
A Full-Length Transcriptome and Analysis of the NHL-1 Gene Family in Neocaridina denticulata sinensis




Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Shrimp Culture and Samples
2.2. RNA Extraction, SMRT Library Preparation, and Sequencing
2.3. Data Processing
2.4. Alternative Splicing, Transcription Factor, and Long Non-Coding RNA Analysis
2.5. Gene Analysis
3. Results
3.1. Overview of Full-Length Transcript Sequencing Data
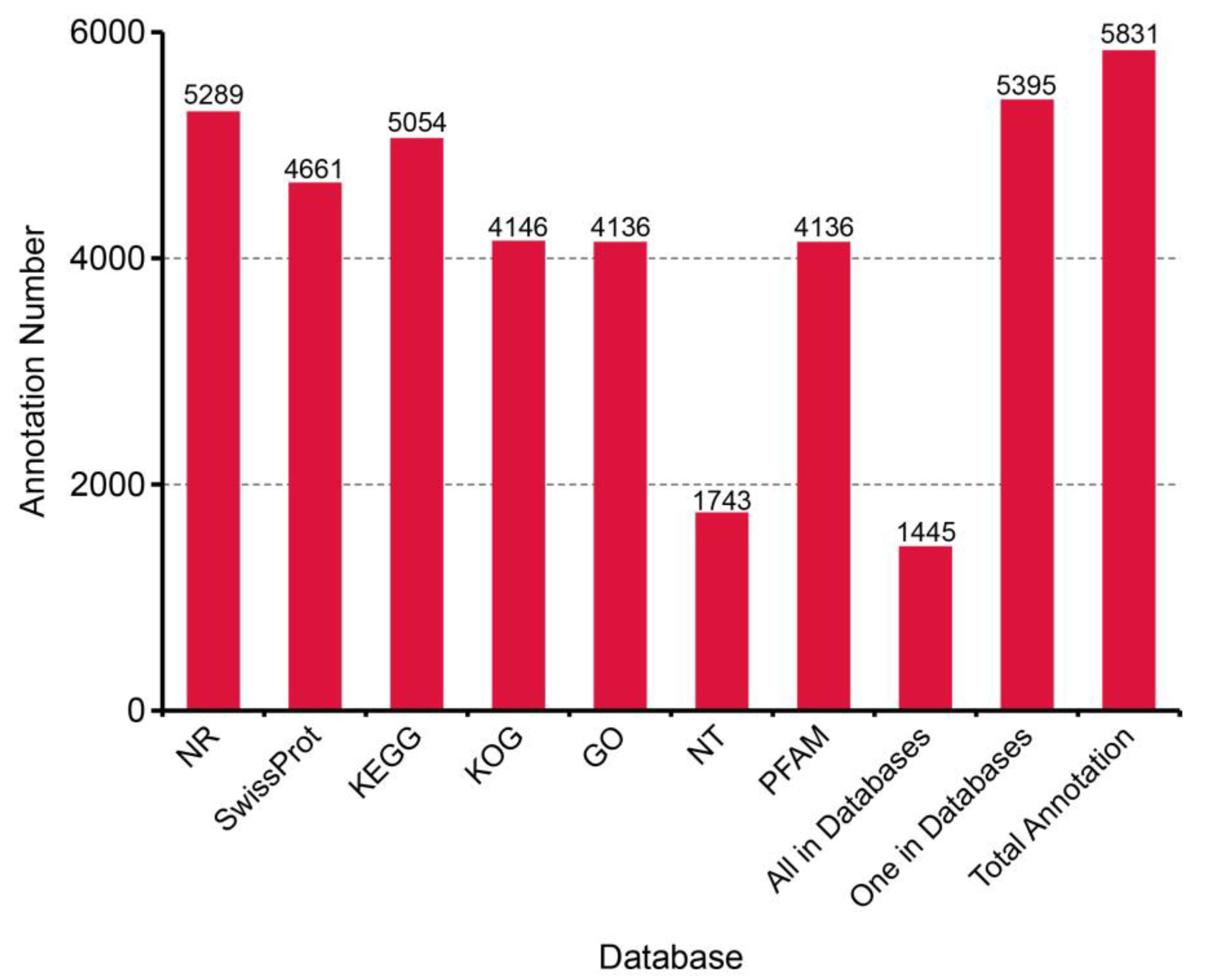
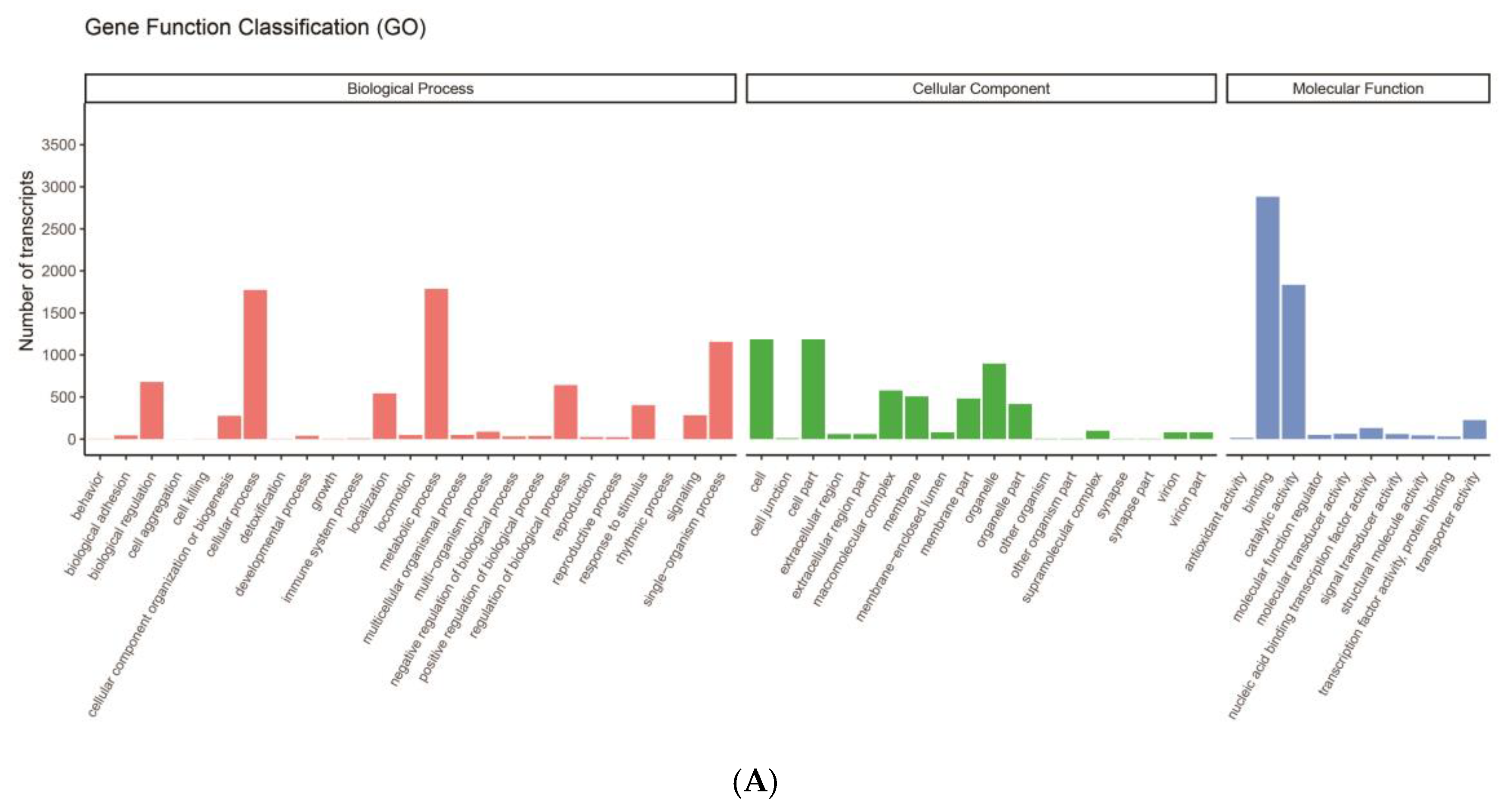
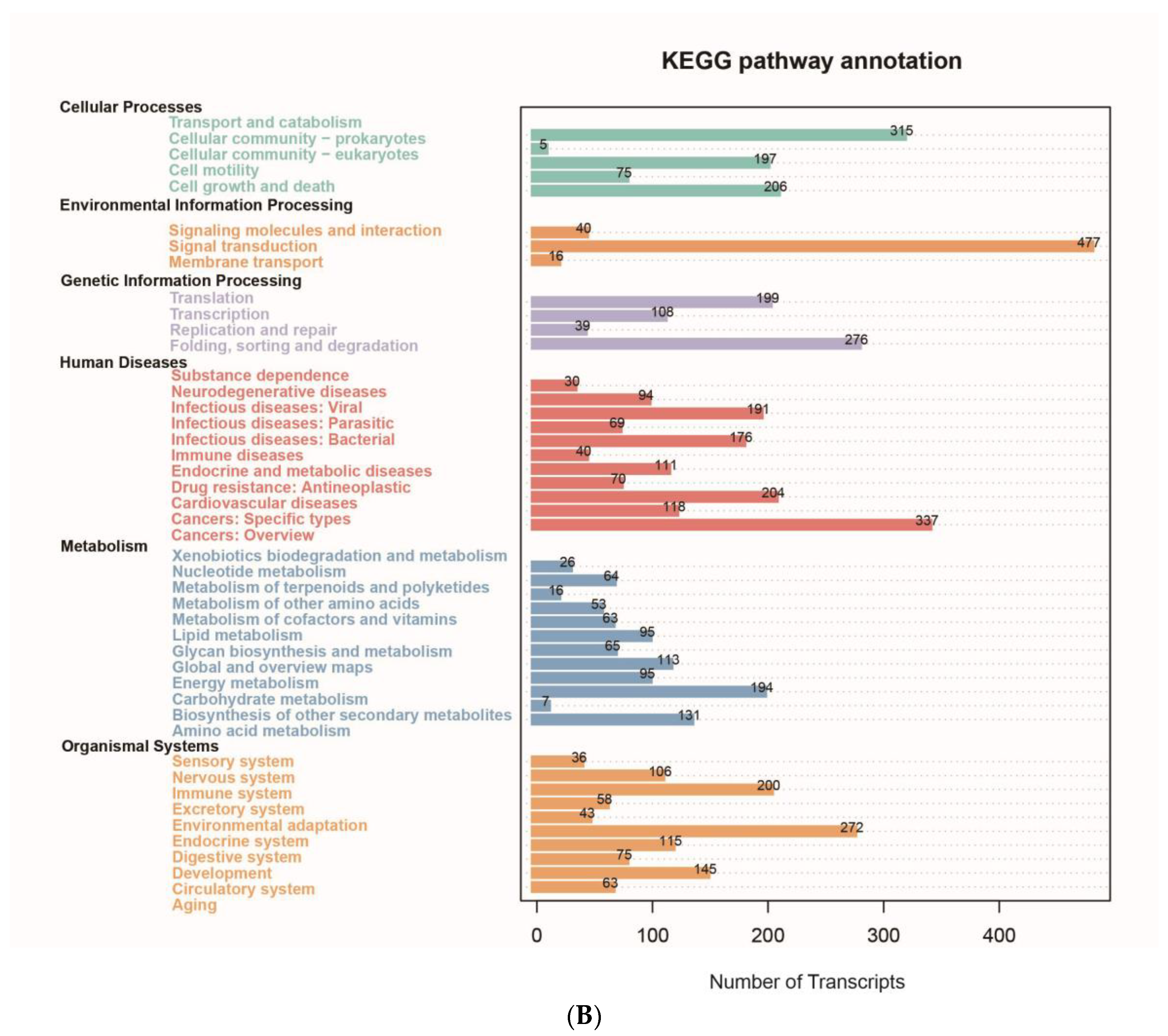
3.2. Classification and Functional Annotation of Transcripts
3.3. Alternative Splicing and Transcription Factor Analysis
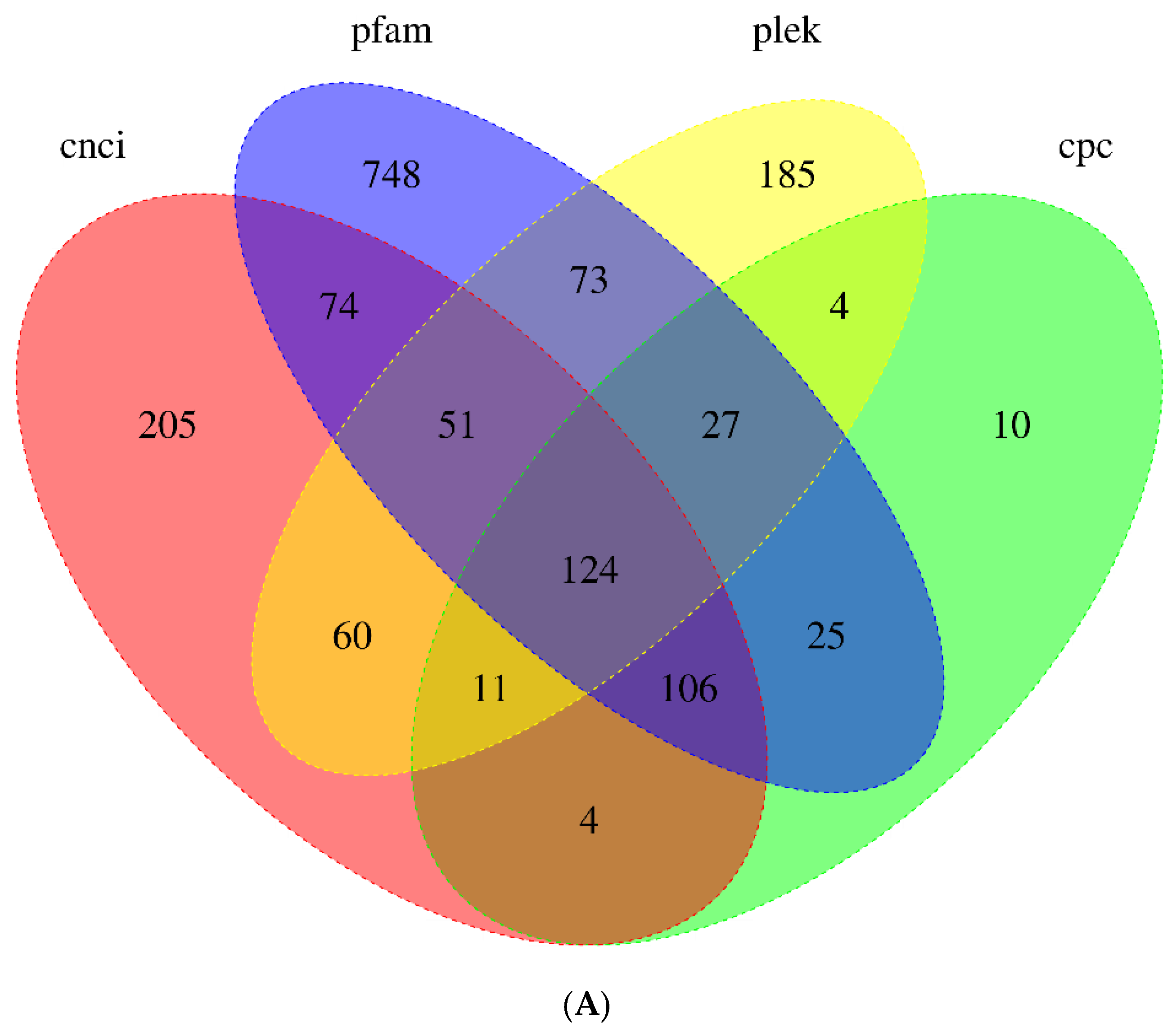
