
On the Importance of Acidity in Cancer Cells and Therapy


Abstract
Simple Summary
Abstract
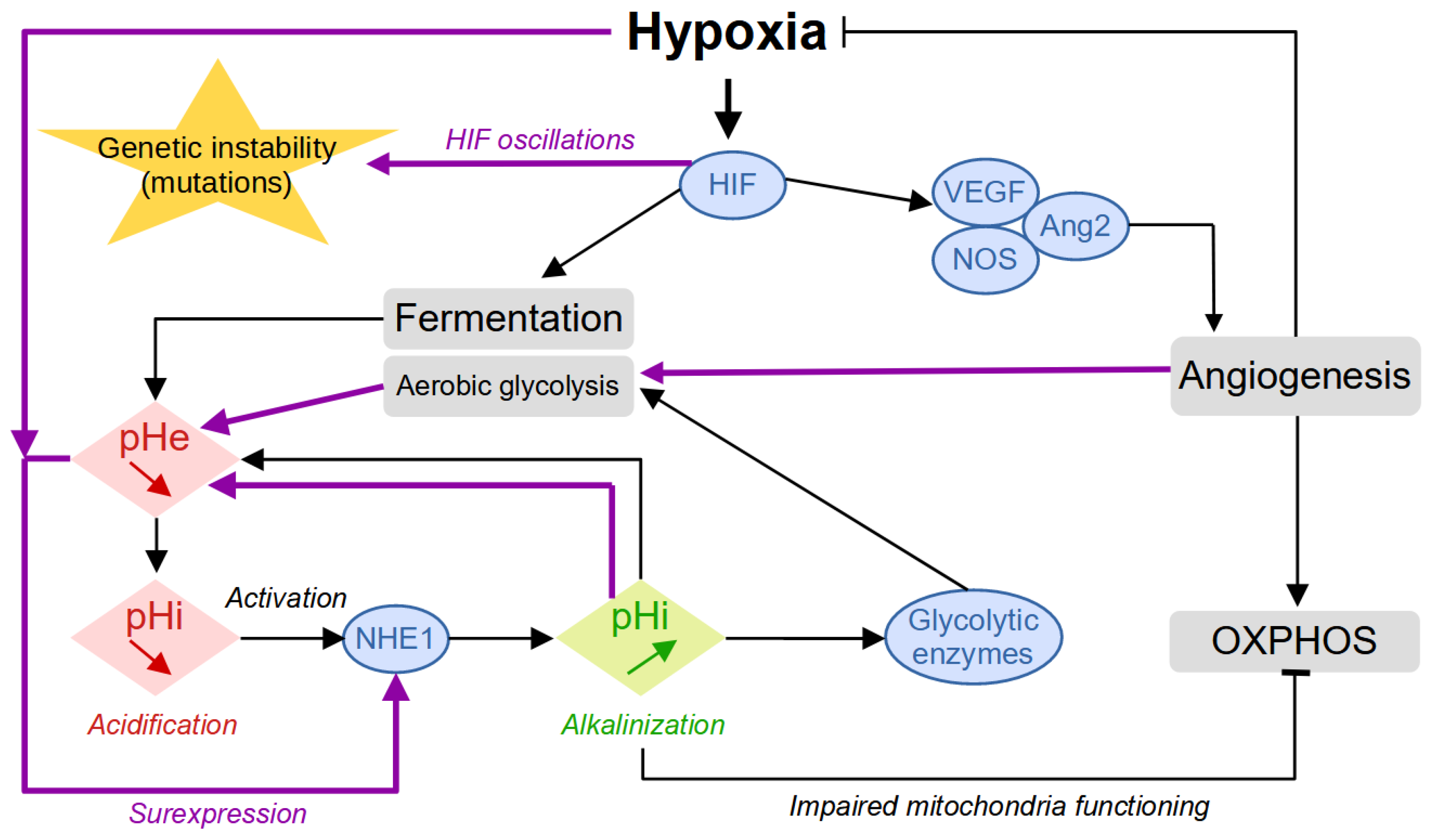
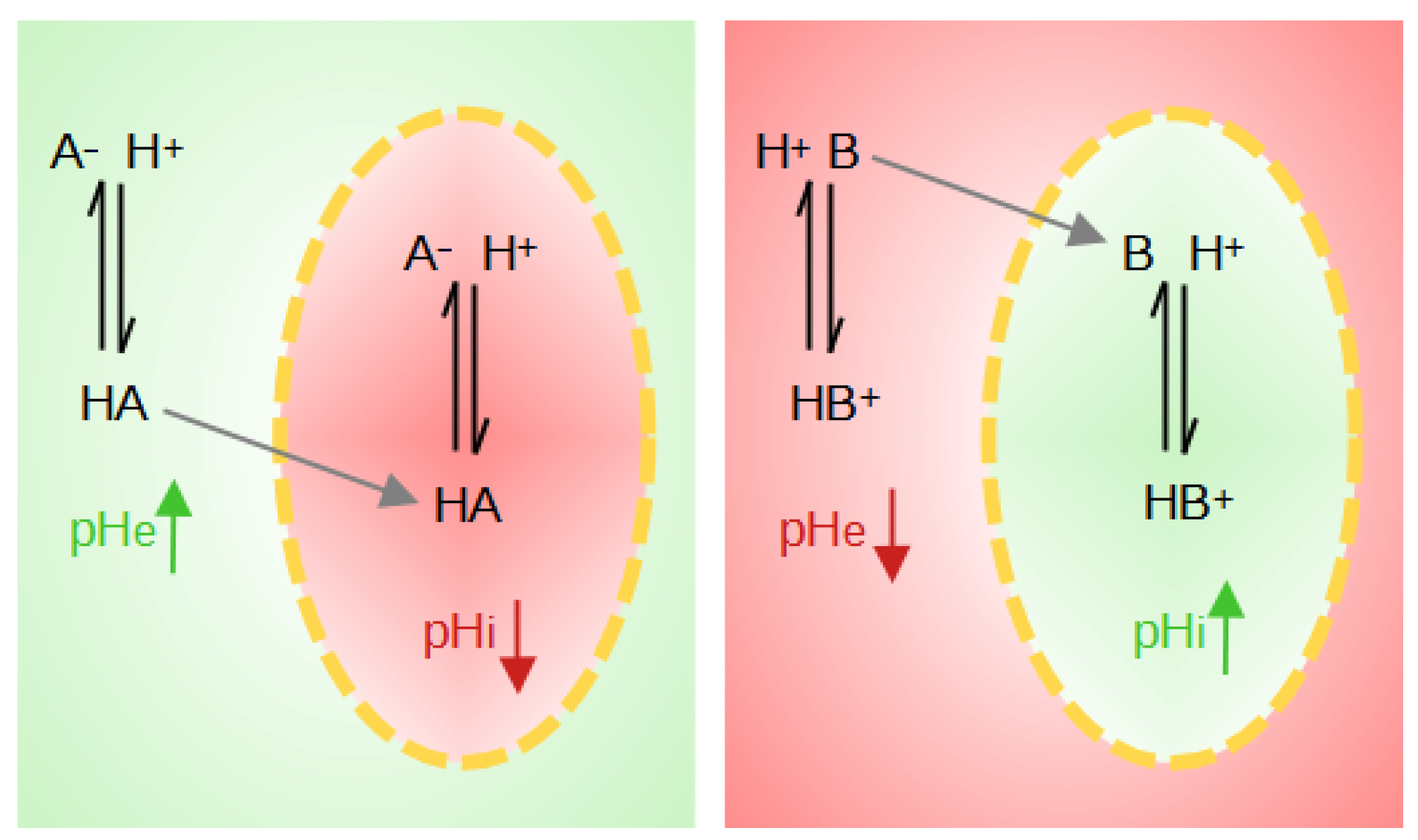
1. Origin of Tumor Acidity

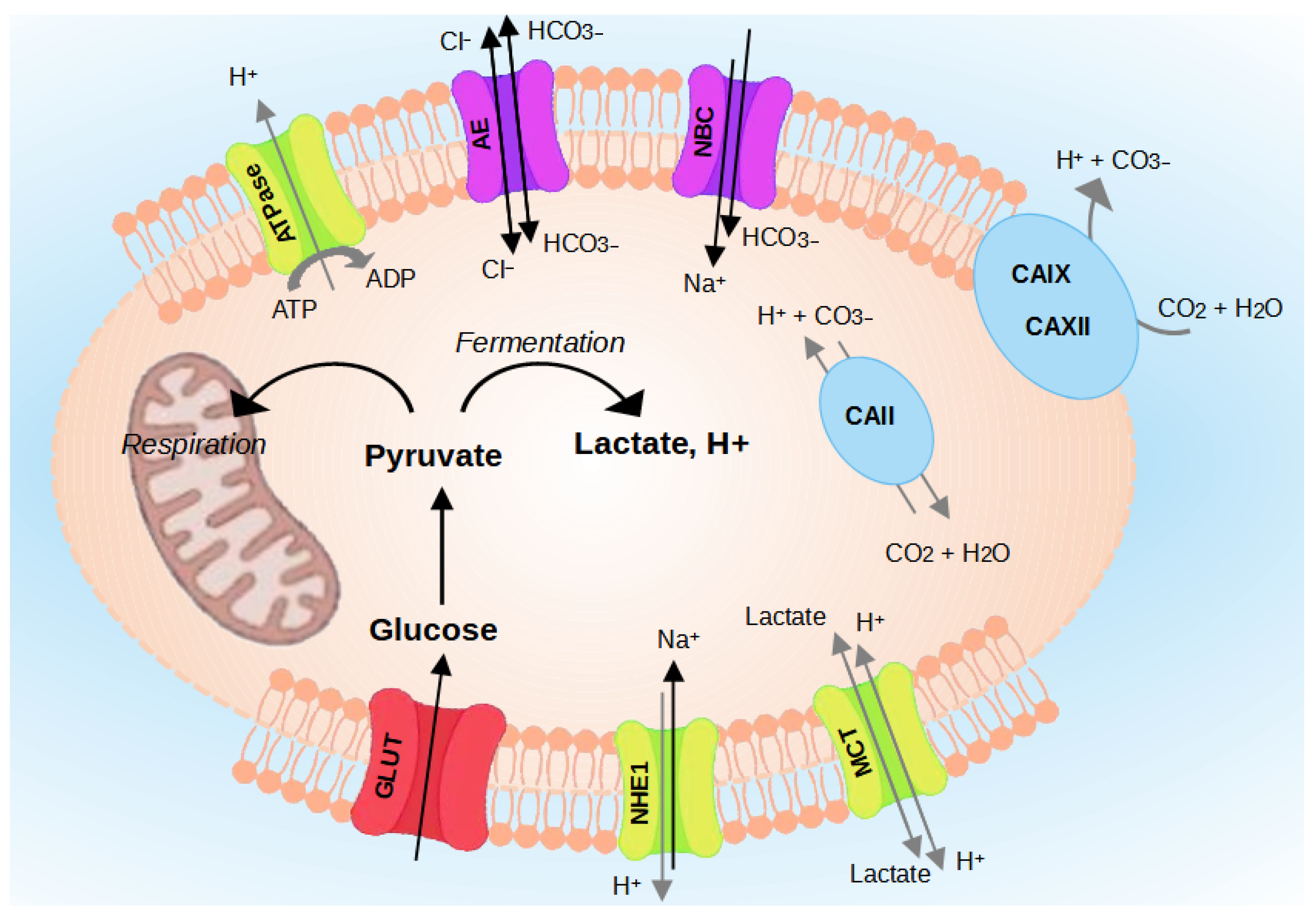
2. Regulation of Intracellular Acidity
2.1. The Family of Na+/H+ Exchangers (NHEs)
2.2. The Enzyme V-ATPase
2.3. Proton-Lactate Transporters (MCTs)
2.4. Carbonic Anhydrases (CA)
2.5. Transporters
3. Consequences of Acidity
3.1. Acidity and Tumor Invasion
3.2. Acidity and Cellular Plasticity
3.3. Acidity and Immunosuppression
3.4. Acidity and Warburg Effect
4. Evolution of the Methods for Intracellular pH Measurements
4.1. pH Microelectrodes
4.2. 31P NMR Spectroscopy
4.3. Weak Acids and Bases
4.4. Fluorescence Microscopy
5. Acidity and Therapy
5.1. Extracellular Acidity and Chemotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Cancer Type | Cell Model | Tested Drug | pH Assessed | Mechanisms Evaluated | Main Results |
|---|---|---|---|---|---|---|
| Mellor and Callaghan (2011) [144] | Colorectal adenocarcinoma (DLD-1), colon adenocarcinoma (HT29), and ovarian adenocarcinoma (NCIADR) | in vitro | Doxorubicin (DOX) | 6.9–7.0 and 7.2–7.3 | Growth of tumor spheroids in normoxia and hypoxia, intracellular accumulation of DOX, and inhibition of Pgp by Tariquidar (XR9576) | The distribution and accumulation of DOX were heterogeneous in all cell lines evaluated. The acidity generated by hypoxia decreased the accumulation of DOX in tumor spheroid. The inibition of Pgp by Tariquidar (XR9576) increased the accumulation of DOX in tumor spheroids. |
| Avnet et al. (2016) [132] | Osteosarcoma | in vivo | Doxorubicin (DOX) | 6.5 and 7.4 | Combined treatment of DOX and omeprazole (a proton pump inhibitor targeting lysosomal acidity) | The combined treatment of DOX with omeprazole showed a higher necrotic areas, smaller tumor volumes, and less body weight loss. |
