
Modulator Effect of AT1 Receptor Knockdown on THP-1 Macrophage Proinflammatory Activity
 , , , and
, , , and
Abstract
Simple Summary
Abstract

1. Introduction
2. Material and Methods
2.1. Reagents
2.2. Proliferation and Differentiation of THP-1
2.3. Culture Conditions with Treatments
2.4. Small Interfering RNA (siRNA)
2.5. RNA Extraction and Genetic Expression of Cytokines
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Statistical Analysis
3. Results
3.1. The Effect of AT1R siRNA
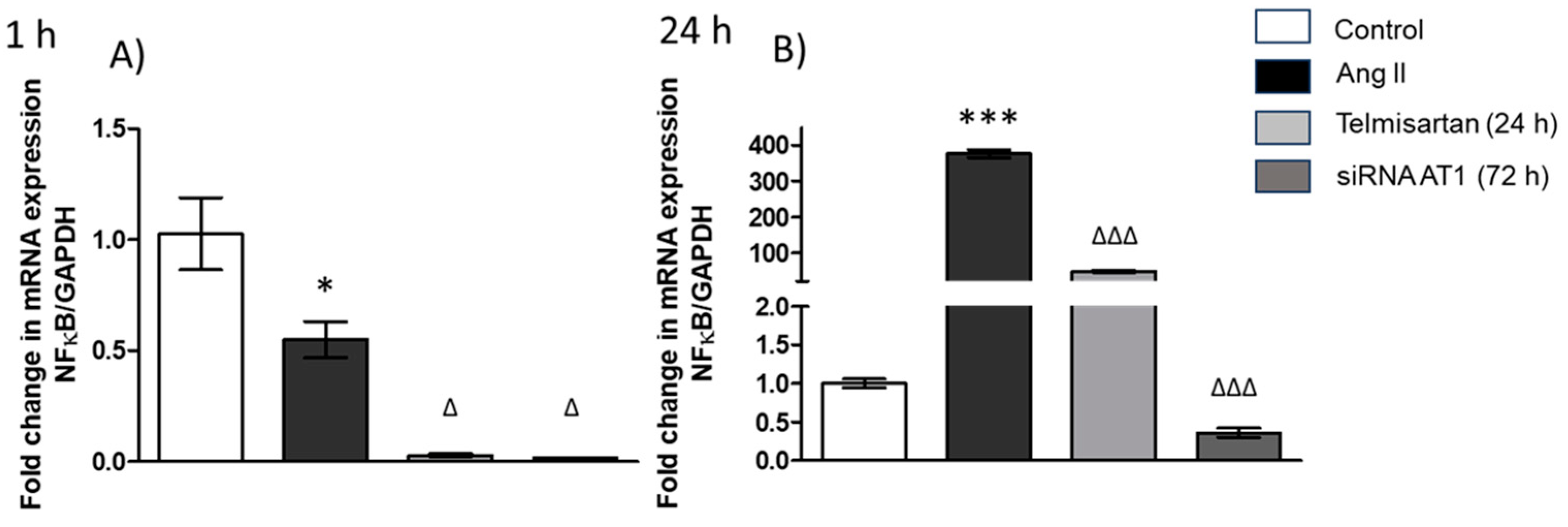
3.2. siRNA AT1R Modulates NF-κB Expression in THP-1 Cells
3.3. AT1R siRNA Changes the Gene Expression of Cytokines in Macrophages
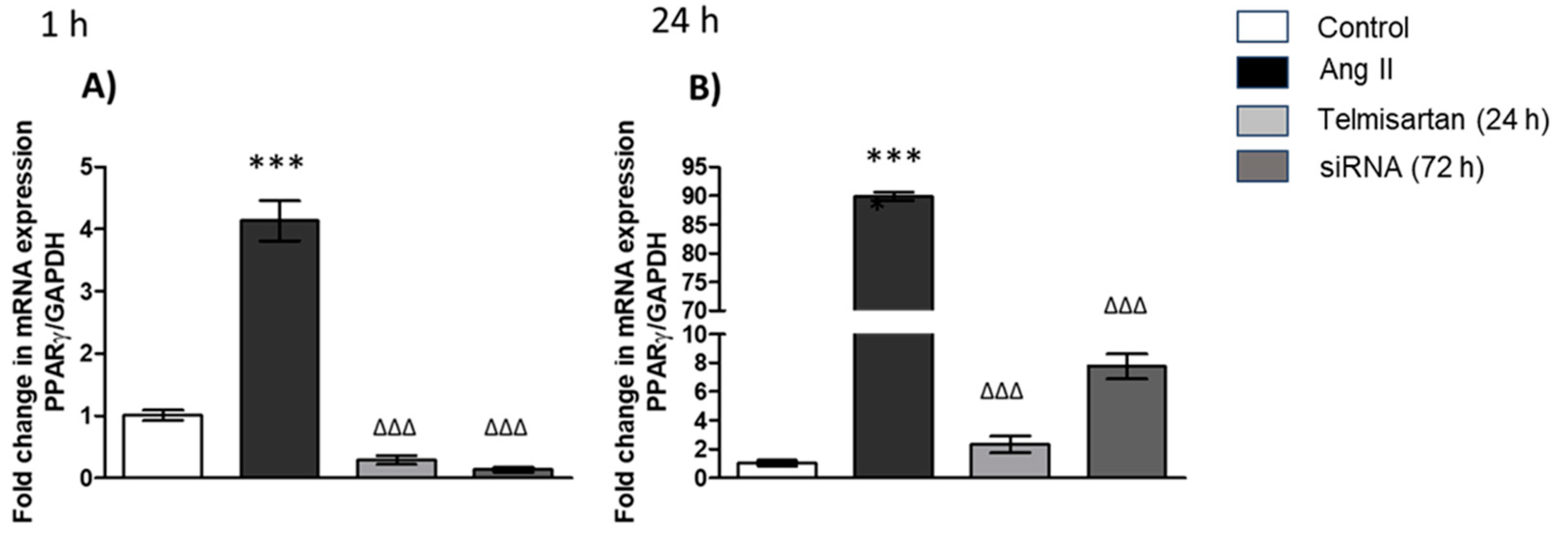
3.4. PPAR-γ Gene Expression in Macrophages
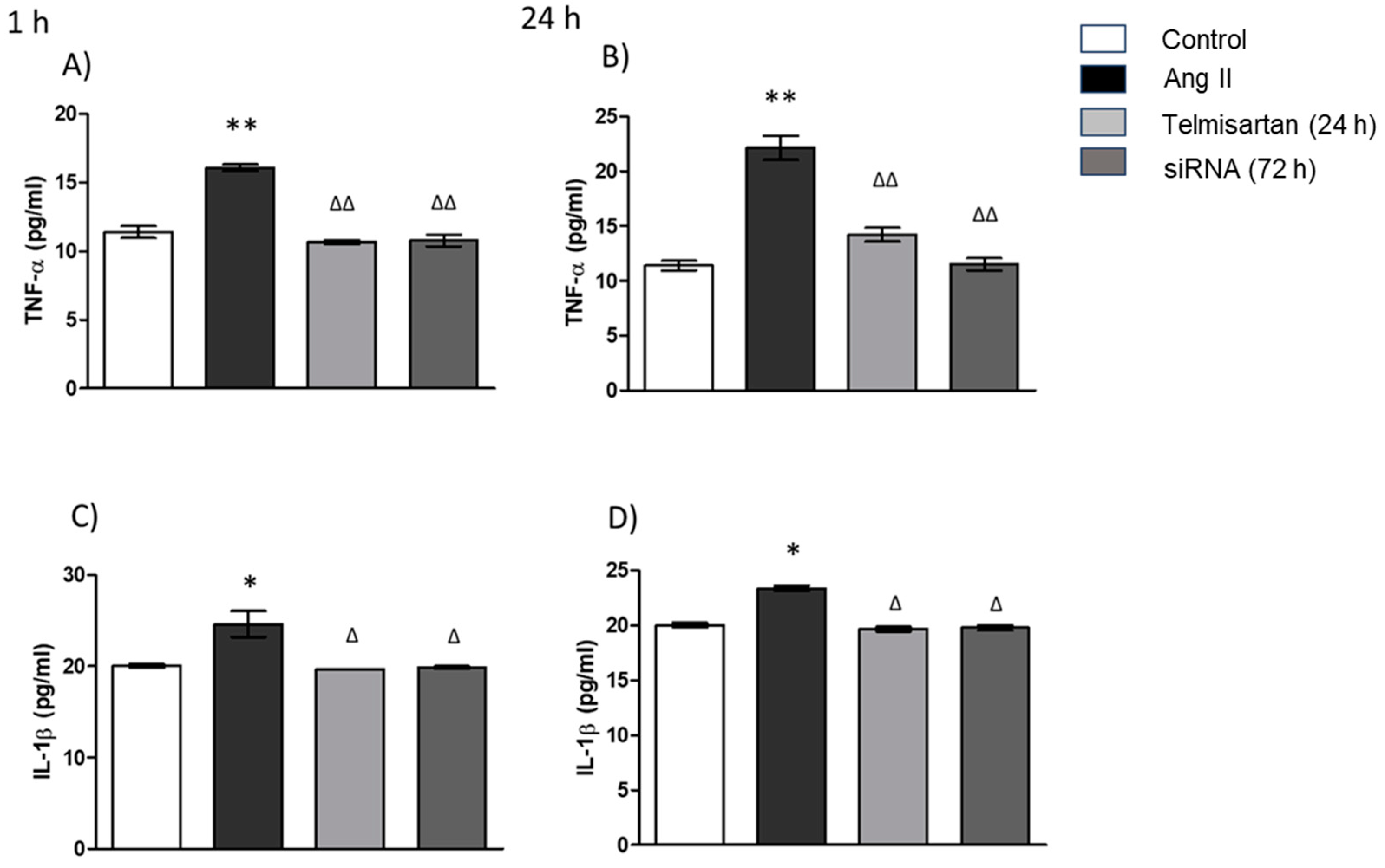
3.5. AT1 siRNA Inhibits Ang II-Induced Inflammation in THP-1 Macrophages by Regulating TNF-α and IL-1β Secretion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cao, J.; Chen, C.; Wang, Y.; Chen, X.; Chen, Z.; Luo, X. Influence of autologous dendritic cells on cytokine-induced killer cell proliferation, cell phenotype and antitumor activity in vitro. Oncol. Lett. 2016, 12, 2033–2037. [Google Scholar] [CrossRef] [PubMed]
- Kiran, S.; Kumar, V.; Murphy, E.A.; Enos, R.T.; Singh, U.P. High Fat Diet-Induced CD8+ T Cells in Adipose Tissue Mediate Macrophages to Sustain Low-Grade Chronic Inflammation. Front. Immunol. 2021, 12, 680944. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef]
- Li, Y.; Wang, R.; Gao, Q. The Roles and Targeting of Tumor-Associated Macrophages. Front. Biosci. 2023, 28, 207. [Google Scholar] [CrossRef]
- Tyurin-Kuzmin, P.A.; Kalinina, N.I.; Kulebyakin, K.Y.; Balatskiy, A.V.; Sysoeva, V.Y.; Tkachuk, V.A. Angiotensin receptor subtypes regulate adipose tissue renewal and remodelling. FEBS J. 2020, 287, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Sanghavi, D.K.; Morris, D.L.; Kahwaji, C.I. Angiotensin II. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Suzuki, Y.; Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Egido, J. Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 2003, 35, 881–900. [Google Scholar] [CrossRef]