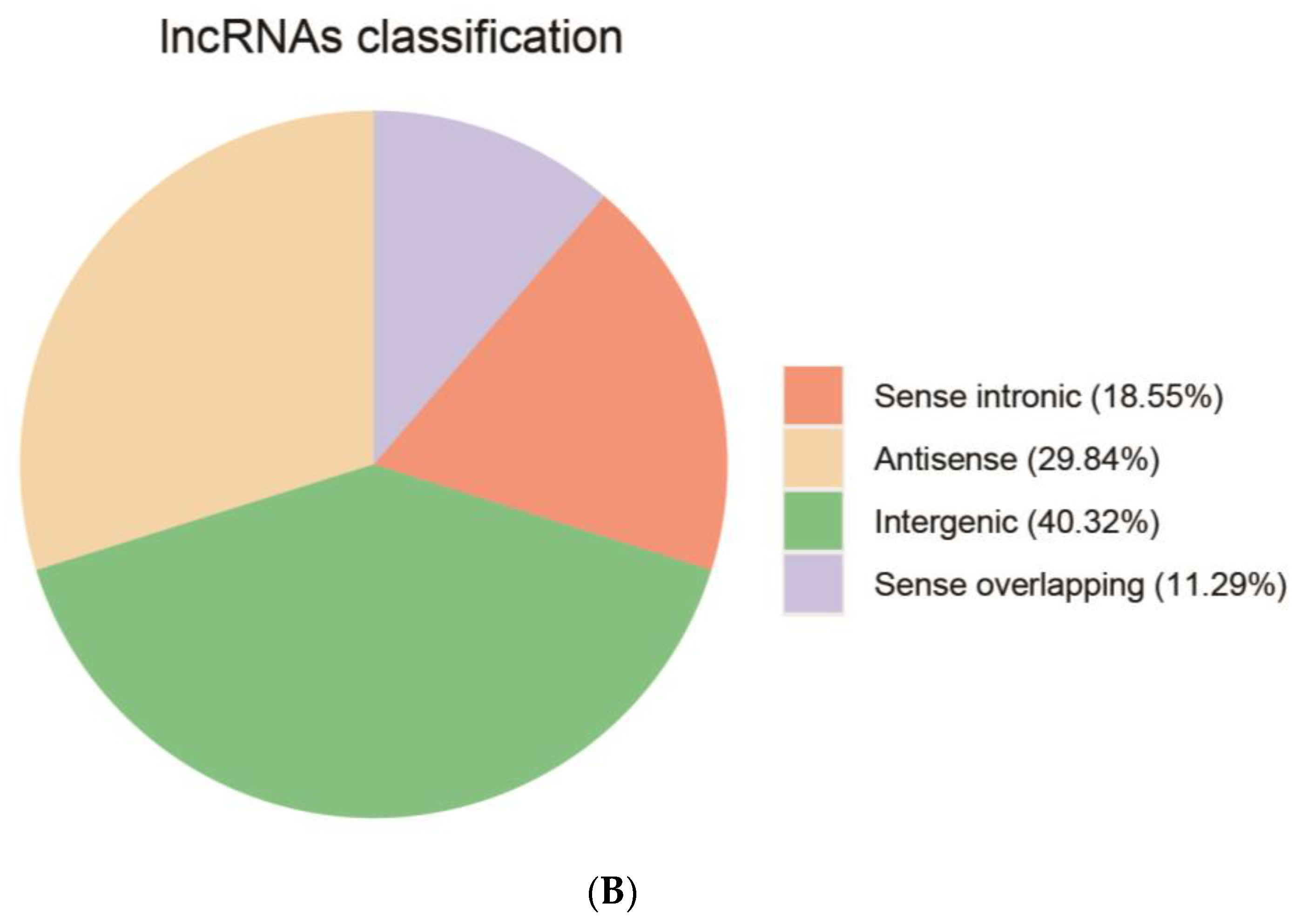
3.4. Identification of LncRNAs
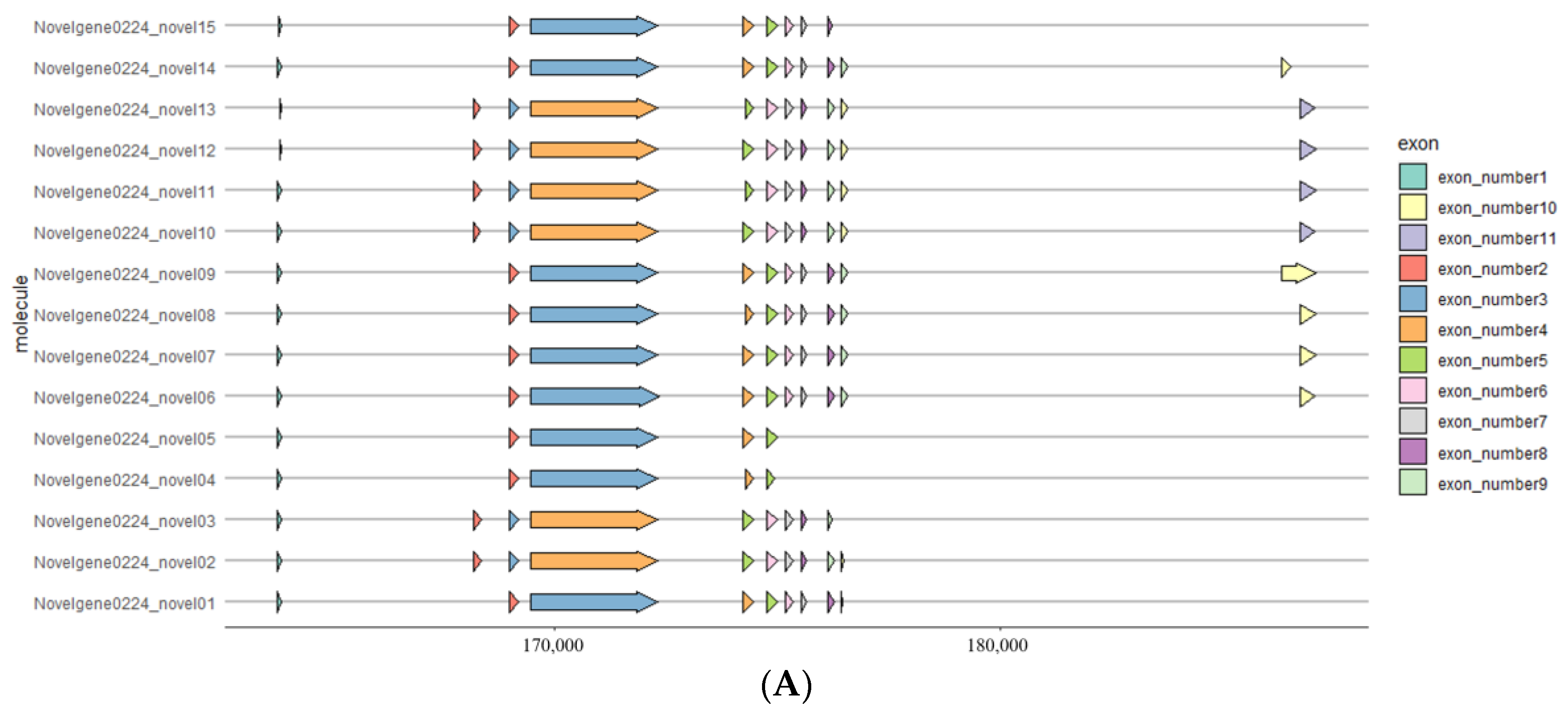
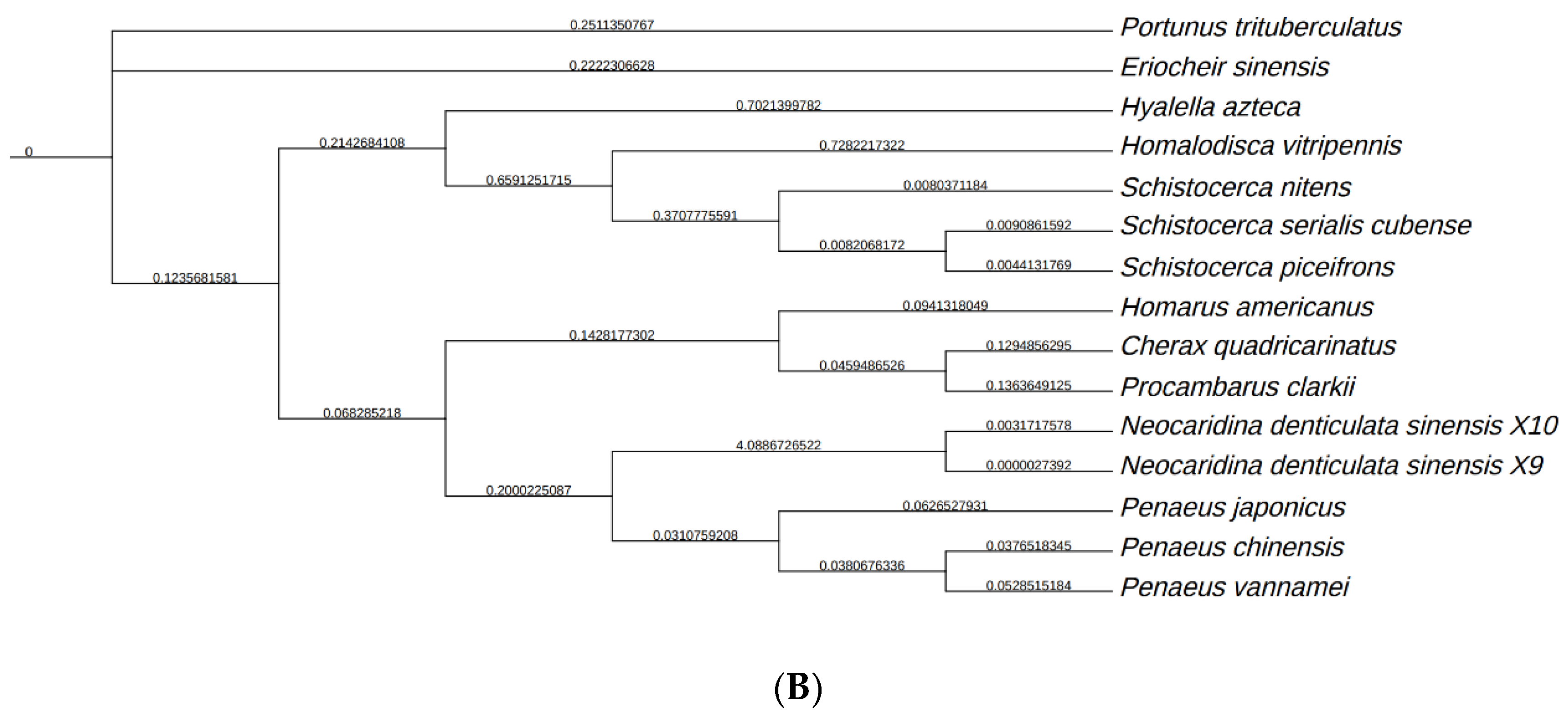
3.5. Analysis of NdNHL-1
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mykles, D.L.; Hui, J.H. Neocaridina denticulata: A Decapod Crustacean Model for Functional Genomics. Integr. Comp. Biol. 2015, 55, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Chen, W.Y.; Wu, C.C.; Lu, E.; Shih, W.Y.; Chen, J.W.; Tsai, J.W. Ecosystem metabolism regulates seasonal bioaccumulation of metals in atyid shrimp (Neocaridina denticulata) in a tropical brackish wetland. Aquat. Toxicol. 2020, 225, 105522. [Google Scholar] [CrossRef]
