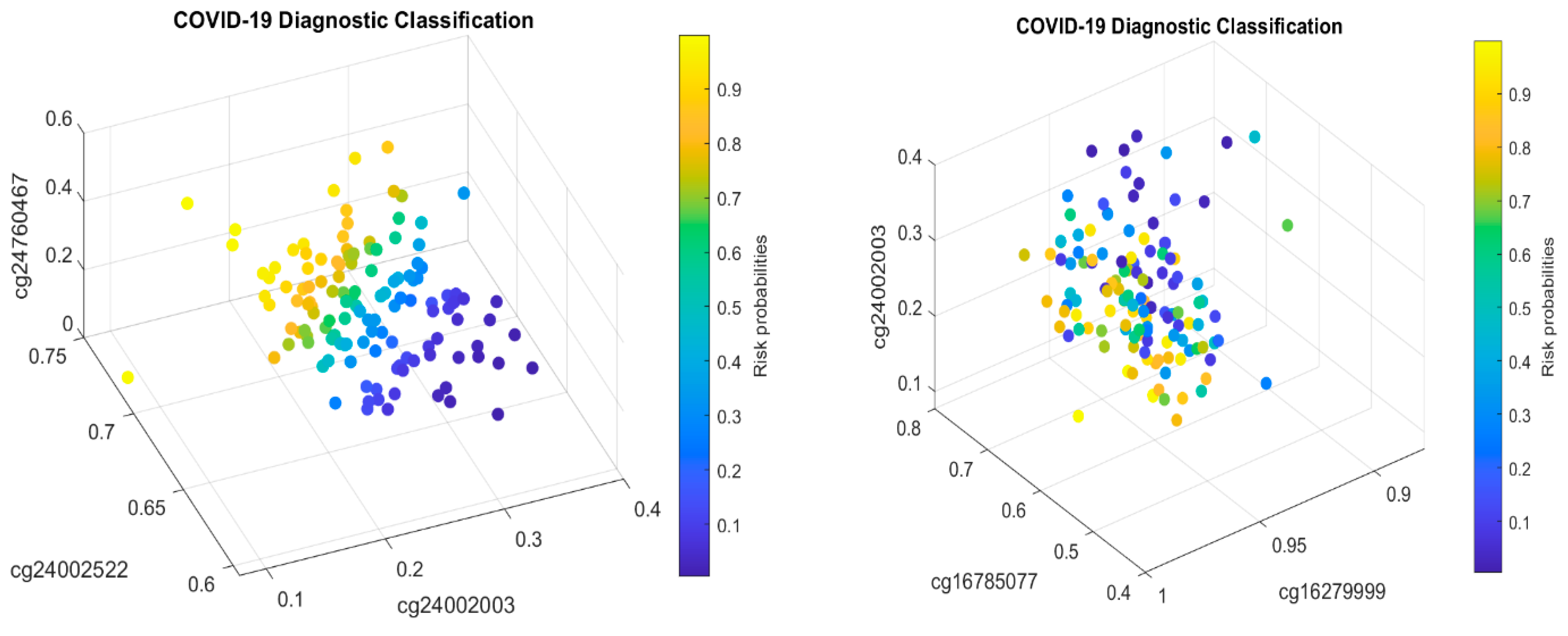
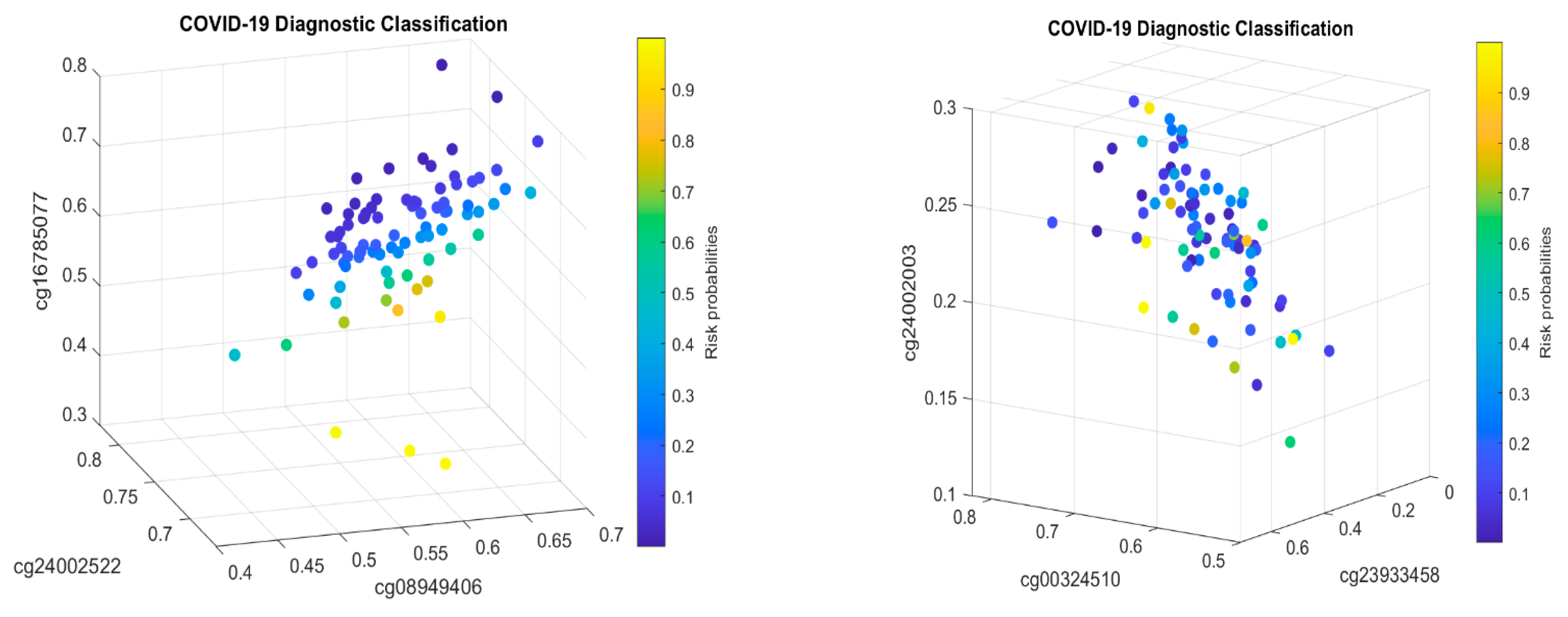
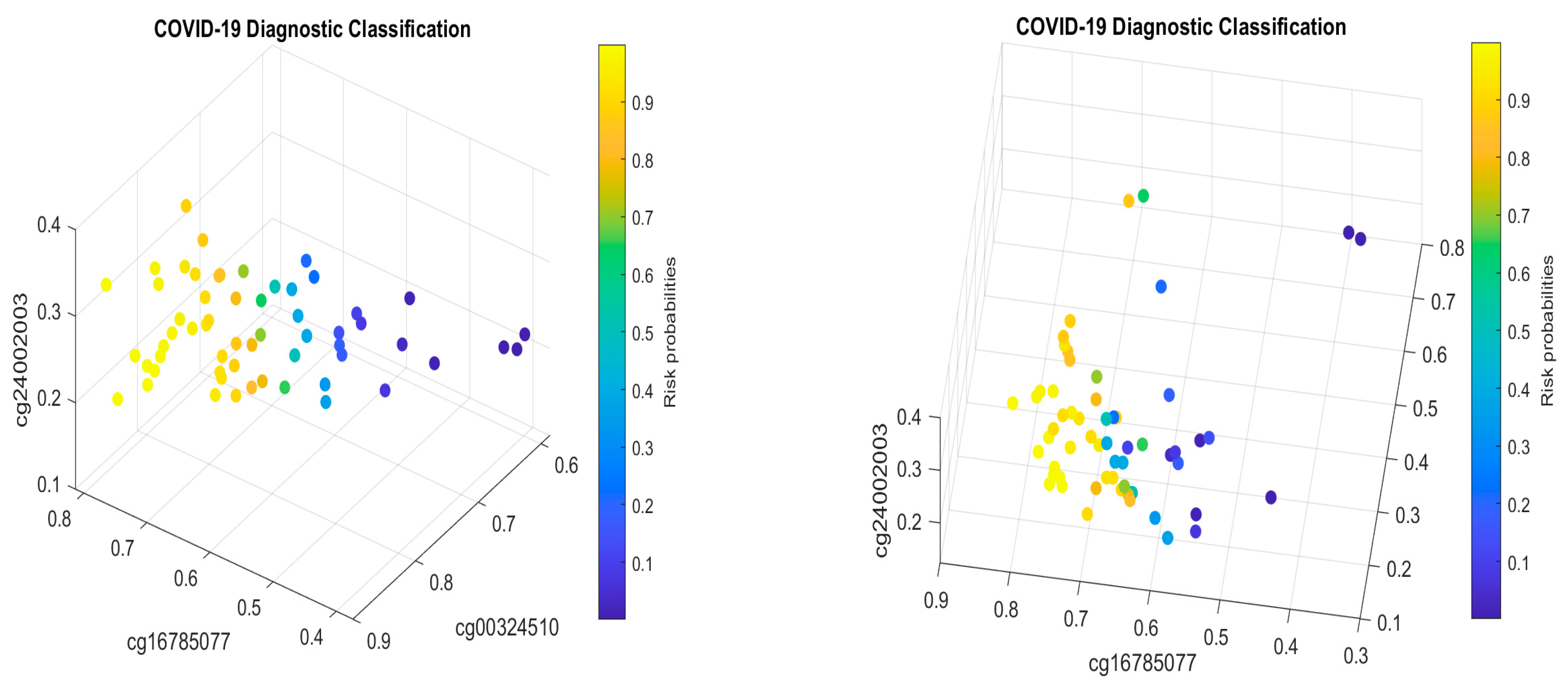
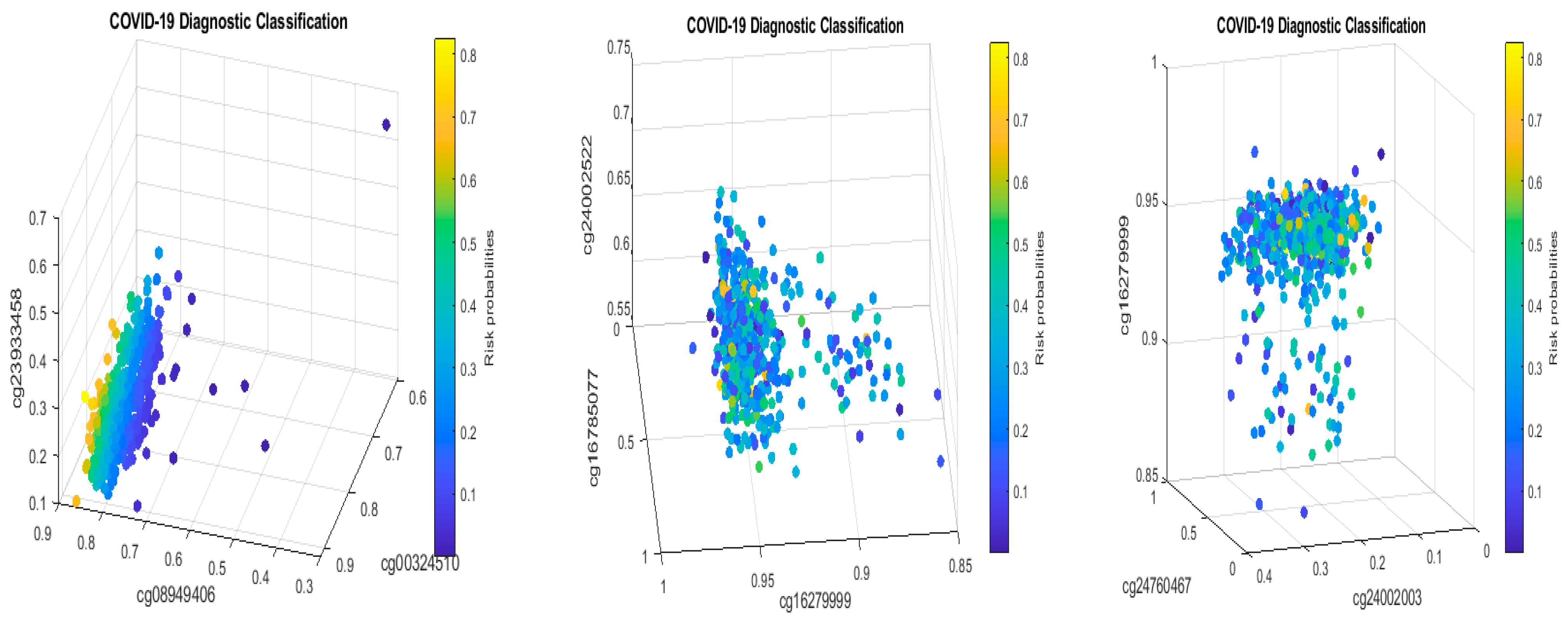
5.1. Discussion
Many COVID-19 research results at the genomic level have been published in the literature. These published results have explored the pathological causes of COVID-19 infection from various aspects. Due to the limitations of research methodology, some of the published results can hardly be