- Shepherd, A.J.; Copits, B.A.; Mickle, A.D.; Karlsson, P.; Kadunganattil, S.; Haroutounian, S.; Tadinada, S.M.; de Kloet, A.D.; Valtcheva, M.V.; McIlvried, L.A.; et al. Angiotensin II triggers peripheral macrophage-to-sensory neuron redox crosstalk to elicit pain. J. Neurosci. 2018, 38, 7032–7057. [Google Scholar] [CrossRef]
- Montezano, A.C.; Nguyen Dinh Cat, A.; Rios, F.J.; Touyz, R.M. Angiotensin II and vascular injury. Curr. Hypertens. Rep. 2014, 16, 1–11. [Google Scholar] [CrossRef]
- Wang, C.H.; Li, S.H.; Weisel, R.D.; Fedak, P.W.; Dumont, A.S.; Szmitko, P.; Li, R.K.; Mickle, D.A.; Verma, S. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation 2003, 107, 1783–1790. [Google Scholar] [CrossRef]
- Skultetyova, D.; Filipova, S.; Riecansky, I.; Skultety, J. The role of angiotensin type 1 receptor in inflammation and endothelial dysfunction. Recent Pat. Cardiovasc. Drug Discov. 2007, 2, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Huang, C.; Lin, Z.; Zhan, S.; Kong, L.; Fang, C.; Li, J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014, 26, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Rafatian, N.; Milne, R.W.; Leenen, F.H.H.; Whitman, S.C. Role of renin-angiotensin system in activation of macrophages by modified lipoproteins. Am. J. Physiol. Circ. Physiol. 2013, 305, H1309–H1320. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.M.A.; Hussain, S.A.; Mirza, R.R. Azilsartan improves the efects of etanercept in patients with active rheumatoid arthritis: A pilot study. Ther. Clin. Risk Manag. 2018, 14, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Chen, X.L.; Wang, F.; Liang, X.; Sun, Y.X.; Wang, Y.J. Role of angiotensin II type 1 receptor in angiotensin II-induced cytokine production in macrophages. J. Interferon Cytokine Res. 2011, 31, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Kranzhöfer, R.; Browatzki, M.; Schmidt, J.; Kübler, W. Angiotensin II activates the proinflammatory transcription factor nuclear factor-κB in human monocytes. Biochem. Biophys. Res. Commun. 1999, 257, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, K.; Xiao, J.; Xin, J.; Zhang, L.; Li, X.; Li, L.; Si, J.; Wang, L.; Ma, K. Angiotensin II induces RAW264. 7 macrophage polarization to the M1-type through the connexin 43/NF-κB pathway. Mol. Med. Rep. 2020, 21, 2103–2112. [Google Scholar] [PubMed]
- Wienen, W.; Hauel, N.; Van Meel JC, A.; Narr, B.; Ries, U.; Entzeroth, M. Pharmacological characterization of the novel nonpeptide angiotensin II receptor antagonist, BIBR 277. Br. J. Pharmacol. 1993, 110, 245–252. [Google Scholar] [CrossRef]