- Liang, M.; Ma, L.; Li, X.; Feng, D.; Zhang, J.; Sun, Y. Identification and characterization of two types of triacylglycerol lipase genes from Neocaridina denticulata sinensis. Fish Shellfish Immun. 2022, 131, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Escarate, C.; Valenzuela-Munoz, V.; Nunez-Acuna, G.; Valenzuela-Miranda, D.; Goncalves, A.T.; Escobar-Sepulveda, H.; Liachko, I.; Nelson, B.; Roberts, S.; Warren, W. Chromosome-scale genome assembly of the sea louse Caligus rogercresseyi by SMRT sequencing and Hi-C analysis. Sci. Data 2021, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, Y.; Kang, K.; Zhang, D. SMRT sequencing of the full-length transcriptome of the white-backed planthopper Sogatella furcifera. PeerJ 2020, 8, e9320. [Google Scholar] [CrossRef]
- Zhang, Y.; Lou, F.; Chen, J.; Han, Z.; Yang, T.; Gao, T.; Song, N. Single-molecule Real-time (SMRT) Sequencing Facilitates Transcriptome Research and Genome Annotation of the Fish Sillago sinica. Mar. Biotechnol. 2022, 24, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.; Gu, L.; Reddy, A.S.N. Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-Based Direct RNA Sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Pootakham, W.; Uengwetwanit, T.; Sonthirod, C.; Sittikankaew, K.; Karoonuthaisiri, N. A novel full-length transcriptome resource for black tiger shrimp (Penaeus monodon) developed using isoform sequencing (Iso-Seq). Front. Mar. Sci. 2020, 7, 172. [Google Scholar] [CrossRef]