cross-validated from cohort to cohort. One exception is that the earlier work [
8,
9,
10] cross-validated thirteen genes across fourteen cohort studies with thousands of patients, heterogeneous ethics, ages, and geographical regions and showed interpretable, reliable, and robust results. The work at the genomic level was a comprehensive study with nearly perfect performance. We did not find any other method that led to 100% accuracy in the literature, not to mention interpretability. In the literature,
MX1 (cg25888371) and
PARP9 (cg22930808) combined with eight other genes (i.e., a total of 10 genes) to reach an overall 78.4% accuracy [
42] which is much lower than 89.84% in CF2 in
Table 2 with
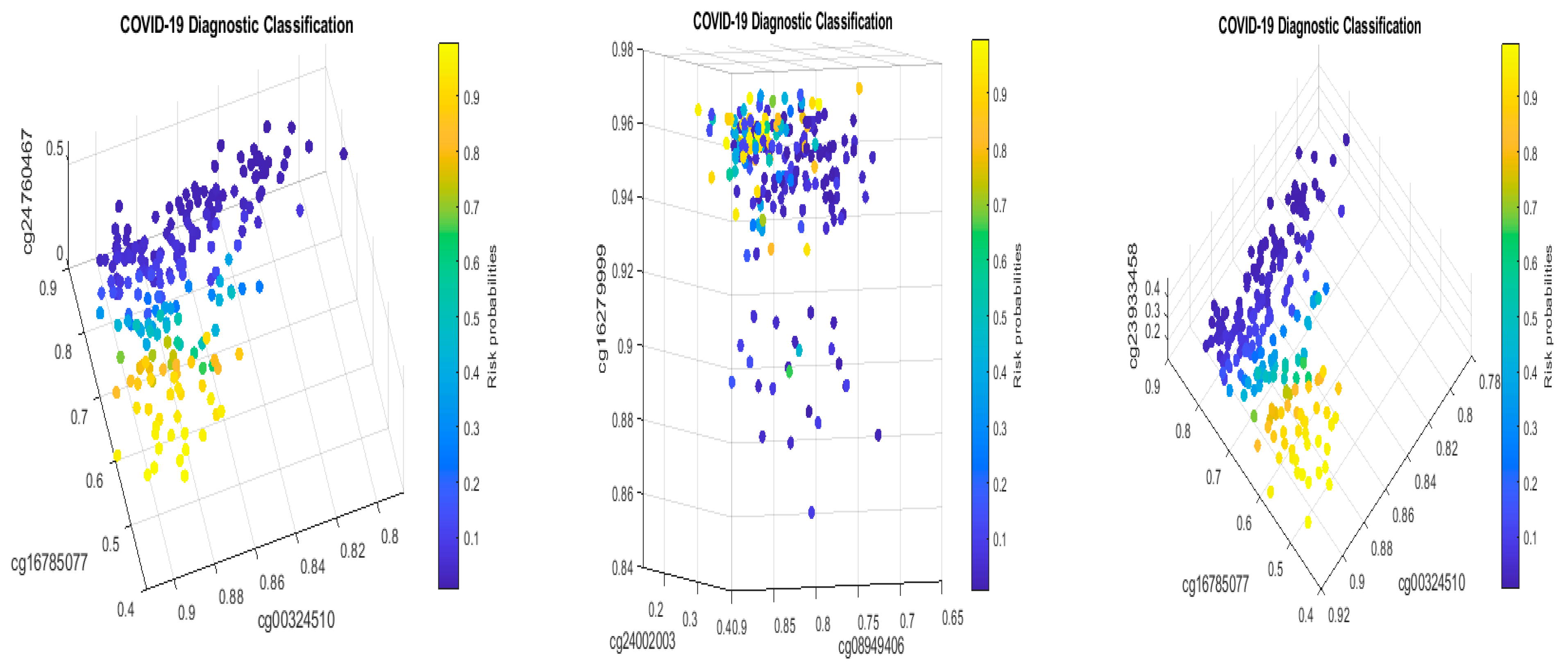
MX1 (cg16785077) combined with only two other genes (i.e., a total of three genes). In addition, many studies have focused on only a single cohort whose representativeness cannot be assessed.