- Deppe, S.; Böger, R.H.; Weiss, J.; Benndorf, R.A. Telmisartan: A review of its pharmacodynamic and pharmacokinetic properties. Expert Opin. Drug Metab. Toxicol. 2010, 6, 863–871. [Google Scholar] [CrossRef]
- Shen, X.; Li, H.; Li, W.; Wu, X.; Sun, Z.; Ding, X. Telmisartan ameliorates adipoR1 and adipoR2 expression via PPAR-γ activation in the coronary artery and VSMCs. Biomed. Pharmacother. 2017, 95, 129–136. [Google Scholar] [CrossRef]
- Wen, J.; Zeng, M.; Liu, Z.; Zhou, H.; Xu, H.; Huang, M.; Zhang, W. The influence of telmisartan on metformin pharmacokinetics and pharmacodynamics. J. Pharmacol. Sci. 2019, 139, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Fakhr, E.; Zare, F.; Teimoori-Toolabi, L. Precise and efficient siRNA design: A key point in competent gene silencing. Cancer Gene Ther. 2016, 23, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Liu, H.; Zeng, Z.; Liang, Z.; Xie, H.; Li, W.; Xiong, L.; Liu, Z.; Chen, M.; Jie, H.; et al. KRT17 Promotes T-lymphocyte Infiltration Through the YTHDF2–CXCL10 Axis in Colorectal Cancer. Cancer Immunol. Res. 2023, 11, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Ranjit, A.; Khajehpour, S.; Aghazadeh-Habashi, A. Update on Angiotensin II Subtype 2 Receptor: Focus on Peptide and Nonpeptide Agonists. Mol. Pharmacol. 2021, 99, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Vasile, S.; Hallberg, A.; Sallander, J.; Hallberg, M.; Åqvist, J.; Gutiérrez-de-Terán, H. Evolution of Angiotensin Peptides and Peptidomimetics as Angiotensin II Receptor Type 2 (AT2) Receptor Agonists. Biomolecules 2020, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Hammer, A.; Stegbauer, J.; Linker, R.A. Macrophages in neuroinflammation: Role of the renin-angiotensin-system. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 431–444. [Google Scholar] [CrossRef]
- Lawrence, T. Macrophages and NF-κB in cancer. Curr. Top. Microbiol. Immunol. 2010, 349, 171–184. [Google Scholar]
- Mao, X.; Chen, Y.; Lu, X.; Jin, S.; Jiang, P.; Deng, Z.; Zhu, X.; Cai, Q.; Wu, C.; Kang, S. Tissue resident memory T cells are enriched and dysfunctional in effusion of patients with malignant tumor. J. Cancer 2023, 14, 1223–1231. [Google Scholar] [CrossRef]
