD70(TNFSF9/TNFSF7)”, and “CD30L (TNFSF8)” by Redmond et al. [
13] are not supported by these new data. It is important to note that they are lumped together in these putative orthogroups sequences that appear in particular branches of their tree, but support for three out of those four critical branches was statistically non-significant: the corresponding bootstrap values were just 93% (GITRL), 67% (OX40L), and 49% (4-1BBL/CD70). In “OX40L”, they grouped together genes belonging to the TNFSF4 and TNFSF-New orthogroups, an association that not only lacked bootstrap support, but also contradicted synteny data. As I already had shown [
14], those two genes are in totally different locations, which in the human genome, respectively, correspond to 6p21 and 1q24. This indicates that Redmond et al. [
13] had no evidence whatsoever to propose that orthogroup. Regarding the other three, when compared with my results, their “GITRL” group includes the
TNFSF18 and
TNFSF-F8 genes which I have discussed above, showing them to be different genes. In the “CD70 and 4-1BBL” group, they lumped together sequences that I have included here in orthogroups TNFSF9, TNFSF7, and TNFSF-F9. The presence of the three genes in
Danio rerio (
Figure 9) directly proves that they are paralogs and that three different orthogroups must be defined. For the fourth one (“CD30L/TNFSF8”), they obtained a significant value of bootstrap (97%) for the branch that includes
TNFSF8 with the chondrichthyan sequences. However, as I already described, that branch was not recovered in my analysis; the shark sequences appear relatively close to the
TNFSF15 genes, while bona fide
TNFSF8 genes (present only in sarcopterygians) are close to
TNFSF7 and
TNFSF9 and are very distant from
TNFSF15 (
Figure 2). In conclusion, in all four cases, there was little to no evidence to propose those putative orthogroups. Significantly, these erroneous associations led Redmond et al. [
13] to assume that the
TNFSF8 and
TNFSF18 genes were present in both chondrichthyans and osteichthyans, leading to an artificial inflation of the number of ancient genes in their model. The main reason for these discrepancies in the definitions of the gnathostome orthogroups are the radical differences in the topologies of the trees in Redmond et al. [
13] and my two studies, which will be explained in the
Section 4.
In summary, 28 orthogroups have been identified, of which 21 are present in both chondrichthyans and osteichthyans, as summarized in
Figure 10 (next page). Very significantly, these 21 ancient orthogroups are not only supported by sequence similarity (
Figure 2), but also by synteny data, given that, for all of them, orthologous genes have been found adjacent to those of the TNF superfamily in at least a chondrichthyan and an osteichthyan species (
Figure 3,
Figure 4,
Figure 5,
Figure 6,
Figure 7,
Figure 8 and
Figure 9;
Supplementary File S3), demonstrating that the position of the genes today found in living species corresponds to the one in the ancestor of all gnathostomes. Therefore, twenty-one is the minimum number of genes present in that ancestor. Of course, additional genes, now lost in all living species, may have existed, but it is more parsimonious to assume that they did not if all data can be explained without recurring to them, which, as we will see, is the case here.
A final technical point concerns the fact that sampling additional sarcopterygian species in this study compared to my previous work [
14] has indicated that genes in one of the orthogroups, which in that work were found only in fish species and thus called
TNFSF-Fish4, are actually absent in eutherian mammals but present in the other tetrapod taxa (
Figure 10). Given this result, it is preferable to give this gene a different name. Considering its high similarity to
TNFSF11 and its presence in some mammals, which allows to use the convention for mammalian genes, I propose naming it
TNFSF11B. However, to allow an easier comparison between this study and my previous work, I will refer to it as
TNFSF11B/F4 throughout this text.
3.3. Deduction of Ancient TNFSF Genes Based on Tree Topology and Common Synteny
The second level of the dissection of the tree shown in
Figure 2 concerns the information it provides regarding ancient genes from which the ones present in modern species are derived. In my previous work, I identified five clusters of closely related genes, which I named TNF, FASL, 1-1BBL, CD40, and EDA. I hypothesized that these clusters descended from five ancestral genes emerged before WGD2 [
14]. It is simple to determine, comparing
Figure 2 above with the trees presented in that work, that these five groups are again identified here. This was obviously expected, given the strong similarity among the genes within each cluster. However, there is a single significant difference in the deepest topology of the tree found here with respect to that obtained in my previous work, substantially altering how these five clusters must be interpreted. This difference concerns the position of the EDA cluster genes (
EDA,
BALM,
TNFSF13, and
TNFSF13B). These genes appeared in my previous work in a long branch, quite separate from the rest of the genes; their closest relatives, according to that topology, were
TNFSF-F2 (a TNF cluster gene) and
TNFSF6/TNFSF14, the only two genes in the FASL cluster (see Figures 1 and 2 in [
14]). However, in this new analysis, the EDA cluster genes appear in a totally different position, close to
TNFSF4 (
Figure 2). This change, no doubt due to the improvement associated with the use of a better alignment algorithm, has a very important implication, which is that the topology found here perfectly agrees with how the genes are distributed across four different gnathostome chromosomes, something that was not the case with the topology obtained in my previous study. This observation allows for us to develop a very simple model of the evolution of the whole TNF superfamily. In
Figure 2, four groups of genes, V11, V12, V21, and V22 (V meaning vertebrate), are indicated. The hypothesis that I will develop is that each of these groups descends from a single ancestral gene, which may be named
TNFSF-V11 to
-V22, already present before WGD1.