- Zhang, Y.; Ni, M.Q.; Bai, Y.H.; Shi, Q.; Zheng, J.B.; Cui, Z.X. Full-length transcriptome analysis provides new insights into the diversity of immune-related genes in Portunus trituberculatus. Front. Immunol. 2022, 13, 843347. [Google Scholar] [CrossRef]
- Wan, H.; Jia, X.; Zou, P.; Zhang, Z.; Wang, Y. The Single-molecule long-read sequencing of Scylla paramamosain. Sci. Rep. 2019, 9, 12401. [Google Scholar] [CrossRef]
- Zhao, J.; He, Z.; Chen, X.; Huang, Y.; Xie, J.; Qin, X.; Ni, Z.; Sun, C. Growth trait gene analysis of kuruma shrimp (Marsupenaeus japonicus) by transcriptome study. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 40, 100874. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.M.; Tang, Y.D.; Zhai, J.B.; Zheng, C.F. The RING finger protein family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 300. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Alamancos, G.P.; Pagès, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Liu, T.; Liu, C.J.; Song, S.Y.; Zhang, X.T.; Liu, W.; Jia, H.B.; Xue, Y.; Guo, A.Y. AnimalTFDB 2.0: A resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015, 43, D76–D81. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Li, A.M.; Zhang, J.Y.; Zhou, Z.Y. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wilkins, D. gggenes: Draw Gene Arrow Maps in ‘ggplot2’. 2023. Available online: https://wilkox.org/gggenes/ (accessed on 15 April 2024).