- López-Bojorquez, L.N. La regulación del factor de transcripción NF-κB. Un mediador molecular en el proceso inflamatorio [Regulation of NF-κB transcription factor. A molecular mediator in inflammatory process]. Rev. Investig. Clin. 2004, 56, 83–92. (In Spanish) [Google Scholar]
- Alique, M.; Sánchez-López, E.; Rayego-Mateos, S.; Egido, J.; Ortiz, A.; Ruiz-Ortega, M. Angiotensin II, via angiotensin receptor type 1/nuclear factor-κB activation, causes a synergistic effect on interleukin-1-β-induced inflammatory responses in cultured mesangial cells. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, T.; Du, Y.; Pan, D.; Wu, W.; Zhu, H.; Zhang, Y.; Li, D. Salvianolic Acid A Attenuates Cell Apoptosis, Oxidative Stress, Akt and NF-κB Activation in Angiotensin-II Induced Murine Peritoneal Macrophages. Curr. Pharm. Biotechnol. 2016, 17, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Suzuki, Y.; Rupérez, M.; Egido, J. Proinflammatory actions of angiotensins. Curr. Opin. Nephrol. Hypertens. 2001, 10, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Dhindsa, S.; Ghanim, H.; Chaudhuri, A. Angiotensin II and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J. Hum. Hypertens. 2007, 21, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Aki, K.; Shimizu, A.; Masuda, Y.; Kuwahara, N.; Arai, T.; Ishikawa, A.; Fujita, E.; Mii, A.; Natori, Y.; Fukunaga, Y.; et al. ANG II receptor blockade enhances anti-inflammatory macrophages in anti-glomerular basement membrane glomerulonephritis. Am. J. Physiol.-Ren. Physiol. 2010, 298, F870–F882. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yuan, L.; Li, J.; Li, H.; Cheng, S. Blockade of renin angiotensin system increased resistance to STZ-induced diabetes in rats with long-term high-fat diet. Exp. Diabetes Res. 2012, 2012, 618923. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yancey, P.G.; Zuo, Y.; Ma, L.J.; Kaseda, R.; Fogo, A.B.; Ichikawa, I.; Linton, M.F.; Fazio, S.; Kon, V. Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; McKarns, S.; DeMarco, V.G.; Jia, G.; Sowers, J.R. Maladaptive immune and inflammatory pathways lead to cardiovascular insulin resistance. Metabolism 2013, 62, 1543–1552. [Google Scholar] [CrossRef]
- Sriramula, S.; Haque, M.; Majid, D.S.; Francis, J. Involvement of tumor necrosis factor-α in angiotensin II–mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 2008, 51, 1345–1351. [Google Scholar] [CrossRef]