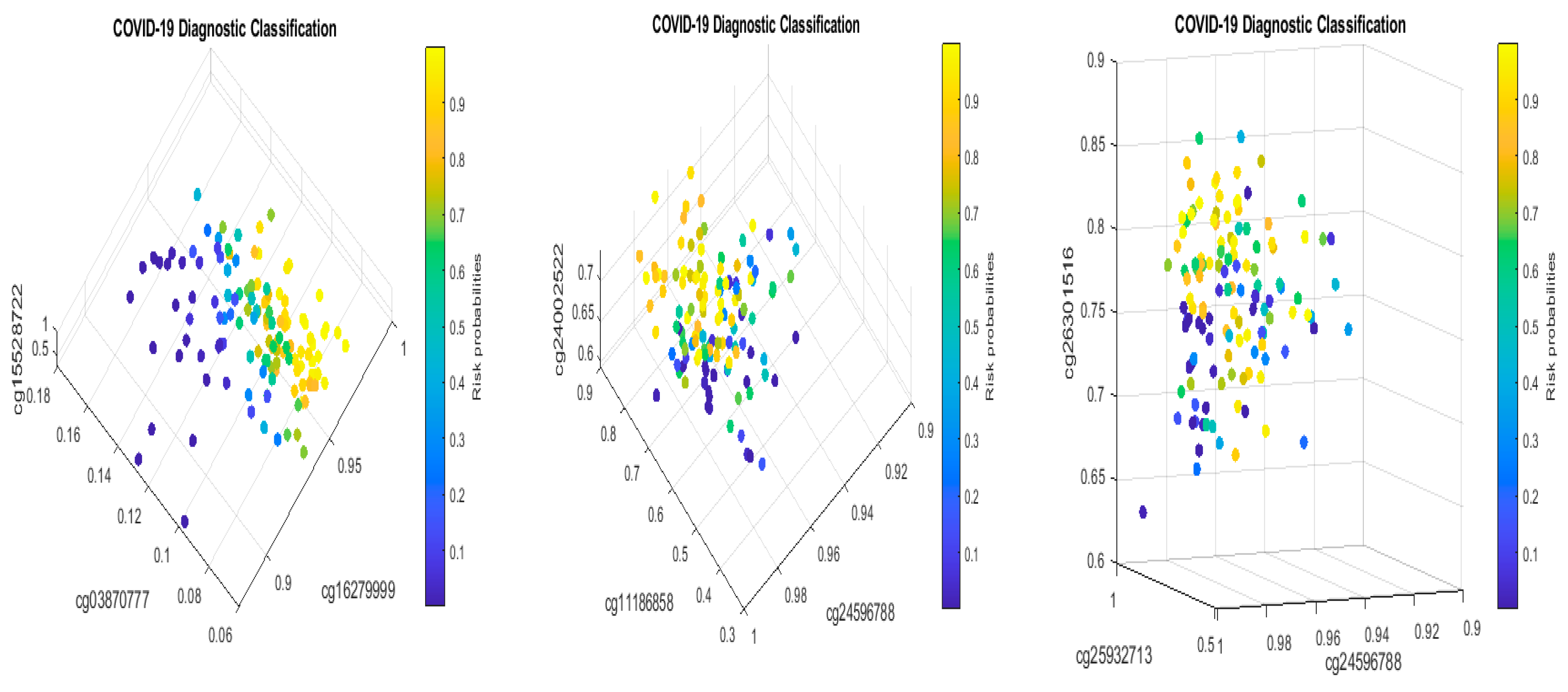
Many published results have studied the functional effects of genes based on single gene expression value changes. They lack an interaction effects study, mainly due to the limitations of the research methods. Using the cg16785077-regulated gene
MX1 as an example, in CF2 in
Table 2, it must be jointly studied with another two genes
TESC and
CHSY1, to fully understand its functional effects on COVID-19, as it does not appear in CF1.
Since COVID-19 started in December 2019, many genes have been reported to be linked to various diseases. However, they lack mathematical proof or biological equivalence. They just happened to be significant in one cohort study. For example, SARS-CoV-2 entering the brain [
47], COVID-19 vaccines complicating mammograms [
48], memory loss and ‘brain fog’ [
49], and COVID-19 endothelial dysfunction causing erectile dysfunction [
50], amongst other symptoms, have been reported. From our findings, cg16785077 (
MX1) may provide a clue for brain-related COVID-19 symptoms; cg08949406 (
FHIT) may lead to an answer about breast cancer-related COVID-19 symptoms; cg24002003 (
CHSY1) may be informative for temtamy preaxial brachydactyly syndrome- and brachydactyly-related COVID-19 symptoms.
In the literature, the gene
MX1 has been reported to affect our response to COVID-19+ [
5,
42,
51,
52,
53,
54,
55,
56,
57].
MX1 becomes a potential druggable target [
51]. We want to note that
MX1 did not appear in the genomic biomarkers with optimum performance for SARS-CoV-2 and COVID-19, though it is regulated by cg16785077 found in this study. Note that multiple sites can regulate a gene, e.g.,
MX1 can be regulated by 188 CpG sites. In addition, from
Table 2, we can see that cg16785077 only appears in CF2, i.e., not in CF1, which tells us that there is a larger portion of COVID-19+ which is not caused by or related to cg16785077 (
MX1).
Recall that the earlier work dealt with genomic biomarkers [
8,
9,
10,
24], and identified genes (
ABCB6,
KIAA1614,
MND1,
SMG1,
RIPK3,
CDC6,
ZNF282, and