Let us consider the data in detail. V11 genes correspond to those included in my previous work in the TNF and FASL clusters. In humans, they are found on chromosomes 1, 6, 9, and 19 in well-characterized regions, containing the MHC and its paralogons, known to be related, coming from a single region present before WGD1 [
41]. Thus, it is reasonable to hypothesize that all these TNF and FASL cluster genes originated from a single ancestral gene,
TNFSF-V11. There are eight different V21 genes: the ancient
TNFSF4 and
TNFSF9, present in both chondrichthyans and osteichthyans,
TNFSF7 and
TNFSF18, present only in tetrapods, and
TNFSF-F5,
-F7,
-F8, and
-F9, found only in fishes (details in
Figure 10).
TNFSF4,
TNFSF7,
TNFSF9, and
TNFSF18 were included in the 4-1BBL cluster, while
TNFSF-F5 was thought to be a TNF cluster gene with an unusually fast evolutionary rate, causing its abnormal position in the tree [
14]. However, a simpler explanation exists for all these genes appearing together in
Figure 2, which is that they all descend from a single progenitor gene,
TNFSF-V21, present before WGD1. These genes are again found in humans on chromosomes 1, 9, and 19, in perfect agreement with what is observed for V11 genes, except that no V21 gene exists on chromosome 6, which may be explained by an early gene loss. Moreover, they not only appear in the same three chromosomes, but also are found in tandem with V11 genes in all three:
TNFSF-F5 is detected in tandem with
TNFSF15;
TNFSF4,
TNFSF18,
TNFSF-F7, and
TNFSF-F8 form a large tandem with
TNFSF6; and
TNFSF7,
TNFSF9, and
TNFSF-F9 are together in another large tandem with
TNFSF14. The simplest hypothesis to explain all these data is that the
TNFSF-V11 and
TNFSF-V21 genes were located in tandem before WGD1, and the tandem underwent two duplications, caused by WGD1 and WGD2, leading to the presence of related genes in four chromosomes of modern gnathostomes. Additionally, still more recent, duplications finally produced the full diversity of genes now detected in modern species on those chromosomes.
The exact same pattern emerges for V12 and V22 genes. V12 genes (
TNFSF5,
TNFSF10,
TNFSF11,
TNFSF11B/F4,
TNFSF12, and
TNFSF-F3) were, in my previous work, due to its strong similarity, all included in the CD40 cluster. They are located in regions that, in humans, correspond to chromosomes X, 3, 13, and 17, which also have a common origin, deriving from a single region present before WGD1 [
15,
16,
21]. On the other hand, the four gnathostome V22 genes, which correspond to those ascribed to the EDA cluster, are found also on chromosomes X, 13, and 17 in humans. Moreover, as described for V11 and V12 genes, V12 and V22 genes may also appear in tandem:
TNFSF12 and
TNFSF13 are adjacent in many species. Therefore, the simplest explanation is again that they all derived from two ancestral genes, let us call them
TNFSF-V12 and
TNFSF-V22, present in tandem before WGD1 and quadruplicated due to WGD1 and WGD2. A few subsequent duplications, losses, and chromosomal inversions are sufficient to explain the observed pattern in modern species.
Now, if we examine the deepest branches of the tree (
Figure 2), it becomes obvious why the change in the position of the EDA cluster genes is so crucial. V11 and V12 genes are grouped together, occupying half of the tree, while V21 and V22 genes form the other half. This is exactly the expected relationship if all V11 and V12 genes derive from a still older gene, let us name it
TNFSF-V1, while V21 and V22 genes come from a second one, originally located adjacent to the first, which may be called
TNFSF-V2. This general explanation therefore postulates that a systematic expansion of two original TNFSF genes, due to the two whole-genome duplications plus several gene-specific duplications and gene losses, resulted in the current set of genes found in all living gnathostomes. Simple and elegant as this new hypothesis is, it still remains to be developed in full detail, gene by gene, and tested against the cyclostome data, which, as previously mentioned, provides direct insights into the situation after WGD1 but before WGD2. We will show below that cyclostome TNFSF genes fit perfectly within this new paradigm. However, before analyzing cyclostome species, it is necessary to consider another aspect of the evolution of the TNF superfamily in gnathostomes, namely how its complexity has evolved in different jawed vertebrate lineages.
3.4. Complexity of the TNF Superfamily Throughout Gnathostome Evolution
Once all the orthogroups in gnathostomes have been defined, it is possible to trace the origin of each one and analyze their patterns of survival or loss in the different lineages. In
Figure 10, above, the number of genes, the number of different orthogroups observed, and the number of ancestral orthogroups, defined as groups that predate the chondrichthyan/osteichthyan split, have been detailed for each species. As already shown [
14], the number of genes is quite variable. Among the model species examined here,
Rhincodon typus and
Acipenser ruthenus have the largest sets, with 33 genes each, and
Gallus gallus, the smallest, with only 11 genes. Most of the genes in the sturgeon
A. ruthenus appear in pairs, due to a whole genome duplication which occurred over 200 million of years ago [
42]. It is noteworthy that the ancestor of all teleost fishes, such as
Danio rerio and
Takifugu rubripes, also underwent a WGD about 270 million years ago according to the latest estimates [
43], but these two species have a relatively low number of TNFSF genes (
Figure 10), indicating that, contrary to what happened in the
Acipenser lineage, most duplicates were subsequently lost.
The number of orthogroups also varies widely across species (
Figure 10).