| Fan et al. (2012) [135] | Breast cancer (MCF-7/ADR) | in vitro and in vivo | Doxorubicin (DOX), Cisplatin (CIS), and 5-fluorouracil (5-FU) | Acidic pH (unknown value) | Induction of LASS2 expression | The overexpression of LASS2 in MCF-7/ADR breast cancer cells increased the effect of several chemotherapeutic agents. LASS2 inhibited the function of V-ATPase. More DOX entered the cells and stayed in the nuclei of cells, inducing increased rates of apoptosis. |
| Visioli et al. 2014 [136] | Endothelial cells from human oral squamous cell carcinomas (OSCC) | in vitro | Sunitinib | 6.0–6.4 | Activation of UPR: quantification of Grp78 in endothelial cells and cytotoxicity using SRB | Extracellular acidity increased expression of the UPR marker (Grp78) and its inhibition reversed the drug sensitivity to Sunitinib. |
| Cheng and To (2012) [134] | Human colon carcinoma HCT-116 and S1 and its ABCG2-overexpressing resistant S1M1-80 cell lines | in vitro | Cisplatin (CIS) and mitoxantrone | 5.00 | Regulation of ABCG2 under adverse conditions within the tumor microenvironment (hypoxia, glucose deprivation, and acidosis) | Glucose depletion, decreased extracellular pH, and hypoxia can all upregulate ABCG2 transcript level leading to multidrug resistance. Acidic pH did not significantly alter the level of ABCG2 in S1 cells. |
| Federici et al. 2014 [139] | MCF7 (human breast cancer), Me30966 and Me501 (human metastatic melanoma), and SW480 (human colon carcinoma) cell lines | both in vitro and in vivo experiments | Cisplatin (CIS) | acidic (5.0–6.0), buffered (7.4), and unbuffered media | Cisplatin cellular resistance and effects of proton pump inhibitor (lansoprazole) on Cisplatin tumor uptake | Treatment with lansoprazole increased the intracellular absorption of Cisplatin and reduced the amount of Cisplatin present in the exosomes. |
| Thews et al. (2006) [133] | Rat prostate cancer (AT1) | in vitro | Daunorubicin (DNR) and Cisplatin (CIS) | 6.6 and 7.4 | Caspase 3 activity and cell toxicity assay | Exposure to acidic extracellular environment doubled Pgp activity and reduced cytotoxic efficacy of CIS and DNR. |