CEP72) at the genomic level as a set of optimally performing interactive COVID-19 biomarkers based on whole blood samples and genes (
ATP6V1B2,
IFI27,
BTN3A1,
SERTAD4, and
EPSTI1) at the genomic level as a set of optimally performing interactive SARS-CoV-2 biomarkers based on nasopharyngeal (NP) and oropharyngeal (OP) PCR swab samples. The genes listed in
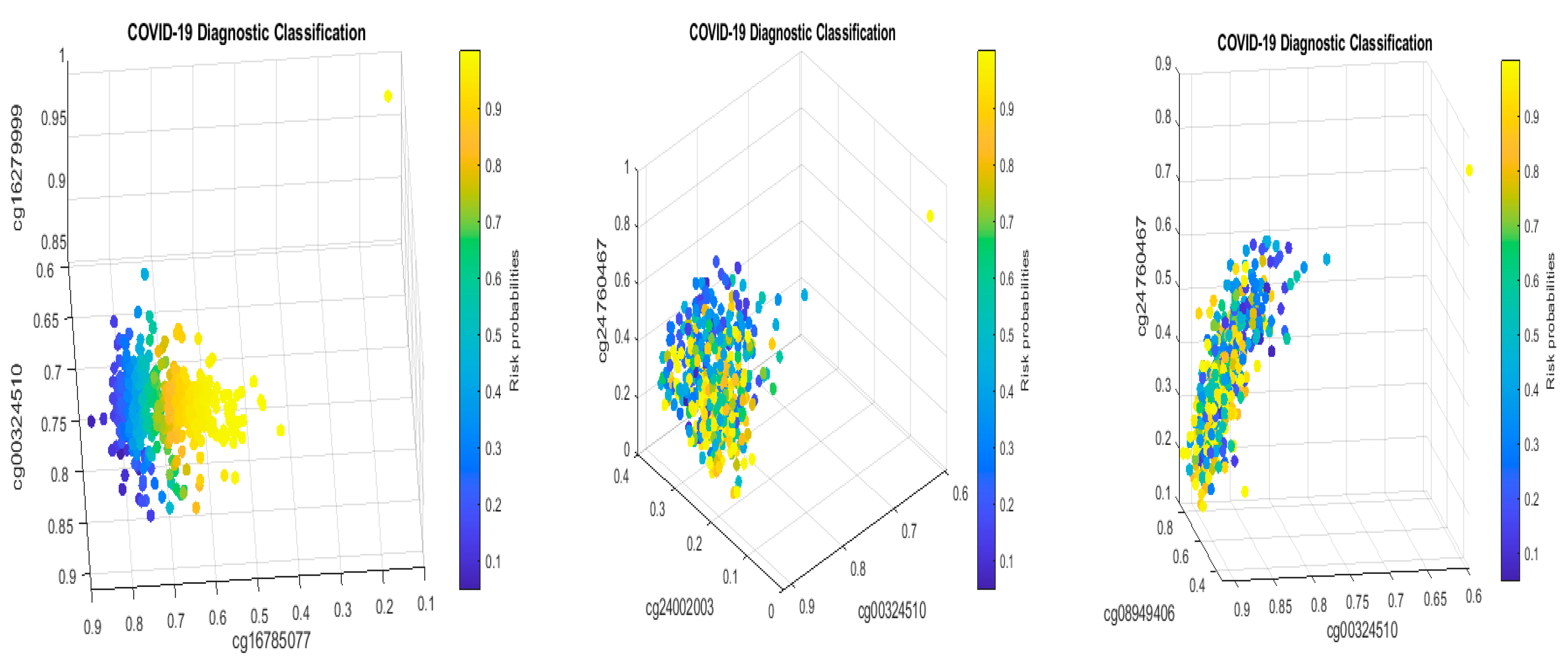
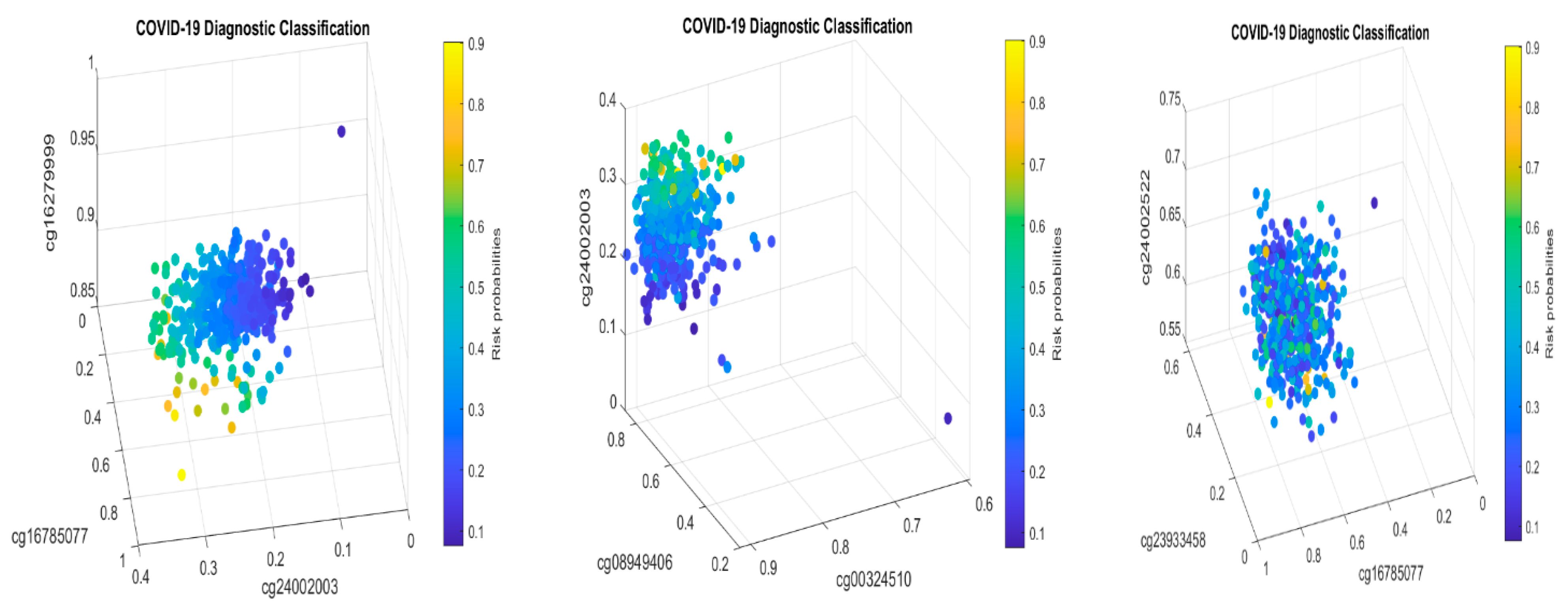
Table 2 in this paper are
TESC,
ALCAM,
PACS1,
FHIT,
MX1,
LINC00456,
CHSY1, and
LZTS2, which are biomarkers at the DNA methylation level or epigenetic level and are different from those optimally performing biomarkers at the genomic level or RNA-seq level. We used basketball players on a team to explain the site–site interactions among sites. In the earlier work [
9,
10], we used the same analogy to interpret gene–gene interactions. Considering the fundamental differences between DNA and RNA in terms of their functions, we can consider, on a basketball team, cg16279999 (
TESC), cg24002522 (
ALCAM), cg00324510 (
PACS1), cg08949406 (
FHIT), cg16785077 (
MX1), cg23933458 (
LINC00456), cg24002003 (
CHSY1), and cg24760467 (
LZTS2) as coaches, managers, trainers, and team doctors, while considering
ABCB6,
KIAA1614,
MND1,
SMG1,
RIPK3,
CDC6,
ZNF282,
CEP72,
ATP6V1B2,
IFI27,
BTN3A1,
SERTAD4, and
EPSTI1 as players. Each gene (site) has a specific role on the team, and they interact. This analogy and the analogies used in earlier sections can help some readers understand the model and site–site interactions, though they do not provide insightful biological implications.
At the genomic level,
MX1 may trigger diseases like influenza and subacute sclerosing panencephalitis (SSPE), a progressive neurological disorder in children and young adults that affects the central nervous system (CNS); SSPE can have up to a 6- to 8-year- long incubation period for youngsters, which can be the most significant concern for COVID-19 infection. Therefore, urgent efforts are needed to investigate this potential. In addition, other symptoms listed in
Section 3.2 are also urgent.