Gallus again has the smallest number (11), while the largest is also found in
Rhincodon typus (22), although followed closely by
Latimeria chalumnae (21) and
Protopterus annectens (20). The notable difference between the number of genes and number of orthogroups in
R. typus indicates that this species must have many duplicates of some genes. Indeed,
Figure 10 indicates that eight genes belonging to the TNFSF1/2 group are found in
Rhincodon, which largely explains this discrepancy. Finally, regarding ancestral orthogroups, present in both chondrichthyans and osteichthyans, 21 have been identified, as noted earlier. Strikingly, all of them are present in
Rhincodon and most in
Latimeria and
Protopterus (18 in each species), but the rest of species have quite less, ranging from just 10 to 16. In my first study [
14], I obtained data suggesting that, in the lineages leading to some modern gnathostome species (
Takifugu rubripes,
Homo sapiens), a significant reduction in the number of TNFSF genes occurred. Here, the analysis of many species allows for a more precise determination of the patterns of duplication and loss of TNFSF genes. Results are summarized in
Figure 11 (next page), where genes have been color-coded according to the hypothesized ancestral genes (
TNFSF-V11 to
-V22) from which they originated.
Figure 11 shows that it is not possible to find two model species with the same set of genes, indicating that the TNF superfamily has had a highly dynamic evolutionary history in gnathostomes. After the split of chondrichthyans and osteichthyans, a few genes emerged in each group, increasing the original number of 21 ancestral genes (
Figure 11). Notably, three of them correspond to the newly described genes
TNFSF-F7 to
-F9. TNFSF-F6 is even older, existing already in the ancestor of all gnathostomes. Therefore, these four genes, absent in tetrapods, may have functions quite different from those described for mammalian TNFSF genes. Later, a trend towards the reduction in the TNF superfamily is observed in osteichthyans. Excluding lineages that underwent WGDs (such as
Danio,
Takifugu, and
Acipenser), a total of 18 duplications and 32 losses are deduced in osteichthyan lineages. In contrast, in chondrichthyans, 16 duplications but only 11 losses are observed, and this number may be even lower, given that several losses were deduced to have occurred in
Callorhinchus milii, but, in
Carcharodon carcharias, a close relative of
C. milii, there are genes of four orthogroups not found in the latter species (TNFSF1/2, TNFSF14, TNFSF-F1, and TNFSF-F6; see
Figure 5 and
Figure 9). These results suggest that some regions of the
C. milii genome may remain uncharacterized, and those genes be present. The occurrence of multiple losses at certain branches is very interesting. A substantial reduction in genes occurred before tetrapod diversification (five genes lost and just one added), a trend that continued in saurian lineages. Before osteichthyan fish diversification, there were also four gene losses. Moreover, several lineages, such as those leading to
Gallus,
Takifugu,
Polypterus,
Leucoraja, and perhaps also
Callorhichus, have undergone significant reductions in their TNF superfamily compared to their closest relatives (
Figure 11).
Considering now the origin of the genes, notable differences are observed. V11 genes have a unique pattern, being duplicated more frequently than lost. This is largely due to the expansions of the TNFSF1/2 orthogroup genes in ten different lineages, in three cases involving multiple duplications, an extreme pattern not observed in any other orthogroup. Also contributing to the increase in V11 genes are two smaller expansions, which involve TNFSF6 genes in chondrichthyans and TNFSF14 in actinopterygian fishes. In contrast, V21 genes show a strong reductive trend, with only six duplications but twenty-one losses. On the other hand, for V12 and V22 genes, losses exceed duplications, but both are relatively rare. In summary, throughout gnathostome evolution the sets of V11 and V21 genes have changed very dynamically, while V12 and V22 genes have evolved more conservatively.
3.5. Characterization of Cyclostome Orthogroups and Relationships Between Cyclostome and Gnathostome Genes
In my initial study [
14], I analyzed the TNFSF genes present in three cyclostome species, established cyclostome TNFSF orthogroups, and proposed a hypothesis linking each cyclostome orthogroup to a single gnathostome orthogroup. At the time, this one-to-one comparison was logical, as it was assumed that WGD2 occurred before the cyclostome/gnathostome split (see
Section 1 Introduction). However, two important facts have since then emerged. First, as already mentioned, it is known that cyclostomes did not undergo WGD2; and, second, it has been established that a genome triplication occurred before the hyperoartia/myxini split, that is, affecting all cyclostomes, both hagfishes and lampreys [
17,
18]. Given this new evidence, a one-to-one comparison is no longer valid. In absence of gene losses, we would expect three similar but distinct cyclostome genes, generated by the cyclostome-specific triplication, to be equally related to two different gnathostome genes, emerged in WGD2. Of course, secondary gene duplications or losses could obscure this basic pattern.
Alongside the gnathostome orthogroups,
Figure 2 indicates the positions of the cyclostome genes, which have been classified into six fundamental orthogroups, TNFSF-C1 to TNFSF-C6 (C = cyclostome), plus an additional one, TNFSF-C5b, which will be analyzed separately. This classification into just seven orthogroups is based on both sequence similarity and synteny data. For example,
TNFSF-C6 genes appear as three distinct branches, but synteny data confirm that they are orthologs; the division into three groups is driven by the combination of two factors, namely the presence of lineage-specific duplicates and the rapid sequence evolution of some genes. Comparing the tree in
Figure 2 with the corresponding one in my initial study [
14] reveals that six of the seven orthogroups, all but TNFSF-C5b, were already identified in my previous work. These six orthogroups appear in the same positions in both trees relative to the gnathostome genes. In summary, despite including three new cyclostome species and doubling the number of cyclostome genes analyzed, the results in both studies show nearly perfect congruence.
The hypothesis developed in the previous section, which proposed that four genes,
TNFSF-V11 to
TNFSF-V22, existed before WGD1, predicts that cyclostomes should possess four distinct classes of genes, positioned in specific positions of the phylogenetic trees.