- Wu, R.; Xiong, J.; Zhou, T.; Zhang, Z.; Huang, Z.; Tian, S.; Wang, Y. Quercetin/Anti-PD-1 Antibody Combination Therapy Regulates the Gut Microbiota, Impacts Macrophage Immunity and Reshapes the Hepatocellular Carcinoma Tumor Microenvironment. Front. Biosci. 2023, 28, 327. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.; Cosarderelioglu, C.; Vajapey, R.; Walston, J.; Abadir, P.M. Losartan Mitigates Oxidative Stress in the Brains of Aged and Inflamed IL-10−/− Mice. J. Gerontol. Ser. A 2022, 77, 1784–1788. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Galan, M.; Partyka, M.; Trebak, M.; Matrougui, K. Interleukin-10 released by CD4+ CD25+ natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Dhande, I.; Ma, W.; Hussain, T. Angiotensin AT2 receptor stimulation is anti-inflammatory in lipopolysaccharide-activated THP-1 macrophages via increased interleukin-10 production. Hypertens. Res. 2015, 38, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Xu, Z.; Chen, L.F.; Wang, W.; Fang, Q.; Yan, X.W. Valsartan and telmisartan abrogate angiotensin II-induced downregulation of ABCA1 expression via AT1 receptor, rather than AT2 receptor or PPARγ activation. J. Cardiovasc. Pharmacol. 2012, 59, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, M.; Sakoda, H.; Kushiyama, A.; Fujishiro, M.; Ohno, H.; Nakatsu, Y.; Fukushima, T.; Kumamoto, S.; Tsuchiya, Y.; Kikuchi, T.; et al. Valsartan, independently of AT1 receptor or PPARγ, suppresses LPS-induced macrophage activation and improves insulin resistance in cocultured adipocytes. Am. J. Physiol.-Endocrinol. Metab. 2012, 302, E286–E296. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Zhang, Y.; Li, W.; Wang, L.; Wu, Y.; Yuan, Z.; Zhou, J. The role of monocyte chemotactic protein-induced protein 1 (MCPIP1) in angiotensin II-induced macrophage apoptosis and vulnerable plaque formation. Biochem. Biophys. Res. Commun. 2019, 515, 378–385. [Google Scholar] [CrossRef]
- Nakaya, K.; Ayaori, M.; Hisada, T.; Sawada, S.; Tanaka, N.; Iwamoto, N.; Ogura, M.; Yakushiji, E.; Kusuhara, M.; Nakamura, H.; et al. Telmisartan enhances cholesterol efflux from THP-1 macrophages by activating PPARγ. J. Atheroscler. Thromb. 2007, 14, 133–141. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 5′ to 3′ | Primers | Accession Number |