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.J.; Kang, X.N.; Liu, S.L. The regulatory roles of long noncoding RNAs in the biological behavior of pancreatic cancer. Saudi J. Gastroenterol. 2019, 25, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Chen, X.; Peng, J.; Yang, C.; Peng, M.; Zhu, W.; Xie, D.; He, P.; Wei, P.; Lin, Y.; et al. Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci. Rep. 2018, 8, 16920. [Google Scholar] [CrossRef]
- Gao, B.Q.; Lv, J.J.; Meng, X.L.; Li, J.T.; Li, Y.K.; Liu, P.; Li, J. Full-length transcriptome construction of the blue crab Callinectes sapidus. Front. Mar. Sci. 2022, 9, 922188. [Google Scholar] [CrossRef]
- Wu, X.; Gong, Q.; Chen, Y.; Liu, Y.; Song, M.; Li, F.; Li, P.; Lai, J. Full-length transcriptome and analysis of bmp-related genes in Platypharodon extremus. Heliyon 2022, 8, e10783. [Google Scholar] [CrossRef]
- Xing, K.F.; Liu, Y.J.; Yan, C.C.; Zhou, Y.Z.; Sun, Y.Y.; Su, N.K.; Yang, F.S.; Xie, S.; Zhang, J.Q. Transcriptome analysis of Neocaridina denticulate sinensis under copper exposure. Gene 2021, 764, 145098. [Google Scholar] [CrossRef]
- Liu, Y.J.; Xing, K.F.; Yan, C.C.; Zhou, Y.Z.; Xu, X.M.; Sun, Y.Y.; Zhang, J.Q. Transcriptome analysis of Neocaridina denticulate sinensis challenged by Vibrio parahemolyticus. Fish Shellfish Immun. 2022, 121, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.M.; Albu, M.; Chen, X.T.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Mackeh, R.; Marr, A.K.; Fadda, A.; Kino, T. C2H2-Type Zinc Finger Proteins: Evolutionarily Old and New Partners of the Nuclear Hormone Receptors. Nucl. Recept. Signal. 2018, 15, 1550762918801071. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, R.; Wang, Q.; Yue, Y.; Gao, Q.; Wang, C.; Zheng, H.; Peng, S. Transcriptomic analysis of large yellow croaker (Larimichthys crocea) during early development under hypoxia and acidification stress. Vet. Sci. 2022, 9, 632. [Google Scholar] [CrossRef]
- Yin, Z.; Nie, H.; Jiang, K.; Yan, X. Molecular Mechanisms Underlying Vibrio Tolerance in Ruditapes philippinarum Revealed by Comparative Transcriptome Profiling. Front. Immunol. 2022, 13, 879337. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Maeda, T. POK/ZBTB proteins: An emerging family of proteins that regulate lymphoid development and function. Immunol. Rev. 2012, 247, 107–119. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Wu, L.L.; Li, J.; Chen, J.Y.; Yu, Q.; Yao, H.; Xu, Y.P.; Liu, L. Identification of ZBTB9 as a potential therapeutic target against dysregulation of tumor cells proliferation and a novel biomarker in Liver Hepatocellular Carcinoma. J. Transl. Med. 2022, 20, 602. [Google Scholar] [CrossRef]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.W.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Morlando, M.; Ballarino, M.; Fatica, A. Long Non-Coding RNAs: New Players in Hematopoiesis and Leukemia. Front. Med. 2015, 2, 23. [Google Scholar] [CrossRef]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Azlan, A.; Obeidat, S.M.; Das, K.T.; Yunus, M.A.; Azzam, G. Genome-wide identification of Aedes albopictus long noncoding RNAs and their association with dengue and Zika virus infection. PLoS Neglected Trop. Dis. 2021, 15, e0008351. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cheng, T.; Liu, C.; Liu, D.; Zhang, Q.; Long, R.; Zhao, P.; Xia, Q. Systematic Identification and Characterization of Long Non-Coding RNAs in the Silkworm, Bombyx mori. PLoS ONE 2016, 11, e0147147. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.J.; Smith, C.W.J.; Jiggins, C.D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 2022, 23, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Mohr, C.; Hartmann, B. Alternative splicing in Drosophila neuronal development. J. Neurogenet. 2014, 28, 199–215. [Google Scholar] [CrossRef]
- Riddell, C.E.; Lobaton Garces, J.D.; Adams, S.; Barribeau, S.M.; Twell, D.; Mallon, E.B. Differential gene expression and alternative splicing in insect immune specificity. BMC Genom. 2014, 15, 1031. [Google Scholar] [CrossRef]