As to
MX1 becoming a potential druggable target, the site cg16785077 should be the true druggable target. In addition, the genes
MND1,
CDC6,
ZNF282,
ATP6V1B2,
IFI27, and
GCKR at the genomic level become potential druggable targets [
10].
A combination of cg16785077 (MX1), ATP6V1B2, and IFI27 can explain many COVID-19 infection symptoms, e.g., influenza-like symptoms (omicron symptoms), and ‘brain fog’. However, the connections of reported diseases in the literature to COVID-19 are not confirmed due to the infection, and the diseases may not be relevant. Our findings can certainly provide useful clues. Again, we should not assume all published literature is true. Many published papers are based on correlation studies. Our new study should not be regarded as a correlation study for the following reason: the identified reliable and nearly perfectly performing DNA methylation biomarkers established a mathematical equivalence between the disease and the site–site interactions. A mathematical equivalence can reveal what a result may be, which can be more informative than unreliable and non-proven published causative studies. Furthermore, ‘causal’ implies invariance, and a mathematical equivalence shows that our new results are invariant. In other words, if an invariance cannot be established, the claimed causal relationships are doubtful. Based on these observations, it can be inferred that our findings at the DNA methylation level are mostly closed to the cause of COVID-19 disease regardless of whether or not all the published literature is true.
All the findings reported in this paper are based on experimental results from published work. One may argue that the conclusions from this paper are too strong to believe. Additional experimental validations are needed. The methodology applied in this paper may not be implementable in new experiments as site–site interactions have never been discussed in any biological literature. On the other hand, a question arises: should we believe published results that may not be cross-validated from different cohort studies, though they were based on experiments? We can definitely question many published results from experiments due to the limitation of analysis methods, where as a result, they may be misleading.
Classical logistic regression models have been widely used as a benchmark and baseline in medical data modeling and inference. However, Teng and Zhang [
58] have pointed out it has a fundamental flaw as it does not directly specify relative treatment effects in the model, and as a result, misleading results have been reported in the literature. A more robust model, called AbRelaTEs, was introduced in [
58], and the classical logistic regression becomes a special case of AbRelaTEs. In scientific research, we first compare the accuracies and then the computational time when applying multiple candidate models. When the accuracies from different models are significantly different, reporting the results from models with lower accuracies becomes meaningless unless we study the properties of the models. In this regard, we do not compare the max-logistic regression with the logistic regression. Interested readers can find many published works using logistic regressions.
Section 4.5 demonstrated a real data example where the max-logistic regression outperforms AI algorithms, machine learning, and probabilistic algorithms. Based on these observations, we argue that the max-logistic regression should be considered as a baseline model and a benchmark model in terms of accuracy.
In medical research, another issue is to avoid overfitting the data, and cross-validation has been applied to many studies. However, as pointed out in [
9], cross-validations should be applied to homogeneous datasets. When data are collected from heterogenous populations, i.e., subtypes in this paper, regular cross-validations will lead to wrong variable selections and miss the most important critical CpG sites and genes. Nevertheless, suppose one really wants to apply cross-validations to the max-logistic regressions in this dataset, given its specificity being 100% accurate. In that case, any 50–50 split of datasets will still lead to 100% specificity and at least 94% sensitivity. The reason is that the S4 classifier in Equation (4) has been mathematically proved to reach the smallest set of genes [
21], the strongest property in the variable selection literature. For this reason, we do not apply the classical cross-validations to our model fitting. Instead, we apply an even more challenging cross-validation procedure: cohort-to-cohort cross-validations. It can be expected that many published works may fail or have very low accuracies when applied to cohort-to-cohort cross-validations. We note that cohort-to-cohort cross-validations are still rather sparse in the literature, and we refer interested readers to the earlier works [
10,
11,
15,
21,
22,
23].
Different from many research papers that considered biological heterogeneity, e.g., the stage of disease, treatment protocol, and inter-individual variation factors as well as demographic and baseline clinical characteristics like ethnicity, age, and sex, as well as containing an absence of pre-pandemic controls and their unknown health status, this paper only focused on DNA methylations CpG sites. In our opinion, those heterogeneities are extrinsic variables; they can provide additional information when studying the severity and duration of the disease. However, the CpG sites are intrinsic factors, and as long as they reach nearly perfect prediction powers, the extrinsic variables can provide little additional information. Interested readers are referred to published work for research on heterogeneity, epigenetic clocks, and surface contamination [
59,
60].
Many published medical research papers do not report the fitted explicit coefficients and interpret the coefficients and the meaning of their associated positive and negative signs, which leave the results in the dark. We reported and interpreted all fitted coefficients, as in
Table 2 and the formula below. We also used different trials and datasets to justify the fitted coefficients. We note that the fitted signs of the coefficients are the most important parts of scientific reasoning. However, many published research papers should have paid more attention to this critical issue and cohort-to-cohort validations.