|---|---|---|---|
| AT1 receptor (AGTR1) | Sense | GCACTGGCTGACTTATGCTT | NM_000685.5 |
| Anti-sense | GTTGAAACTGACGCTGGCTG | ||
| NF-κB | Sense | GGGCAGACCAGTGTCATTGA | NM_001077494.3 |
| Anti-sense | GTTGGTGAGGTTGACAACGC | ||
| TNF-α | Sense | GTGCTTGTTCCTCAGCCTCT | NM_000594.4 |
| Anti-sense | TAGAGAGAGGTCCCTGGGGA | ||
| IL-1β | Sense | AACGAGGCTTATGTGCACGA | NM_000576.3 |
| Anti-sense | TATCCTGTCCCTGGAGGTGG | ||
| IL-10 | Sense | CCAGTCTGAGAACAGCTGCA | NM_000572.3 |
| Anti-sense | TCCTCCAGCAAGGACTCCTT | ||
| PPAR-γ | Sense | CCAGAAGCCTGCATTTCTGC | NM_001330615.4 |
| Anti-sense | CACGGAGCTGATCCCAAAGT | ||
| GAPDH | Sense | CATCCTGGGCTACACTGAGC | NM_001256799.3 |
| Anti-sense | GTCAAAGGTGGAGGAGTGGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acevedo-Villavicencio, L.N.; López-Luna, C.E.; Castillo-Cruz, J.; Gutiérrez-Rojas, R.A.; Paredes-González, I.S.; Villafaña, S.; Huang, F.; Vargas-De-León, C.; Romero-Nava, R.; Aguayo-Cerón, K.A. Modulator Effect of AT1 Receptor Knockdown on THP-1 Macrophage Proinflammatory Activity. Biology 2024, 13, 382. https://doi.org/10.3390/biology13060382
Acevedo-Villavicencio LN, López-Luna CE, Castillo-Cruz J, Gutiérrez-Rojas RA, Paredes-González IS, Villafaña S, Huang F, Vargas-De-León C, Romero-Nava R, Aguayo-Cerón KA. Modulator Effect of AT1 Receptor Knockdown on THP-1 Macrophage Proinflammatory Activity. Biology. 2024; 13(6):382. https://doi.org/10.3390/biology13060382
Chicago/Turabian StyleAcevedo-Villavicencio, Lourdes Nallely, Carlos Enrique López-Luna, Juan Castillo-Cruz, Rocío Alejandra Gutiérrez-Rojas, Iris Selene Paredes-González, Santiago Villafaña, Fengyang Huang, Cruz Vargas-De-León, Rodrigo Romero-Nava, and Karla Aidee Aguayo-Cerón. 2024. "Modulator Effect of AT1 Receptor Knockdown on THP-1 Macrophage Proinflammatory Activity" Biology 13, no. 6: 382. https://doi.org/10.3390/biology13060382
APA StyleAcevedo-Villavicencio, L. N., López-Luna, C. E., Castillo-Cruz, J., Gutiérrez-Rojas, R. A., Paredes-González, I. S., Villafaña, S., Huang, F., Vargas-De-León, C., Romero-Nava, R., & Aguayo-Cerón, K. A. (2024). Modulator Effect of AT1 Receptor Knockdown on THP-1 Macrophage Proinflammatory Activity. Biology, 13(6), 382. https://doi.org/10.3390/biology13060382