Figure 2 confirms this prediction: cyclostome genes are indeed found within each one of the regions of the trees hypothesized to derive from these ancestral genes. For instance,
TNFSF-C1 and
-C2 are located adjacent to the gnathostome genes deduced to come from
TNFSF-V11,
TNFSF-C3 is located adjacent to gnathostome V12 genes,
TNFSF-C4 is close to gnathostome V21 genes and
TNFSF-C5, and
-C6 is adjacent to the proposed V22 gnathostome genes. If these associations are due to shared, ancestral orthology, a second prediction is that the cyclostome TNFSF genes should be flanked by particular gene sets. Around V11 and V21 genes, one would expect to find genes whose orthologs in humans are located on chromosomes 1, 6, 9, and 19, while around V12 and V22 genes, the adjacent genes should have human orthologs on chromosomes 3, 13, 17, and X. This is confirmed in
Figure 12 and
Figure 13 (next page), which display synteny data for the regions containing cyclostome TNFSF genes.
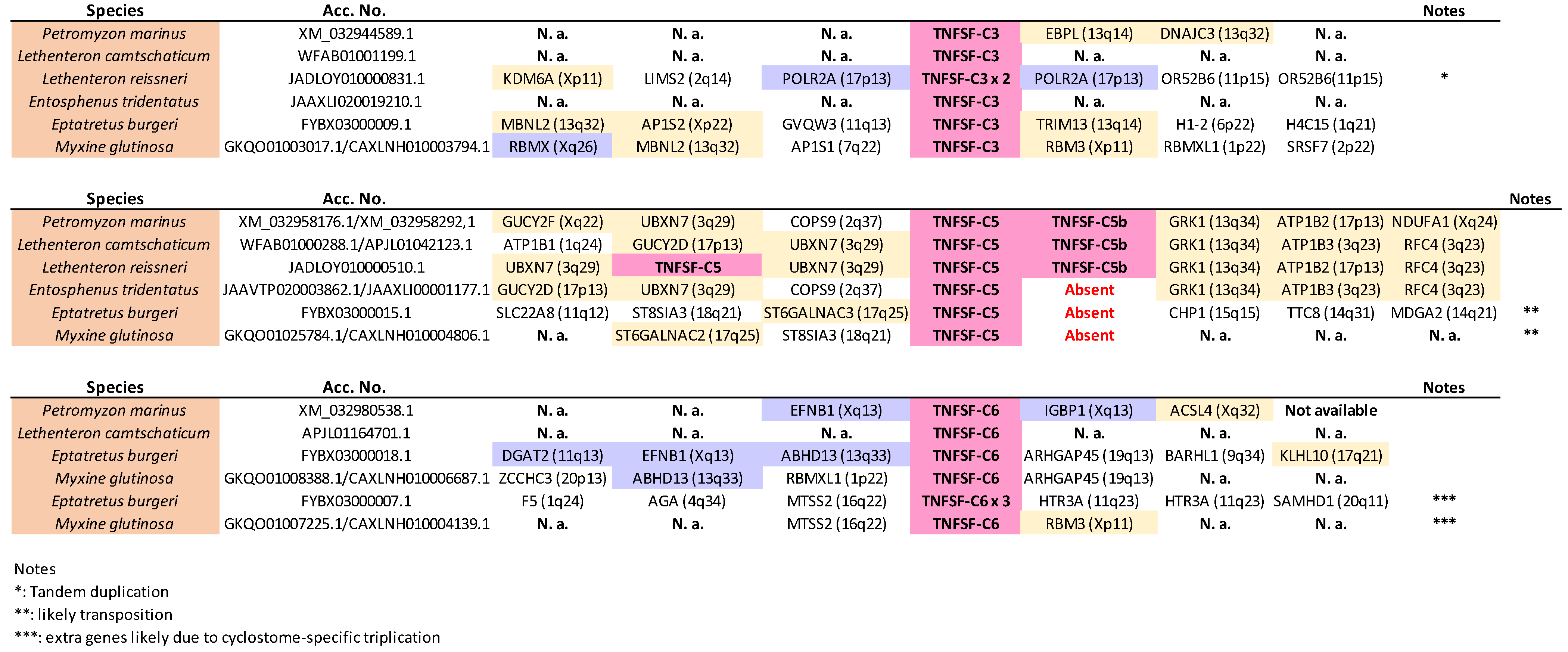
TNFSF-C1 and
TNFSF-C2 are found in tandem in lampreys but separated in the only hagfish for which synteny data are available,
Eptatretus burgeri (
Figure 12). However, in all cases, they are surrounded by genes whose most likely orthologs in humans are on chromosomes 1, 9, and 19, as predicted. Moreover, two of those adjacent genes show strong similarity to
RGS3 and
DNM1, which are adjacent to the V11 genes
TNFSF3 and
TNFSF15 in some gnathostomes (see
Figure 5 and
Figure 7). On the other hand,
TNFSF-C4 genes were detected in at least two different places in the genomes of lampreys (probably in three in
Lethenteron camtschaticum, but data are inconclusive; they are not found in hagfishes), probably due to the cyclostome-specific triplication (see details in the next section). In one of those locations, one to three very similar duplicates in tandem are found, while in the other, single
TNFSF-C4 genes are observed (
Figure 12). Again, as expected, most of the surrounding genes have their most likely human orthologs located on chromosomes 1, 6, 9, and 19. Moreover, one of them is very similar to the human gene
C3, which is adjacent to the
TNFSF9 and
TNFSF14 genes in several gnathostomes (
Figure 9). These results fully agree with the results of the tree, which suggested that
TNFSF-C4 genes descend from the ancestral
TNFSF-V21 gene. If we now turn our attention to the rest of cyclostome TNFSF genes, the situation is again as expected according to our hypothesis.
Most genes adjacent to TNFSF-C3, -C5, -C5b, and -C6 likely have human orthologs on chromosomes X, 3, 13, and 17 (
Figure 13). Moreover, adjacent to the TNFSF-C3 genes, POLR2A and RBMX orthologs can likely be found, which are close in some gnathostomes to, respectively, TNFSF12 and TNFSF5, both TNFSF-V12-derived genes. This agrees with the close sequence similarity observed between TNFSF-C3 and gnathostome V12 genes (
Figure 2). On the other hand, adjacent to TNFSF-C6 genes related to human EFNB1, IGBP1, DGAT2, and ABHD13 can be detected, placed in some gnathostomes close to the V22 genes EDA, BALM, or TNFSF13B (
Figure 8,
Supplementary File S3). Hence, all the available data indicate that TNFSF-C6 is a descendant of the ancestral TNFSF-V22 gene. We are finally left to consider the TNFSF-C5 and -C5b genes. The very strong similarity of TNFSF-C5 with gnathostome EDA (
Figure 2), as well as its adjacency to genes whose human orthologs are most often found on chromosomes X, 3, 13, and 17, indicate that TNFSF-C5 also derives from TNFSF-V22. On the other hand, TNFSF-C5b, which is present only in lampreys in tandem with TNFSF-C5, is quite an anomaly. In principle, it could simply be a recent TNFSF-C5 duplicate. However, its sequence is more similar to gnathostome V21 genes than to V22 genes (
Figure 2). My hypothesis to explain the origin of this gene is based on what I described above for the fast-evolving gnathostome gene TNFSF4. The regions shown at the interface between V21 and V22 genes in the tree in
Figure 2 may be considered to be filled up with fast-evolving genes that “escape” from the positions where they should be found. For example, in that tree, gnathostome TNFSF4 should be adjacent to TNFSF-Fish7 and close to TNFSF7 and TNFSF9, as discussed in a previous section, but it is distant from them. Similarly, I propose that TNFSF-C5b is closely related to TNFSF-C5, but its fast pace of evolution has generated sequences that appear to be quite separated in the tree. This argument may be extended to other genes. It may provide an explanation of why the branches that include all V12 or all V22 genes have significant statistical support (97% and 99%, respectively), but V11 and V21 genes are not similarly grouped (
Figure 2). If we look at the positions of the main V11 and V21 cyclostome genes (i.e., TNFSF-C4 in the first case and TNFSF-C5 and -C6 in the second) and their three respective closest gnathostome relatives, we see that the branches that include them, which may be defined as “central” in the sense that they approximately indicate the place in the tree where the ancestral genes TNFSF-V11 and TNFSF-V21, would be placed if their sequences were available, have relatively high bootstrap support (83% and 89%, respectively;
Figure 2). However, the rest of gnathostome V11 and V21 genes, as well as cyclostome TNFSF-C5b, become distributed progressively away from these branches. This can be interpreted through most V11 and V21 genes having quite fast evolutionary rates, which leads to their relationships becoming obscured, the topology being somewhat altered, and the support values, even for the central branches, being lower than expected. The rapid changes observed in the sets of V11 and V21 genes present in different species (see previous section) and this fast pace of sequence evolution may certainly have related causes.
These findings refute two previous claims: (1) My inference that, in early cyclostome evolution, the number of TNFSF genes was drastically reduced [
14] cannot be sustained. Instead, the results shown above indicate that most genes present before the cyclostome/gnathostome split have been conserved in cyclostomes. In the next section, I will develop the simplest model to explain the early cyclostome data, showing that a few early gene losses have been compensated by some duplications. (2) Contrary to the model proposed by Redmond et al. [
13] which hypothesized as many as 15 TNFSF genes after WGD1, the information obtained from cyclostome genomes points to a much smaller number. In the next section, I will show that the available data support the presence of only eight genes at that time, corresponding to four pairs, derived from the four ancestral genes,
TNFSF-V11 to
TNFSF-V22, as a result of WGD1.
3.6. A Model for TNFSF Evolution in All Vertebrate Lineages
The general conclusion of the previous sections is that all the available data for both cyclostome and gnathostome TNFSF genes are compatible with the hypothesis of four ancestral genes existing very early in vertebrate evolution, before WGD1. This hypothesis is developed in full detail in the evolutionary model shown in
Figure 14 and
Figure 15 (see next pages).
Figure 14 depicts the predicted genes in early vertebrate evolution and cyclostomes, and
Figure 15 refers to gnathostome species. This model has two strong constraints: (1) it must account for all the available data from living species; and (2) it must be maximally parsimonious, minimizing the number of rare events, such as genome duplications, gene duplications, gene losses, transpositions of genes to new locations, etc.
The proposed evolutionary history begins with a single TNFSF gene which became duplicated in tandem very early in animal evolution. Later, this pair of genes duplicated again and transposed, resulting in four genes, precisely the ones which we have called
TNFSF-V11 to
-V22, present in two different chromosomes (
Figure 14, top). Comparing the current situation in living species with the available reconstructions of the ancestral vertebrate chromosomal set, these two chromosomes correspond to ancestral chromosomes 9 and 16 in Sacerdot et al. [
21], CLGM and CLGN in Simakov et al. [
15], or PVC15 and PVC6 in Nakatami et al. [
16]. These four genes are the only ones which have to be hypothesized before WGD1 to explain all the other results. When WGD1 occurred, it led to a set of eight TNFSF genes in four different chromosomes in the common ancestor of all vertebrates. These genes may be called, following the same nomenclature,
TNFSF-V111 to
-V222 (
Figure 14, center). To account for the genes present in cyclostomes, it is sufficient to postulate that three of those genes (
TNFSF-V211,
-V112, and
-V122) were lost, while
TNFSF-V111 was duplicated in tandem. This led to a set of six genes, which correspond to the ones named above
TNFSF-C1 to
-C6. That basic set of TNFSF genes has been slightly modified in different cyclostome lineages; due to additional tandem duplications, some gene losses plus the effect of the cyclostome-specific triplication occurred. For instance, in
Petromyzon marinus, nine genes are found, the three additional ones corresponding to
TNFSF-C5b, adjacent to
TNFSF-C5, plus two additional
TNFSF-C4 genes (
Figure 14, bottom). Two of the three
TNFSF-C4 genes are located in tandem on chromosome 5 of that species, while the third one is found on chromosome 15. This distribution in two different chromosomes in
Petromyzon and other species (
Figure 12) may be explained if
TNFSF-C4 genes became multiplied in the cyclostome-specific triplication, and some of them have been retained thereafter. The overall impact of that triplication on the set of cyclostome TNFSF genes has, however, been minimal, as no other TNFSF duplicates can be attributed to it (
Figure 12 and
Figure 13). This indicates that most TNFSF genes were lost following the genome triplication.
Figure 15 presents the simplest hypothesis to explain the evolution of TNFSF genes in gnathostomes. Following WGD1, four pairs of genes were present, each on a different chromosome (
Figure 15, top). WGD2 doubled that number, resulting in 16 genes, which were later further amplified to generate the gene set present in the ancestor of all gnathostomes. It is deduced that seven duplications and two losses occurred since WGD1 that explain the final set of twenty-one genes in the gnathostome ancestor. Later, changes have been extensive, as summarized in
Figure 11. Two particular examples have been shown on
Figure 15 (bottom): first,
Rhincodon typus, where, exceptionally, no gene has been lost since the ancestor of all gnathostomes and twelve duplications have occurred, largely increasing the total number of TNFSF genes; second, our species, in which seven losses have been partially compensated by four duplications, leading to the current set of eighteen TNFSF genes.
In summary, we can conclude that a quite simple model accounts for all the available data. This indicates that the evolution of the TNF superfamily in vertebrates has been largely determined by the impact of WGD1 and, in gnathostomes, WGD2, but not by the cyclostome genome triplication. Additionally, a considerable number of lineage-specific gene duplications and losses has led to a very variable set of genes, slightly different in each species. On the other hand, the models developed by Collette et al. [
6] and Redmond et al. [
13] cannot explain the available data. In the first case, the general outline is correct (four genes before WGD1, eight before WGD2), but their ancient genes only partially correspond to the ones deduced here. On the other hand, the Redmond et al. model is incompatible with the cyclostome data and also, as already demonstrated, contains some erroneous gnathostome orthogroups, which makes it also unable to explain some of the gnathostome results obtained here. Most significantly, their proposal of a very complex TNF superfamily already before WGD1 is untenable. On the other hand, my previous model [
14] is quite similar to the one developed here, except in two particular aspects. First, my former model is less parsimonious, requiring some assumptions about the rates of evolution and the independent, asynchronous emergence of genes, which the current model avoids due to the new interpretation of the relationships of the genes of the EDA cluster with the rest of TNFSF genes and also the new identification of
TNFSF-F5 as a V21 gene. Second, as already indicated, the discovery of new orthogroups that contain genes present in both chondrichthyan and osteichthyan imply that the ancestor of gnathostomes possessed a somewhat more complex set of TNFSF genes than previously suggested. While I previously estimated 19 genes, current data indicate at least 21, which correspond to the ones that I then proposed plus
TNFSF3 and
TNFSF-F6. It is formally possible but unlikely that this number will increase when more species are sampled, given that this work has explored all main gnathostome groups and also because Redmond et al. [
13] examined four additional shark species without finding other candidates.
3.7. Functional Implications of the Evolutionary Data
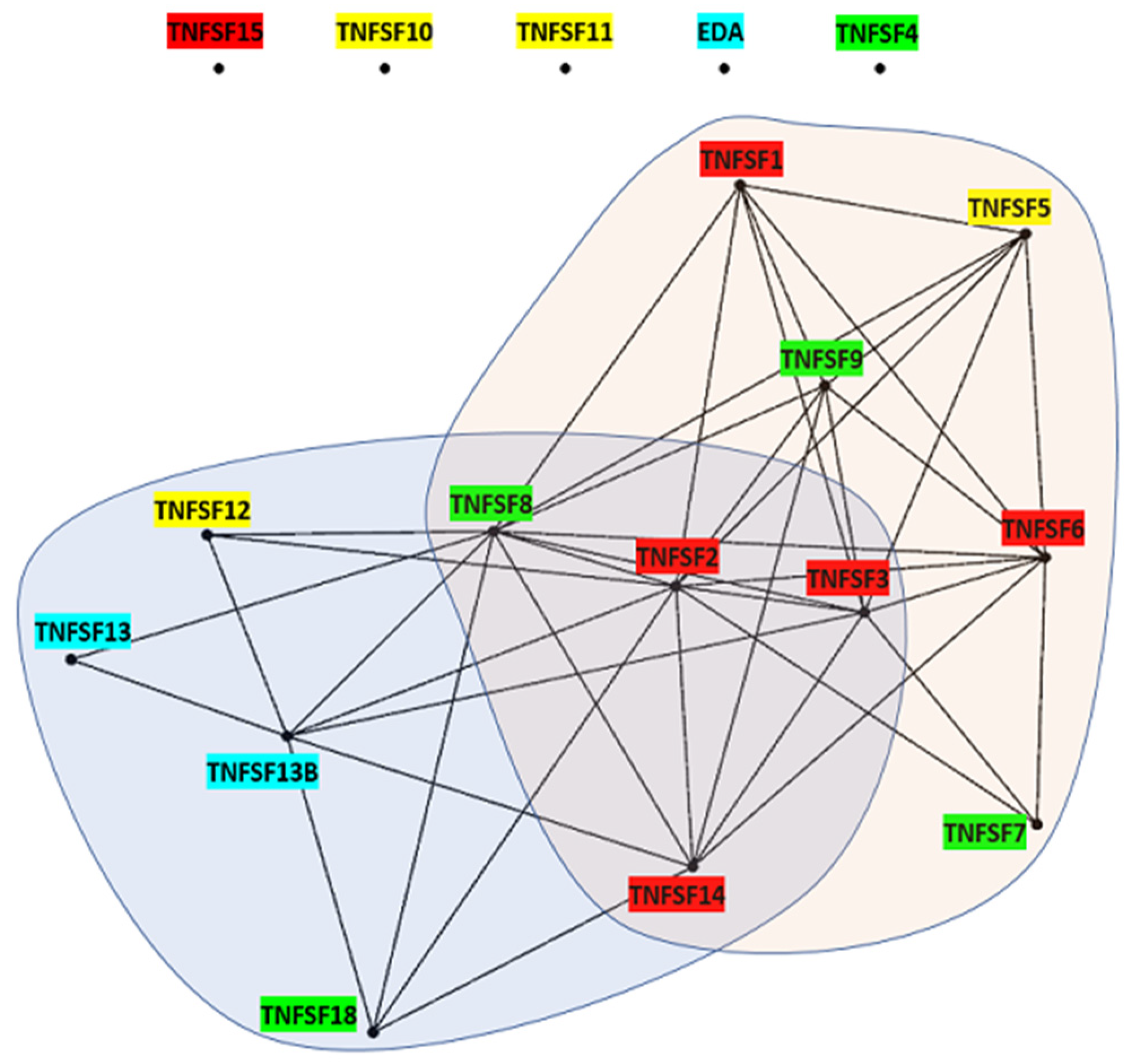
A solid knowledge of the evolution of a gene family allows for a better understanding of its functions. Based on the model discussed above, several aspects of the functions of the TNFSF genes acquire a new meaning. One example is the fact that different TNFSF genes encode proteins which are able to bind to the same receptors of the TNFR superfamily. Sequence similarity alone does not fully explain some of these promiscuous interactions, given that some TNFSF proteins that share receptors are distantly related. For instance, TNFSF6, TNFSF14, and TNFSF15 products can all bind the decoy receptor TNFRSF6B/DcR3 [
5] and, although
TNFSF6 and
TNFSF14 are closely related,
TNFSF15 is not (
Figure 2). However, knowing the origin of these genes helps make sense of these observations. It turns out that all the TNFSF ligands that share receptors are derived from the same ancestral genes (
Figure 16). Thus, although independent cooptions of unrelated ligands to interact with the same receptor cannot be ruled out, a more straightforward explanation is that, along the evolution of the superfamily, there have been episodes, e.g., after the WGDs, where the products of multiple TNFSF genes were able to interact with related receptors encoded also by different genes, many of them generated also in those WGDs, and that such an ambiguous situation has still not been completely sorted out.
Another example illustrating how a sound evolutionary model may illuminate functional aspects refers to the fact that there are four different ligands, the TNFSF4, TNFSF7, TNFSF9, and TNFSF18 proteins, which are expressed in dendritic cells and contribute to the priming of CD8+ T cells [
46]. This multiplicity is difficult to interpret unless we consider that those four ligands are encoded by V21 genes. It is reasonable to hypothesize that
TNFSF-V21 itself had related functions in some cells present in the common vertebrate ancestor, which have been retained by its descendant genes despite hundreds of millions of years of divergent evolution. The presence of both lymphocytes [
47] and cells which express dendritic cell markers [
48] in cyclostomes suggests that these interactions could exist in the common ancestor of all vertebrates. The alternative explanation, that four related but quite different ligands and their corresponding receptors have been independently coopted to perform the same function, is quite less likely. No doubt, many other fine details of the functions of TNF superfamily members would be better understood within the evolutionary framework developed here.
In my earlier work, I provided evidence indicating that the involvement of TNFSF members in the regulation of apoptosis, through interactions with TNF receptors containing death domains, as well as their role in the activation of the NF-KB pathway, potentially leading to cell survival and to inflammatory responses, is very ancient, probably predating vertebrate emergence. However, given that those conclusions were based on a model of TNFSF evolution which has been here modified, it is pertinent to reconsider these issues. As in my previous study [
14], I will conduct a retrograde analysis, starting with the evidence available for mammals and assuming that, if two genes share both a common ancestor and a given function, it is more parsimonious to consider that the ancestral gene already performed that function rather than the alternative of the function evolving independently twice. Applying this concept to the interaction with TNF receptors, it turns out that some V11, V12, and V22 genes encode proteins which interact with death-domain-containing TNF superfamily receptors (data for interactions between ligands and receptors obtained from [
5]). Those are, respectively,
TNFSF1,
TNFSF2,
TNFSF6, and
TNFSF15 (V11 genes),
TNFSF10 (V12 gene), and
EDA (V22 gene). The simplest explanation for these six interactions is that the activation of death-domain receptors by TNFSF proteins predates the divergence of all these genes, implying that the ancestral gene from which all vertebrate TNFSF genes derived already possessed this capability. Later, V21 genes would have secondarily lost that ability. Given that exactly the same argument applies to the interactions of TNFSF proteins with TNF superfamily receptors lacking a death domain, we can conclude that the products of the original TNFSF gene, from which all the ones present in vertebrates emerged, most likely had the possibility of interacting at the same time with both types of receptors, with and without death domains. This is not a far-fetched hypothesis, given that it still occurs today. In the six cases mentioned above in which a TNF superfamily member encodes ligands able to bind death-domain-containing receptors, products of these same genes are also able to bind to one or several TNF superfamily receptors lacking those domains [
5]. The same ancient origin can be inferred for the activation of the NF-KB pathway. All mammalian V11 and V22 genes as well as
TNFSF11 and
TNFSF12 (V12 genes) plus
TNFSF4 and
TNFSF9 (V21 genes) are involved in such regulatory functions [
5]. Thus, a role in NF-KB pathway activation can also be traced back to the ancestral gene from which all current vertebrate TNFSF genes evolved. This type of backward analysis has the obvious problem that it can be affected by functional convergence. However, in all these cases, it is extremely unlikely that the products of so many, very different, genes independently acquired the same functions.
The patterns of expression of TNFSF genes may be examined in order to understand how the evolution of this superfamily relates with the development of the different systems in which it functions, particularly the immune system, in which TNFSF proteins have fundamental roles.
Figure 17 shows the cases in which pairs of TNFSF genes have patterns of expression in 81 human cell types that are significantly correlated (Spearman’s correlation coefficient > 0.38;
p < 0.05; see
Section 2 Methods). A first interesting result is that there are four genes,
TNFSF4,
TNFSF11,
TNFSF18, and
EDA, for which no significant correlation was found. In the case of
EDA, correlations were actually slightly negative for seven out of the seventeen comparisons. A second significant result is that eight genes, shown within a circle in
Figure 17, are highly interconnected. Out of 28 possible positive correlations among them, 22 were found. The meaning of this group of genes becomes clear when the same analysis is repeated, but excluding the 11 cell types in the dataset which belong to the immune system. Then, only eight of those links persist, indicating that the correlations detected for these eight genes are largely due to similar patterns of expression in immune system cells. Finally, there are six genes which have only one or two links with the genes of the main group, so they appear in the periphery of the network (
Figure 17).
Considering the origin of all these genes, it becomes obvious that V12 and V22 genes, shown in yellow and blue, respectively, in
Figure 17, have fewer connections compared to V11 and V21 genes (in red and green, respectively). Genes belonging to the first two sets have, on average, only 2.6 links, while the latter two have a mean of 4.5 links. Also, only one V12 gene,
TNFSF5, appears in the central core of highly interconnected genes, the other seven being V11 and V21 genes. This result suggests that the different dynamics observed along the evolution of gnathostomes (
Figure 11) may be related to a different degree of involvement in immune functions. The emergence of new V11 and V21 genes, as well as the disappearance of some of them, could be due to their roles in rapidly evolving functions associated with the refinement of the system of adaptive immunity in gnathostome lineages. Conversely, V12 and V22 genes likely evolved more conservatively due to them often having more conventional functions in other systems. In this context, it is interesting to point out that, when knockout mice for these genes were generated, only two of them showed phenotypes which are unrelated with the immune system. They are
EDA (a V22 gene) and
TNFSF11 (a V12 gene).
EDA null mutants show very general anomalies, such as abnormal bones and skin, a lack of vibrissae and certain glands, among others [
49,
50,
51,
52].
TNFSF11 mutants suffer severe osteopetrosis, tooth anomalies, and mammary gland defects [
53,
54]. Significantly, neither gene shows significant correlations with other TNFSF genes in our analysis (
Figure 17).
A second approach, focused on the patterns of expression in immune cells, allows us to refine the interpretation of the results just presented.
Figure 18 (see next page) indicates the specific links among TNFSF genes when only immune system cells are considered. These links derive from a qualitative analysis in which two genes are considered similar if they have high levels of expression in three identical cell types out of the eleven immune system cells for which data were available. This type of analysis allows us to establish from which particular cell types the connections derive. In
Figure 18, the network of TNFSF genes has been divided into two groups. Genes in the first group, encircled in blue in that figure, are connected due to them having high levels of expression in macrophages and related cells (such as monocytes, Langerhans cells, Hofbauer cells, etc.). These cell types can be considered to be part of the innate immune system. Cells similar in function to mammalian macrophages are very ancient, being present in invertebrates [
55]. On the other hand, the connections of genes of the second group (enclosed in pink in
Figure 18) are due to high expression levels in B and T lymphocytes and NK cells, closely related cell types of the lymphoid lineage, specific to vertebrate adaptive immunity. Included in both groups are four genes,
TNFSF2,
TNFSF3,
TNFSF8, and
TNFSF14, which have high expression levels in both macrophage-related and lymphocyte-related cells. This new analysis demonstrates that almost all TNFSF genes highly expressed in cells of the lymphoid lineage descend from either
TNFSF-V11 (red) or
TNFSF-V21 (green). Following with the idea developed in the previous paragraph, this may be interpreted as V11 and V21 genes having a central role in the refinement of the adaptive immune system, which may imply rapid changes, not only in their protein sequences or their functions but also in which genes remain critical and which ones become dispensable, when immune challenges are modified. The fact that the eight highly connected genes shown in
Figure 17 are included in the set of genes connected by them having high levels of expression specifically in lymphoid cells (
Figure 18) points to a deep relationship in the functions of all of them. It is also significant that two couples of genes in this set,
TNFSF1/TNFSF2 and
TNFSF7/TNFSF9, derive from recent duplications (
Figure 15), suggesting an advantage for increased complexity linked to refinements of the adaptive immune system in the mammalian lineage.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}