| Thews et al. (2014) [143] | Rat prostate cancer (AT1) | in vitro and in vivo | Daunorubicin (DNR), Cisplatin (CIS), and docetaxel (DOC) | 6.6 and 7.4 | Measurement of apoptosis induction (caspase 3) and cell survival | Acidity reduced the cytotoxicity of DAU, CIS, and DOC. The Pgp inhibitor (verapamil) reversed the acidosis-induced chemoresistance against DNR and DOC. |
5.2. The Case of Temozolomide
5.3. Targeting the Membrane Transporters
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Kallinowski, F.; Vaupel, P. pH distributions in spontaneous and isotransplanted rat tumours. British J. Cancer 1988, 58, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Ueber den Stoffwechsel der Tumoren: Arbeiten aus dem Kaiser Wilhelm-Institut für Biologie, Berlin-Dahlem. JAMA 1926, 87, 1671. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Kerbel, R.; Folkman, J. Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2002, 2, 727–739. [Google Scholar] [CrossRef]
- Serres, S.; O’Brien, E.R.; Sibson, N.R. Imaging Angiogenesis, Inflammation, and Metastasis in the Tumor Microenvironment with Magnetic Resonance Imaging. In Tumor Microenvironment and Cellular Stress. Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Gupta, P.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Reshkin, S.; Bellizzi, A.; Cardone, R.; Tommasino, M.; Casavola, V.; Paradiso, A. Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na+/H+ exchanger (NHE) NHE isoform 1 in human breast cancer cells. Clin. Cancer Res. 2003, 9, 2366–2373. [Google Scholar]
- Bedessem, B.; Stéphanou, A. Role of Compartmentalization on HiF-1a Degradation Dynamics during Changing Oxygen Conditions: A Computational Approach. PLoS ONE 2014, 9, e110495. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Boron, W.F. Intracellular pH. Physiol. Rev. 1981, 61, 296–434. [Google Scholar] [CrossRef] [PubMed]
- Madshus, I.H. Regulation of intracellular pH in eukaryotic cells. Biochem. J. 1988, 250, 1–8. [Google Scholar] [CrossRef]
- Zhuang, Y.X.; Cragoe, E.J.; Shaikewitz, T.; Glaser, L.; Cassel, D. Characterization of potent sodium/proton exchange inhibitors from the amiloride series in A431 cells. Biochemistry 1984, 23, 4481–4488. [Google Scholar] [CrossRef] [PubMed]
- Druzhkova, I.; Shirmanova, M.; Lukina, M.; Dudenkova, V.; Sergeeva, T.; Belousov, V.; Lukyanov, S.; Zagaynova, E. Registration of intracellular pH in cancer cells with genetically encoded ratiometric sensor. In Proceedings of the European Conference on Biomedical Optics 2015, Munich, Germany, 21–25 June 2015. [Google Scholar] [CrossRef]
- Shirmanova, M.V.; Druzhkova, I.N.; Lukina, M.M.; Matlashov, M.E.; Belousov, V.V.; Snopova, L.B.; Prodanetz, N.N.; Dudenkova, V.V.; Lukyanov, S.A.; Zagaynova, E.V. Intracellular pH imaging in cancer cells in vitro and tumors in vivo using the new genetically encoded sensor SypHer2. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 1905–1911. [Google Scholar] [CrossRef]
- Gillies, R.J.; Raghunand, N.; Karczmar, G.S.; Bhujwalla, Z.M. MRI of the tumor microenvironment. J. Magn. Reson. Imaging 2002, 16, 430–450. [Google Scholar] [CrossRef] [PubMed]
- Tafech, A.; Jacquet, P.; Beaujean, C.; Fertin, A.; Usson, Y.; Stéphanou, A. Characterization of the Intracellular Acidity Regulation of Brain Tumor Cells and Consequences for Therapeutic Optimization of Temozolomide. Biology 2023, 12, 1221. [Google Scholar] [CrossRef] [PubMed]
- Barathova, M.; Takacova, M.; Holotnakova, T.; Gibadulinova, A.; Ohradanova, A.; Zatovicova, M.; Hulikova, A.; Kopacek, J.; Parkkila, S.; Supuran, C.T.; et al. Alternative splicing variant of the hypoxia marker carbonic anhydrase IX expressed independently of hypoxia and tumour phenotype. Br. J. Cancer 2007, 98, 129–136. [Google Scholar] [CrossRef]
- Hinton, A.; Sennoune, S.R.; Bond, S.; Fang, M.; Reuveni, M.; Sahagian, G.G.; Jay, D.; Martinez-Zaguilan, R.; Forgac, M. Function of a Subunit Isoforms of the V-ATPase in pH Homeostasis and in Vitro Invasion of MDA-MB231 Human Breast Cancer Cells. J. Biol. Chem. 2009, 284, 16400–16408. [Google Scholar] [CrossRef]
- Enerson, B.E.; Drewes, L.R. Molecular Features, Regulation, and Function of Monocarboxylate Transporters: Implications for Drug Delivery. J. Pharm. Sci. 2003, 92, 1531–1544. [Google Scholar] [CrossRef]
- Supuran, C.T.; Fiore, A.D.; Alterio, V.; Montib, S.M.; Simone, G.D. Recent Advances in Structural Studies of the Carbonic Anhydrase Family: The Crystal Structure of Human CA IX and CA XIII. Curr. Pharm. Des. 2010, 16, 3246–3254. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Fuster, D.; Moe, O.W.; Hilgemann, D.W. Lipid- and mechanosensitivities of sodium/hydrogen exchangers analyzed by electrical methods. Proc. Natl. Acad. Sci. USA 2004, 101, 10482–10487. [Google Scholar] [CrossRef] [PubMed]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Orive, G.; Pedraz, J.L.; Paradiso, A.; Reshkin, S.J. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin—One single nature. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2005, 1756, 1–24. [Google Scholar] [CrossRef]
- Stock, C.; Schwab, A. Protons make tumor cells move like clockwork. Pflügers Archiv.-Eur. J. Physiol. 2009, 458, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Gluck, S.; Hartwig, J. Structure of the novel membrane-coating material in proton-secreting epithelial cells and identification as an H+ATPase. J. Cell Biol. 1987, 105, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Forgac, M. Structure and function of vacuolar class of ATP-driven proton pumps. Physiol. Rev. 1989, 69, 765–796. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zaguilan, R.; Gillies, R.J. A Plasma Membrane V-type H+-ATPase May Contribute to Elevated Intracellular pH (pHin) in Some Human Tumor Cells. Ann. N. Y. Acad. Sci. 1992, 671, 478–480. [Google Scholar] [CrossRef]
- Martinez-Zaguilan, R.; Lynch, R.M.; Martinez, G.M.; Gillies, R.J. Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Cell Physiol. 1993, 265, C1015–C1029. [Google Scholar] [CrossRef]
- Martínez-Zaguilán, R.; Martinez, G.M.; Gomez, A.; Hendrix, M.J.C.; Gillies, R.J. Distinct regulation of pHin and [Ca2+]in in human melanoma cells with different metastatic potential. J. Cell. Physiol. 1998, 176, 196–205. [Google Scholar] [CrossRef]
- Sennoune, S.R.; Bakunts, K.; Martínez, G.M.; Chua-Tuan, J.L.; Kebir, Y.; Attaya, M.N.; Martínez-Zaguilán, R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: Distribution and functional activity. Am. J. Physiol.-Cell Physiol. 2004, 286, C1443–C1452. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; Saedeleer, C.J.D.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Takahashi, M.; Uramoto, H.; Nakayama, Y.; Oyama, T.; Wang, K.Y.; Sasaguri, Y.; Nishizawa, S.; Kohno, K. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011, 102, 1007–1013. [Google Scholar] [CrossRef]
- Gallagher, S.M.; Castorino, J.J.; Wang, D.; Philp, N.J. Monocarboxylate Transporter 4 Regulates Maturation and Trafficking of CD147 to the Plasma Membrane in the Metastatic Breast Cancer Cell Line MDA-MB-231. Cancer Res. 2007, 67, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Froberg, M.K.; Gerhart, D.Z.; Enerson, B.E.; Manivel, C.; Guzman-Paz, M.; Seacotte, N.; Drewes, L.R. Expression of monocarboxylate transporter MCT1 in normal and neoplastic human CNS tissues. Neuroreport 2001, 12, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, K. Perspectives on carbonic anhydrase. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2010, 157, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Review Article. J. Enzym. Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Bartošová, M.; Parkkila, S.; Pohlodek, K.; Karttunen, T.J.; Galbavý, Š.; Mucha, V.; Harris, A.L.; Pastorek, J.; Pastoreková, S. Expression of carbonic anhydrase IX in breast is associated with malignant tissues and is related to overexpression of c-erbB2. J. Pathol. 2002, 197, 314–321. [Google Scholar] [CrossRef]
- Ivanov, S.; Liao, S.Y.; Ivanova, A.; Danilkovitch-Miagkova, A.; Tarasova, N.; Weirich, G.; Merrill, M.J.; Proescholdt, M.A.; Oldfield, E.H.; Lee, J.; et al. Expression of Hypoxia-Inducible Cell-Surface Transmembrane Carbonic Anhydrases in Human Cancer. Am. J. Pathol. 2001, 158, 905–919. [Google Scholar] [CrossRef]
- Švastová, E.; Hulıková, A.; Rafajová, M.; Zat’ovičová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2008, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. pH control mechanisms of tumor survival and growth. J. Cell. Physiol. 2010, 226, 299–308. [Google Scholar] [CrossRef]
- