
The Mechanism of the Development and Maintenance of Sexual Dimorphism in the Dioecious Mulberry Plant (Morus alba)

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and Illumina Sequencing
2.3. Quality Control of Transcriptome Data and Expression Analysis
2.4. RT-qPCR Verification
2.5. Analysis of Adaptive Evolution of Sex-Biased and Unbiased Genes
3. Results
3.1. Sequencing Quality Control and Reference Genome Mapping
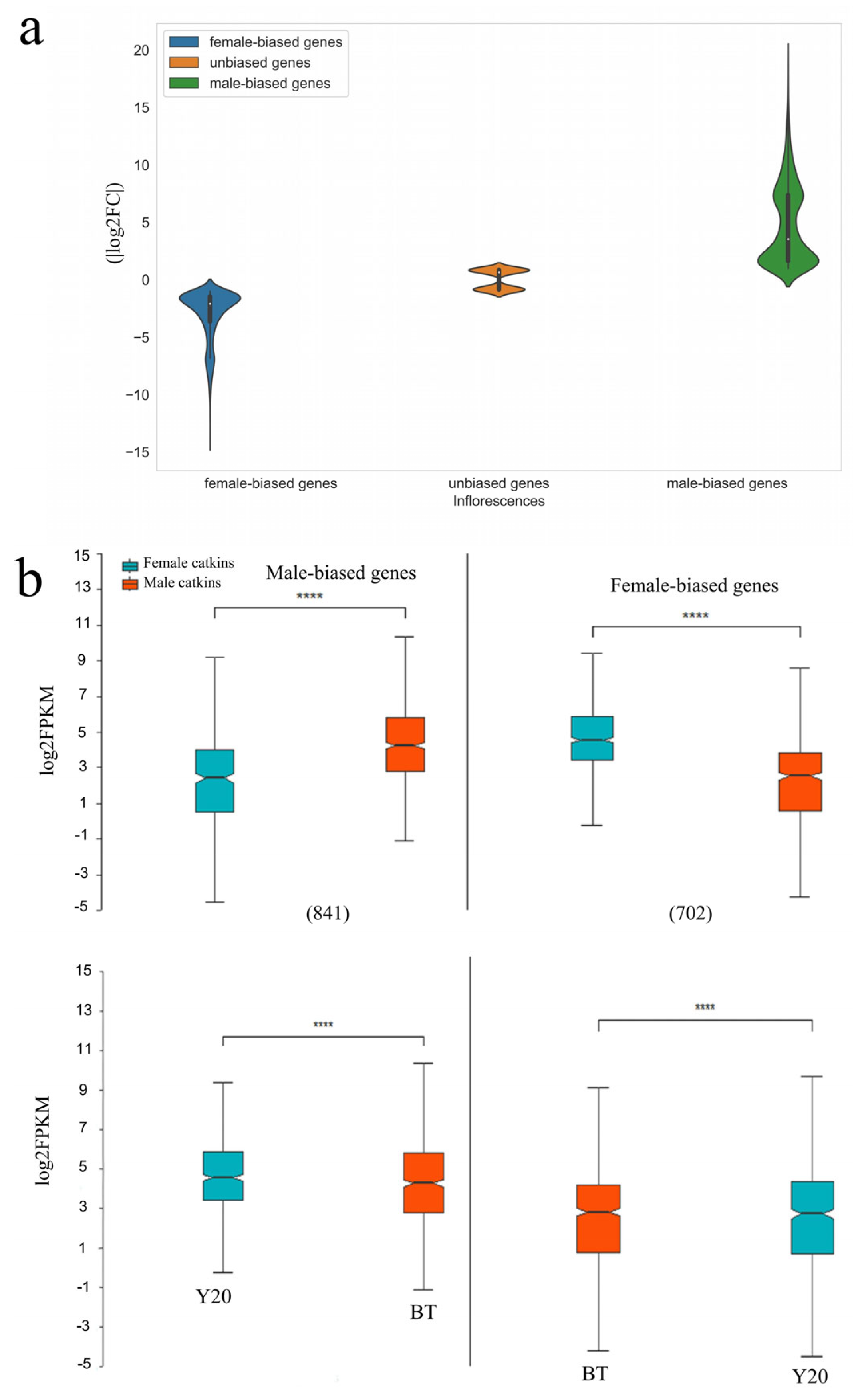
3.2. Differentially Expressed Genes between Male and Female Flower Buds
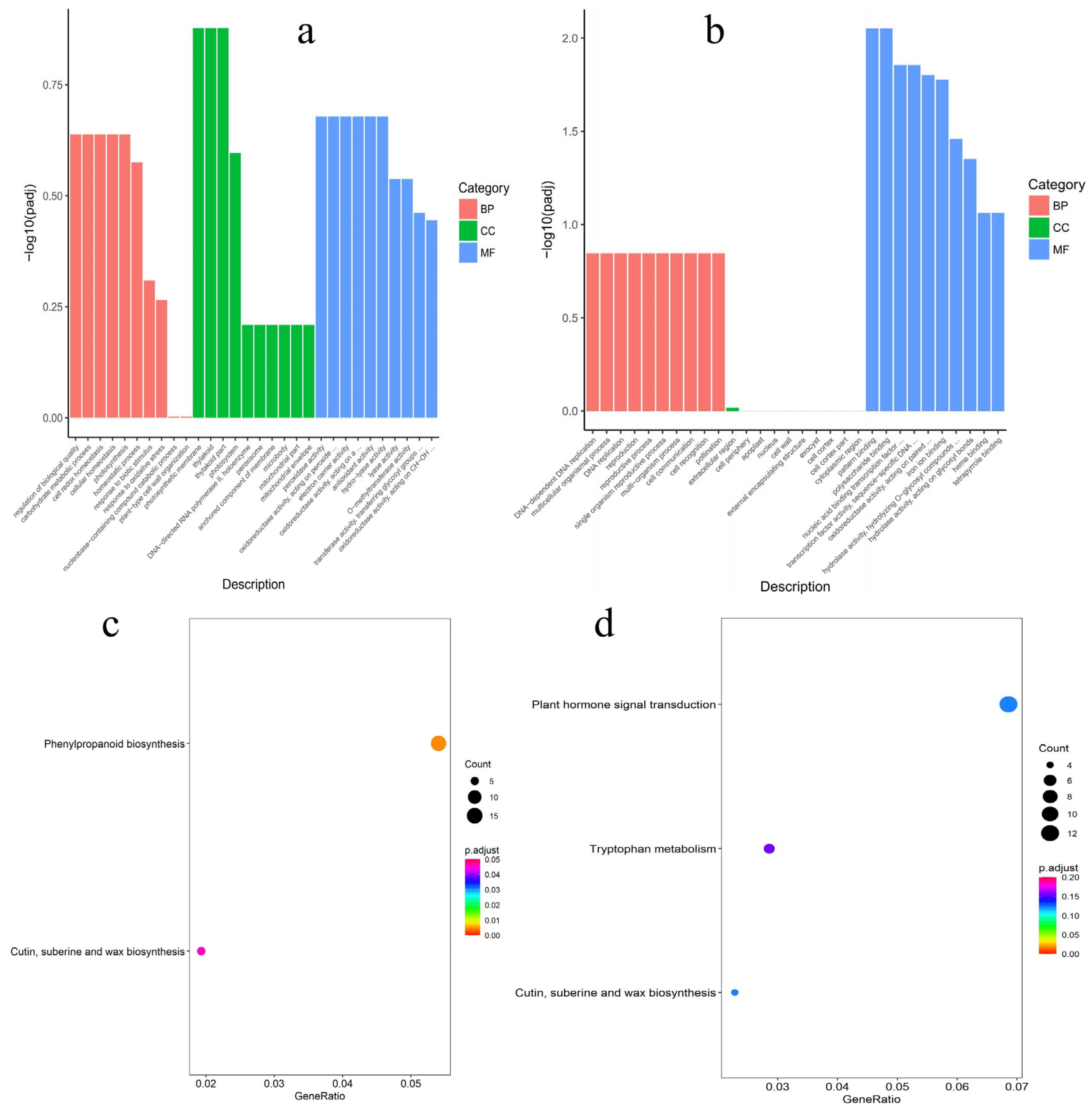
3.3. Functional Annotation Associated with DEGs
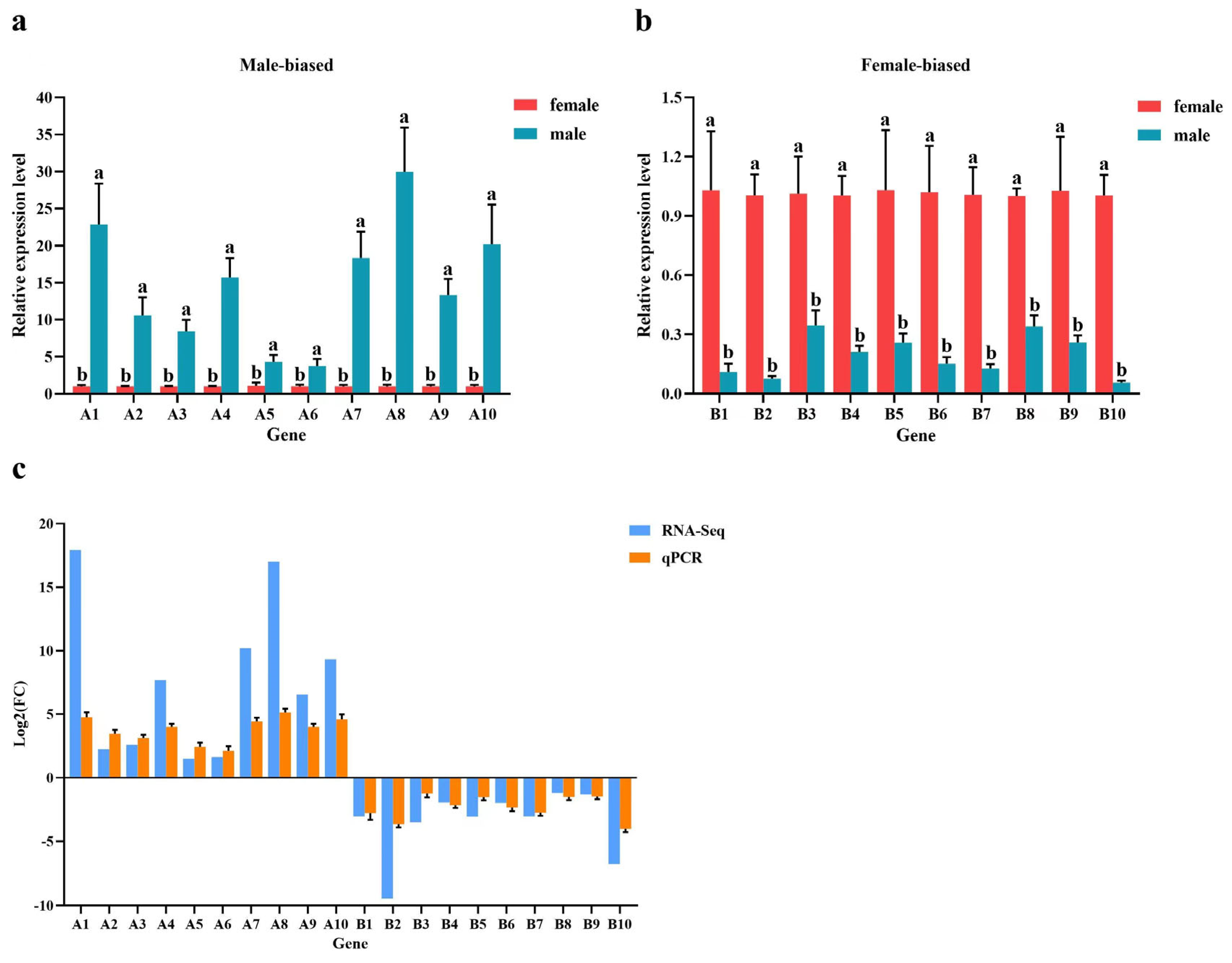
3.4. Verification of DEGs via Real-Time Quantitative PCR
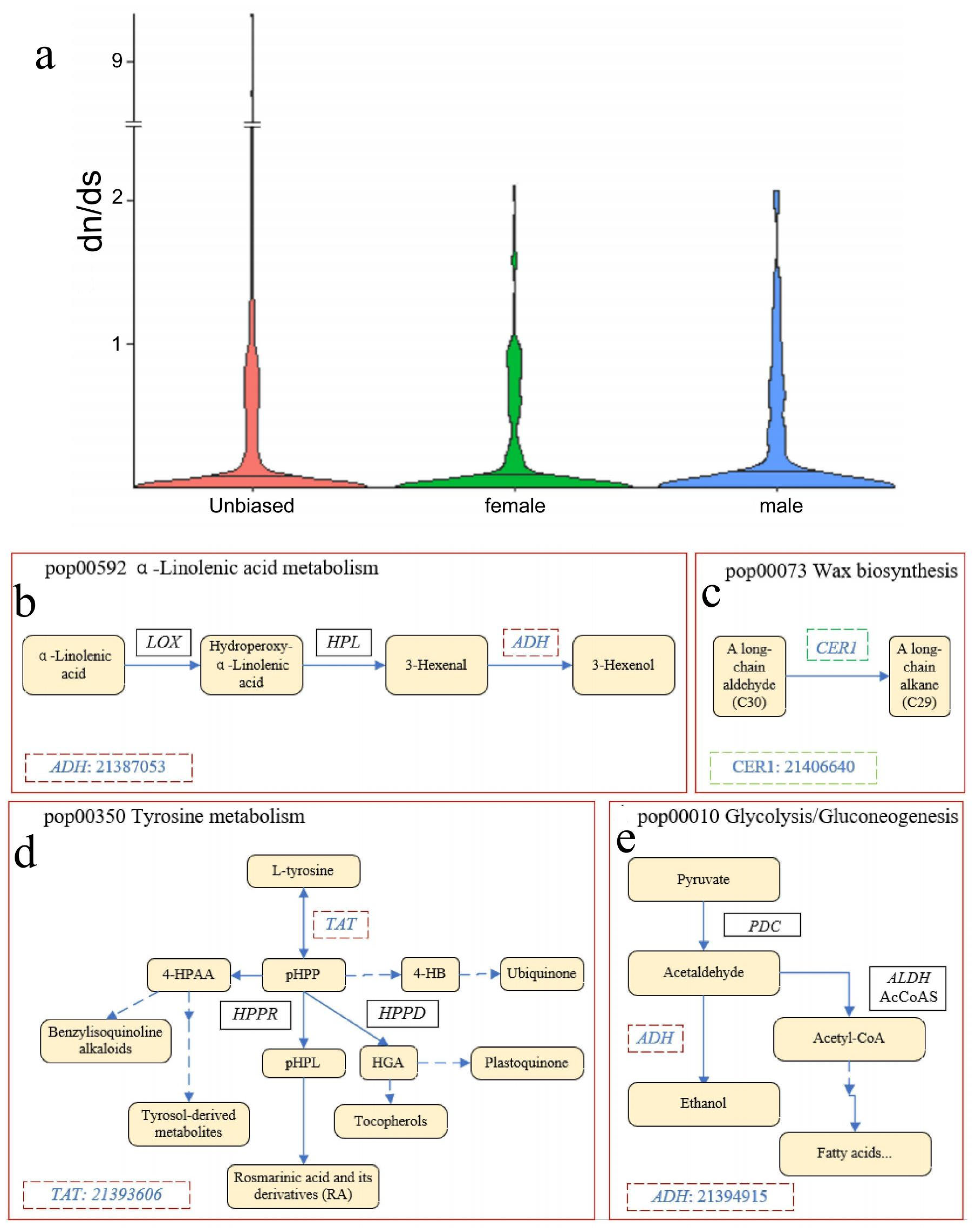
3.5. Evolution of Sex-Limited and Sex-Biased Expressed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Franklin, J.; Serra-Diaz, J.M.; Syphard, A.D.; Regan, H.M. Global change and terrestrial plant community dynamics. Proc. Natl. Acad. Sci. USA 2016, 113, 3725–3734. [Google Scholar] [CrossRef]
- Lamers, J.; Van Der Meer, T.; Testerink, C. How plants sense and respond to stressful environments. Plant Physiol. 2020, 182, 1624–1635. [Google Scholar] [CrossRef]
- Darolti, I.; Wright, A.E.; Pucholt, P.; Berlin, S.; Mank, J.E. Slow evolution of sex-biased genes in the reproductive tissue of the dioecious plant Salix viminalis. Mol. Ecol. 2018, 27, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H.; Parsch, J. The evolution of sex-biased genes and sex-biased gene expression. Nat. Rev. Genet. 2007, 8, 689–698. [Google Scholar] [CrossRef]
- Harrison, P.W.; Wright, A.E.; Zimmer, F.; Dean, R.; Montgomery, S.H.; Pointer, M.A.; Mank, J.E. Sexual selection drives evolution and rapid turnover of male gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 4393–4398. [Google Scholar] [CrossRef]
- Zemp, N.; Tavares, R.; Muyle, A.; Charlesworth, D.; Marais, G.A.B.; Widmer, A. Evolution of sex-biased gene expression in a dioecious plant. Nat. Plants 2016, 2, 16168. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, B.J.; Wang, L.; Tiffin, P.; Wu, Z.; Olson, M.S. Sex-biased gene expression in flowers, but not leaves, reveals secondary sexual dimorphism in Populus balsamifera. New Phytol. 2019, 221, 527–539. [Google Scholar] [CrossRef]
- Barrett, S.C.; Hough, J. Sexual dimorphism in flowering plants. J. Exp. Bot. 2013, 64, 67–82. [Google Scholar] [CrossRef]
- Zajitschek, F.; Bonduriansky, R.; Zajitschek, S.R.; Brooks, R.C. Sexual dimorphism in life history: Age, survival, and reproduction in male and female field crickets Teleogryllus commodus under seminatural conditions. Am. Nat. 2009, 173, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, C.; Alvarez, A.; Cuevas, E. Small but attractive: Female-biased nectar production and floral visitors in a dimorphic shrub. Plant Biol. 2018, 20, 160–164. [Google Scholar] [CrossRef]
- Tsuji, K.; Kobayashi, K.; Hasegawa, E.; Yoshimura, J. Dimorphic flowers modify the visitation order of pollinators from male to female flowers. Sci. Rep. 2020, 10, 9965. [Google Scholar] [CrossRef]
- Gershenzon, J.; Ullah, C. Plants protect themselves from herbivores by optimizing the distribution of chemical defenses. Proc. Natl. Acad. Sci. USA 2022, 119, e2120277119. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Oguro, M.; Itagaki, T.; Sakai, S. Floral-induced and constitutive defense against florivory: A comparison of chemical traits in 12 herb species. Plant Ecol. 2018, 219, 985–997. [Google Scholar] [CrossRef]
- Gao, L.; Yu, G.; Hu, F.; Li, Z.; Li, W.; Peng, C. The patterns of male and female flowers in flowering stage may not be optimal resource allocation for fruit and seed growth. Plants 2021, 10, 2819. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Auxin Biosynthesis and Its Role in Plant Development. Annu. Rev. Plant Biol. 2010, 61, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, L.-Y.; Zhou, P.; Liao, Z.; Lin, H.; Yu, Q.; Ming, R. Sex biased expression of hormone related genes at early stage of sex differentiation in papaya flowers. Hortic. Res. 2021, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Schenck, C.A.; Maeda, H.A. Tyrosine biosynthesis, metabolism, and catabolism in plants. Phytochemistry 2018, 149, 82–102. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, R.; Josephs, E.B.; Williamson, R.J.; Wright, S.I. Pollen-specific, but not sperm-specific, genes show stronger purifying selection and higher rates of positive selection than sporophytic genes in Capsella grandiflora. Mol. Biol. Evol. 2013, 30, 2475–2486. [Google Scholar] [CrossRef]
- Xuan, Y.; Ma, B.; Li, D.; Tian, Y.; Zeng, Q.; He, N. Chromosome restructuring and number change during the evolution of Morus notabilis and Morus alba. Hortic. Res. 2022, 9, uhab030. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Zhang, C.; Qi, X.; Zhao, S.; Tao, Y.; Yang, G.; Lee, T.-H.; Wang, X.; Cai, Q.; Li, D.; et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 2013, 4, 2445. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Luo, R.; Dai, X.; Liu, H.; Yu, G.; Han, S.; Lu, X.; Su, C.; Chen, Q.; Song, Q.; et al. Chromosome-level reference genome and population genomic analysis provide insights into the evolution and improvement of domesticated mulberry (Morus alba). Mol. Plant 2020, 13, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Kumar, A. Activity and isozymes of peroxidase in Morus nigra L. during sex differentiation. Z. Pflanzenphysiol. 1981, 102, 299–302. [Google Scholar] [CrossRef]
- Lal, M.; Jaiswal, V.S. Modification of flower sex and acid phosphatase activity by phthalimides in female plants of Morus nigra L. Plant Growth Regul. 1988, 7, 29–37. [Google Scholar] [CrossRef]
- Jolly, M.S.; Dandin, S.B.; Ravindran, S.; Kumar, R. Sexual polymorphism in the genus Morus L. Proc. Plant Sci. 1986, 96, 315–320. [Google Scholar] [CrossRef]
- Atsumi, R.; Nishihara, R.; Tarora, K.; Urasaki, N.; Matsumura, H. Identification of dominant genetic markers relevant to male sex determination in mulberry (Morus alba L.). Euphytica 2019, 215, 187. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010, 1. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- International Peach Genome Initiative; Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Molecular phylogenetics: Principles and practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Devani, R.S.; Sinha, S.; Banerjee, J.; Sinha, R.K.; Bendahmane, A.; Banerjee, A.K. De novo transcriptome assembly from flower buds of dioecious, gynomonoecious and chemically masculinized female Coccinia grandis reveals genes associated with sex expression and modification. BMC Plant Biol. 2017, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhang, D. Roles of jasmonate signalling in plant inflorescence and flower development. Curr. Opin. Plant Biol. 2015, 27, 44–51. [Google Scholar] [CrossRef] [PubMed]
- McKown, A.D.; Klápště, J.; Guy, R.D.; Soolanayakanahally, R.Y.; La Mantia, J.; Porth, I.; Skyba, O.; Unda, F.; Douglas, C.J.; El-Kassaby, Y.A.; et al. Sexual homomorphism in dioecious trees: Extensive tests fail to detect sexual dimorphism in Populus. Sci. Rep. 2017, 7, 1831. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Delhomme, N.; Mähler, N.; Schiffthaler, B.; Önskog, J.; Albrectsen, B.R.; Ingvarsson, P.K.; Hvidsten, T.R.; Jansson, S.; Street, N.R. Populus tremula (European aspen) shows no evidence of sexual dimorphism. BMC Plant Biol. 2014, 14, 276. [Google Scholar] [CrossRef] [PubMed]
- Harkess, A.; Mercati, F.; Shan, H.; Sunseri, F.; Falavigna, A.; Leebens-Mack, J. Sex-biased gene expression in dioecious garden asparagus (Asparagus officinalis). New Phytol. 2015, 207, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Bourdenx, B.; Bernard, A.; Domergue, F.; Pascal, S.; Léger, A.; Roby, D.; Pervent, M.; Vile, D.; Haslam, R.P.; Napier, J.A.; et al. Overexpression of Arabidopsis ECERIFERUM1 promotes wax very-long-chain alkane biosynthesis and influences plant response to biotic and abiotic stresses. Plant Physiol. 2011, 156, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Strommer, J. The plant ADH gene family. Plant J. 2011, 66, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dong, Q.; Duan, D.; Zhao, S.; Li, M.; van Nocker, S.; Ma, F.; Mao, K. Comprehensive genomic analysis of the TYROSINE AMINOTRANSFERASE (TAT) genes in apple (Malus domestica) allows the identification of MdTAT2 conferring tolerance to drought and osmotic stresses in plants. Plant Physiol. Biochem. 2018, 133, 81–91. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Raw Reads | Clean Reads | Clean Bases | Error Rate | Q30 | GC% | Mapping Ratio |
|---|---|---|---|---|---|---|---|
| MBT-1 | 52,365,500 | 50,926,984 | 7.64 G | 0.03 | 93.12 | 44.31 | 64.6 |
| MBT-2 | 44,385,448 | 41,784,712 | 6.27 G | 0.03 | 93.55 | 42.81 | 58.4 |
| MBT-3 | 49,325,358 | 46,960,136 | 7.04 G | 0.03 | 92.19 | 40.73 | 72.8 |
| FY20-1 | 56,900,778 | 54,807,454 | 8.22 G | 0.03 | 92.27 | 44.23 | 55.8 |
| FY20-2 | 48,518,050 | 47,476,836 | 7.12 G | 0.03 | 93.46 | 44.66 | 66.9 |
| FY20-3 | 54,147,582 | 52,488,884 | 7.87 G | 0.03 | 92.38 | 43.44 | 68.5 |
| Gene ID | Gene Description | Gene ID | Gene Description |
|---|---|---|---|
| Female-Biased | Male-Biased | ||
| 21400326 | auxin-responsive protein IAA32 | 21398828 | stamen-specific protein FIL 1 |
| 21407698 | auxin transporter-like protein 2 | 21393606 | probable aminotransferase TAT2 |
| 21402398 | auxin-induced protein AUX22 | 21396084 | pollen-specific leucine-rich repeat extension-like protein 3 |
| 21400361 | auxin efflux carrier component 3 | 21401585 | pollen receptor-like kinase 1 |
| 21399379 | auxin-responsive protein IAA4 | 21391819 | vesicle-associated protein 2-2 |
| 21406640 | protein eceriferum 2C transcript variant X2 | 21394915 | alcohol dehydrogenase-like 2C transcript variant X1 |
| 21390461 | protein PIN-LIKES | 21387539 | PHD finger protein male sterility1 |
| 21400414 | transcriptional regulator SUPERMAN | 21408700 | transcription factor DYT1 |
| 21397544 | auxin response factor 2C transcript variant X1 | 21387503 | plant UBX domain-containing protein 2 |
| 21387910 | nudix hydrolase 2C transcript variant X2 | 21390125 | endoglucanase 2C transcript variant X1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Ackah, M.; Amoako, F.K.; Zhao, M.; van der Puije, G.C.; Zhao, W. The Mechanism of the Development and Maintenance of Sexual Dimorphism in the Dioecious Mulberry Plant (Morus alba). Biology 2024, 13, 622. https://doi.org/10.3390/biology13080622
Shi Y, Ackah M, Amoako FK, Zhao M, van der Puije GC, Zhao W. The Mechanism of the Development and Maintenance of Sexual Dimorphism in the Dioecious Mulberry Plant (Morus alba). Biology. 2024; 13(8):622. https://doi.org/10.3390/biology13080622
Chicago/Turabian StyleShi, Yisu, Michael Ackah, Frank Kwarteng Amoako, Mengdi Zhao, Grace C. van der Puije, and Weiguo Zhao. 2024. "The Mechanism of the Development and Maintenance of Sexual Dimorphism in the Dioecious Mulberry Plant (Morus alba)" Biology 13, no. 8: 622. https://doi.org/10.3390/biology13080622
APA StyleShi, Y., Ackah, M., Amoako, F. K., Zhao, M., van der Puije, G. C., & Zhao, W. (2024). The Mechanism of the Development and Maintenance of Sexual Dimorphism in the Dioecious Mulberry Plant (Morus alba). Biology, 13(8), 622. https://doi.org/10.3390/biology13080622