- Salz, H.K. Sex determination in insects: A binary decision based on alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 395–400. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, Y.; Zheng, J.; Cui, Z. Alternative splicing derived invertebrate variable lymphocyte receptor displays diversity and specificity in immune system of crab Eriocheir sinensis. Front. Immunol. 2022, 13, 1105318. [Google Scholar] [CrossRef]
- Gibilisco, L.; Zhou, Q.; Mahajan, S.; Bachtrog, D. Alternative splicing within and between Drosophila species, sexes, tissues, and developmental stages. PLoS Genet. 2016, 12, e1006464. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Y.; Pan, L.X.; Cheng, F.P.; Jin, M.J.; Wang, Z.L. A global survey of the full-length transcriptome of Apis mellifera by single-molecule long-read sequencing. Int. J. Mol. Sci. 2023, 24, 5827. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.X.; Pan, L.Q.; Zhang, X.; Li, Y.F. Neuroendocrine-immune regulation mechanism in crustaceans: A review. Rev. Aquac. 2022, 14, 378–398. [Google Scholar] [CrossRef]
- Zhang, X.; Pan, L.Q.; Tong, R.X.; Li, Y.F.; Tian, Y.M.; Li, D.Y.; Si, L.J. PacBio full length transcript sequencing and Illumina transcriptome insight into immune defense mechanism of Litopenaeus vannamei under ammonia-N stress. Aquaculture 2021, 536, 736457. [Google Scholar] [CrossRef]
- Xu, Z.; Wei, Y.; Wang, G.; Ye, H. B-type allatostatin regulates immune response of hemocytes in mud crab Scylla paramamosain. Dev. Comp. Immunol. 2021, 120, 104050. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.A.; Wilkinson, B.D.; Wharton, R.P.; Aggarwal, A.K. Model of the brain tumor-Pumilio translation repressor complex. Genes Dev. 2003, 17, 2508–2513. [Google Scholar] [CrossRef]
- Slack, F.J.; Ruvkun, G. A novel repeat domain that is often associated with RING finger and B-box motifs. Trends Biochem. Sci. 1998, 23, 474–475. [Google Scholar] [CrossRef]
- Volovik, Y.; Moll, L.; Marques, F.C.; Maman, M.; Bejerano-Sagie, M.; Cohen, E. Differential regulation of the heat shock factor 1 and DAF-16 by neuronal nhl-1 in the nematode C. elegans. Cell Rep. 2014, 9, 2192–2205. [Google Scholar] [CrossRef]
- Xu, X.J.; Hao, L.L.; Zhu, J.W.; Zhou, Q.; Song, F.H.; Chen, T.T.; Zhang, S.S.; Dong, L.L.; Lan, L.; Wang, Y.Q.; et al. Database Resources of the BIG Data Center in 2018. Nucleic Acids Res. 2018, 46, D14–D20. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Number | Minimum Length | Maximum Length | Mean Length | N50 |
|---|---|---|---|---|---|
| Polymerase read | 548,606 | 119,673 | 192,219 | ||
| Subread | 23,201,449 | 2757 | 3237 | ||
| CCS | 193,468 | 74 | 16,527 | 3243 | 3640 |
| FLNC read | 154,402 | 52 | 14,041 | 3145 | 3559 |
| Consensus sequence | 13,082 | 83 | 9788 | 3262 | 3749 |
| Isoform sequence | 5831 | 3166.76 | 3697 |
| Rank | Species | Transcript Number | % |
|---|---|---|---|
| 1 | Hyalella azteca | 2224 | 42.05 |
| 2 | Zootermopsis nevadensis | 359 | 6.79 |
| 3 | Limulus polyphemus | 164 | 3.1 |
| 4 | Litopenaeus vannamei | 124 | 2.34 |
| 5 | Daphnia magna | 112 | 2.12 |
| 6 | Penaeus monodon | 88 | 1.66 |
| 7 | others | 2200 | 41.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, K.; Li, H.; Wang, X.; Sun, Y.; Zhang, J. A Full-Length Transcriptome and Analysis of the NHL-1 Gene Family in Neocaridina denticulata sinensis. Biology 2024, 13, 366. https://doi.org/10.3390/biology13060366
Xing K, Li H, Wang X, Sun Y, Zhang J. A Full-Length Transcriptome and Analysis of the NHL-1 Gene Family in Neocaridina denticulata sinensis. Biology. 2024; 13(6):366. https://doi.org/10.3390/biology13060366
Chicago/Turabian StyleXing, Kefan, Huimin Li, Xiongfei Wang, Yuying Sun, and Jiquan Zhang. 2024. "A Full-Length Transcriptome and Analysis of the NHL-1 Gene Family in Neocaridina denticulata sinensis" Biology 13, no. 6: 366. https://doi.org/10.3390/biology13060366
APA StyleXing, K., Li, H., Wang, X., Sun, Y., & Zhang, J. (2024). A Full-Length Transcriptome and Analysis of the NHL-1 Gene Family in Neocaridina denticulata sinensis. Biology, 13(6), 366. https://doi.org/10.3390/biology13060366