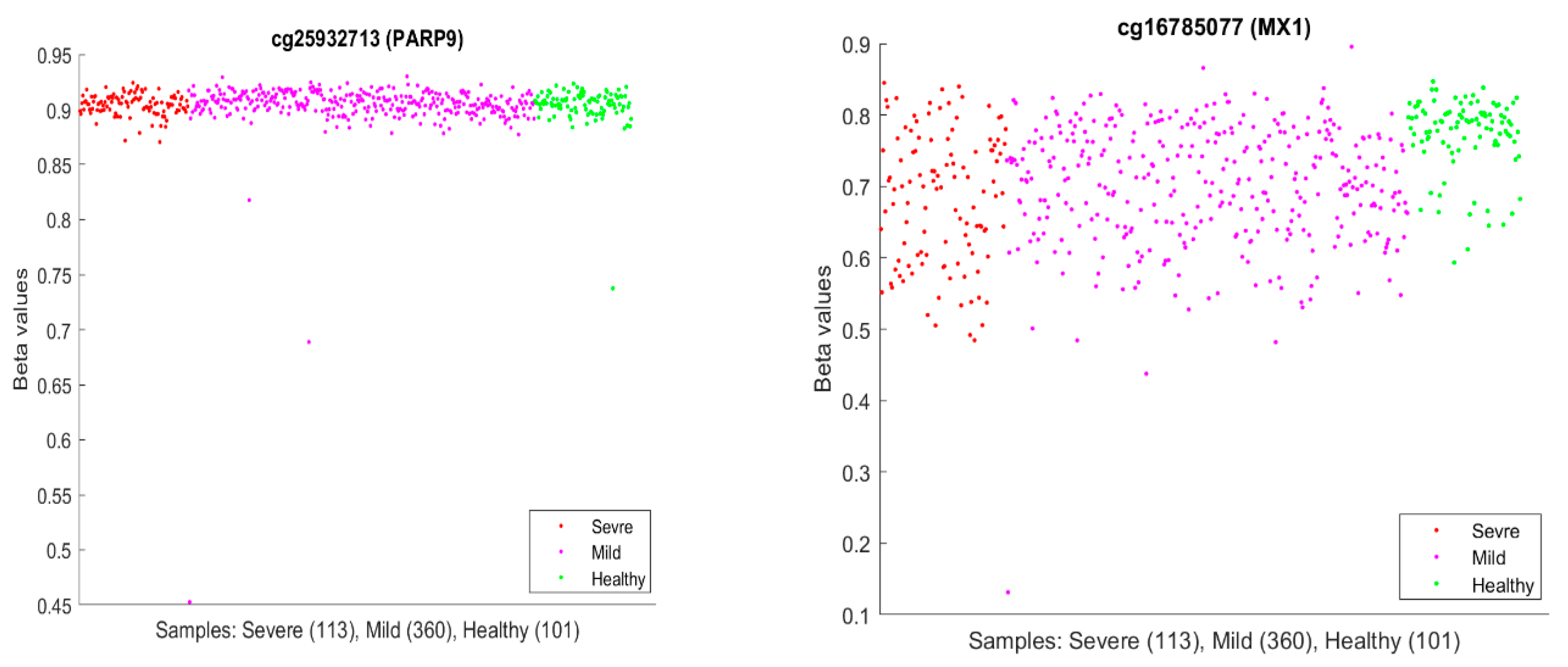
It is usual to see epigenetic changes [
61], including DNA methylation changes, in the host (patient) blood and solid tissue after the infection of DNA/RNA viruses and bacteria, which may be partially attributed to the systemic inflammation brought by the host–virus interaction and anti-infection immunity. In our work, our hypotheses are based on CpG site–site interactions and site–subtype interactions with nearly perfect accuracy. We note that the existing literature has seldom addressed these features and, more importantly, has lacked convincing accuracies. In the literature, we discussed earlier that
PARP9 and MX1 had been linked to COVID-19.
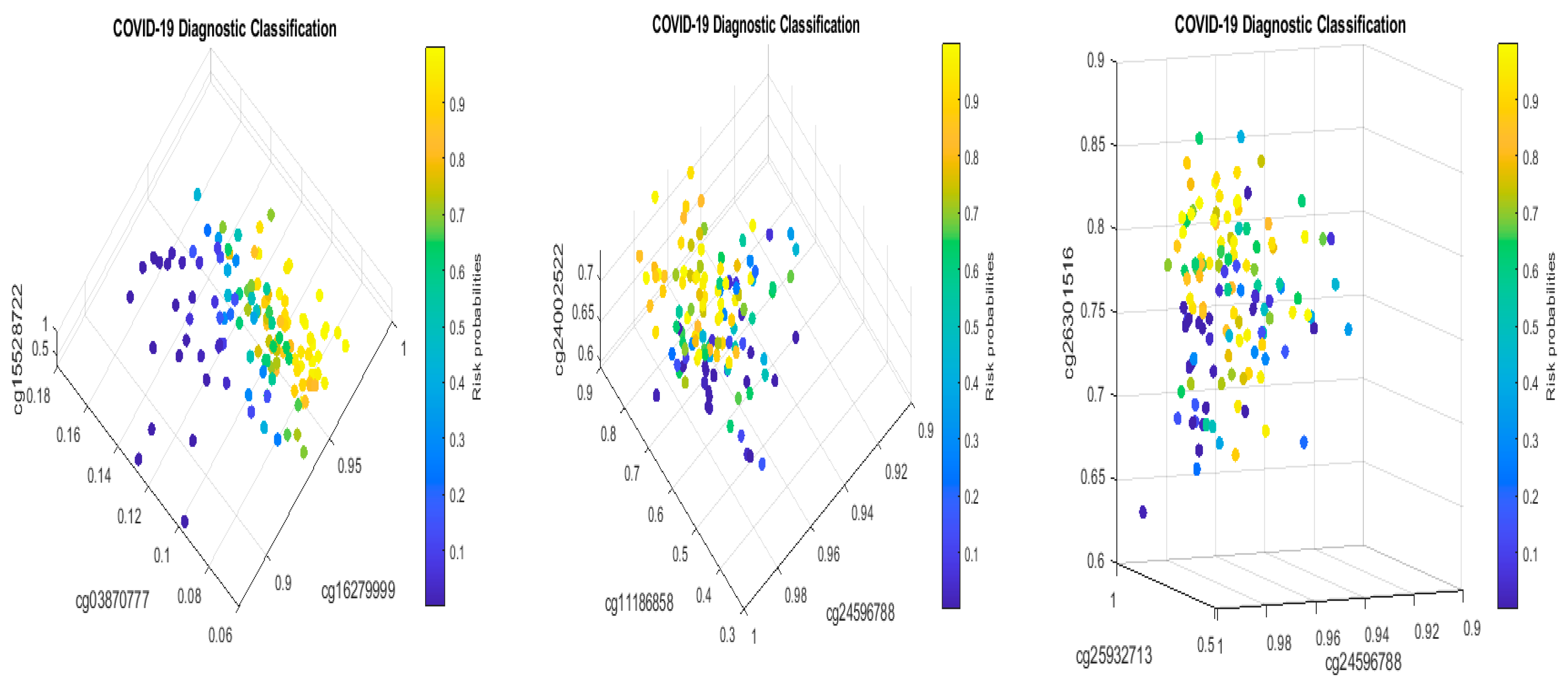
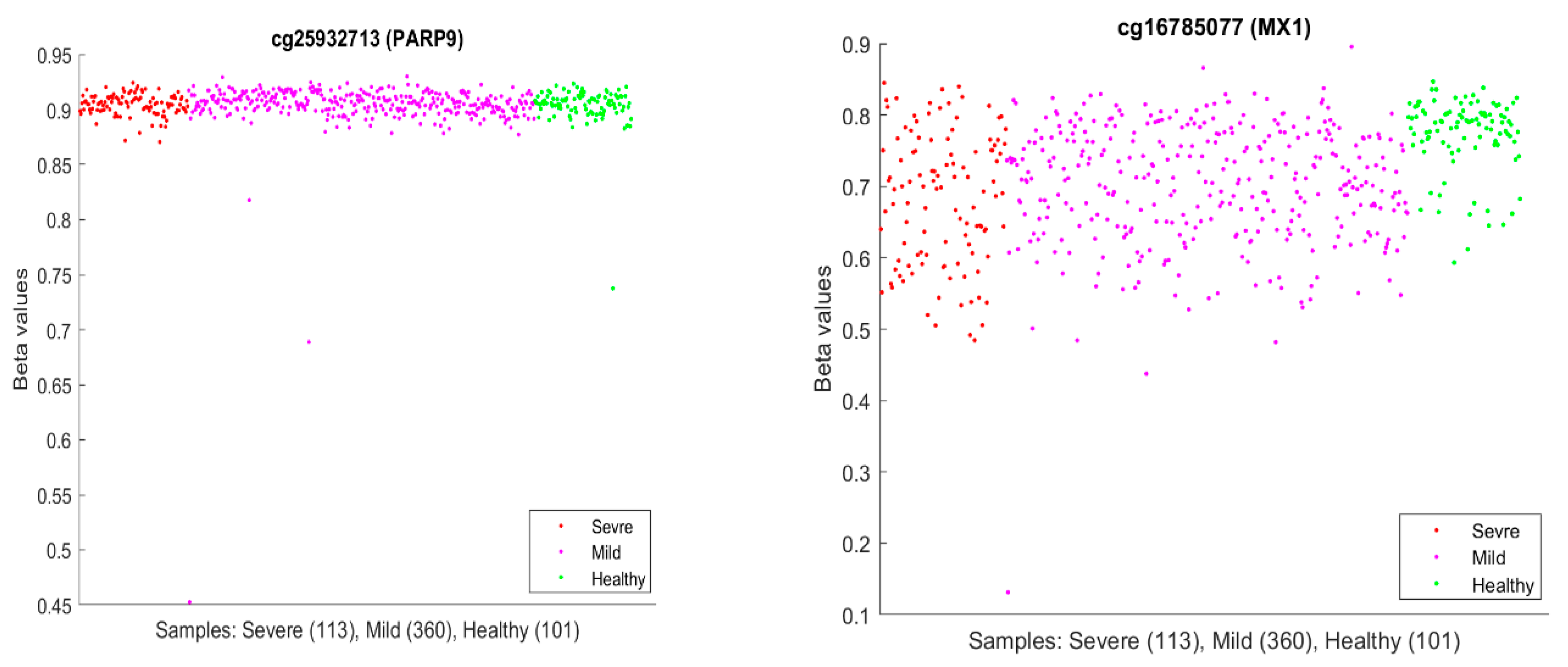
Figure 10 plots beta values of cg25932713 (
PARP9) and cg16785077 (
MX1).
In
Figure 10, individual beta values clearly cannot distinguish severe, mild, and healthy cases. Such a phenomenon again confirmed that CpG site–site interactions are the key to uncovering the truth. We further note that in GSE174818, all patients are hospitalized patients with either COVID-19 or other respiratory diseases. As a result, the CpG sites identified are COVID-19-specific.
In their model of methylation clocks, the authors [
33] identified cg26312951 (
MX1) and cg00959259 (
PARP9) to be effective. These two CpGs are different from the CpGs in
Table 2 and
Table 11a. This phenomenon needs further studying.
We have shown that pediatric COVID-19 cases have different DNA methylation signatures from adults. As a result, treatments for children should be evaluated with additional care.
Finally, methylation analysis can provide more comprehensive and detailed information, particularly in understanding the interplay between viral infection and the host genome. Notably, recent studies have reported associations between DNA methylation changes and the worsening of SARS-CoV-2 infection [
32,
62]. Additionally, this method has shown promise in identifying mild cases [
32], predicting outcomes [
14,
63], and assessing treatment strategies [
64], thus offering valuable insights for personalized medicine. The aim of our research was to identify epigenetic changes associated with COVID-19 symptoms or infection status and to explore the causality of the disease.
5.2. Conclusions
Our work is the first to study COVID-19 site–site interactions at the epigenetic level. We discovered a miniature set of nearly perfect interactive COVID-19 DNA methylation biomarkers. Unlike much other research, this paper advances the exploration of site interaction relationships based on competing risk models. We indicate significant differences in DNA methylation data in identifying critical CpG sites. We identify cg16279999 (
TESC), cg00324510 (
PACS1), cg08949406 (
FHIT), cg16785077 (
MX1), cg24002003 (
CHSY1), cg25932713, and cg22930808 (
PARP9) as potential diagnostic and druggable targets. In addition, the genes
MND1,
CDC6,
ZNF282,
ATP6V1B2,
IFI27, and
GCKR at the genomic level become potential druggable targets [
10]. Here, potential druggable targets mean that these CpG sites are either over-methylated or under-methylated, or their site–site interactions undergo changes, and the genes are either over-expressed or knocked down, or the gene–gene interactions undergo changes, which point toward directions for developing antiviral drugs.
This new work, together with the earlier work [
8,
9,
10,
11,
24], systematically and accurately describes both SARS-CoV-2 and COVID-19 at the genetic level.
We have discussed that the sites (genes) at the DNA methylation level are different from optimum genomic biomarker genes at the genomic level. We hypothesize that the initial SARS-CoV-2 was a DNA virus and then was transcribed to an RNA virus. Such findings open the door to understanding the pathology of the SARS-CoV-2 infection and COVID-19 disease.
It would be more significant if the interactions between methylation and phosphorylation, ubiquitination, acetylation, and their respective interactions in epigenetics could be studied. It is worth studying how the impact of COVID-19 would change in actively differentiating cells vs. terminally differentiated cells based on the CpG interactions. In addition, pathway analysis merits further study. We leave these aspects for a future project once relevant data can be obtained.
Finally, the most innovative work in this paper are the site–site interactions, gene–gene interactions, and site–gene interactions, which have been missed in published papers that applied other non-interactive variable analysis methods.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}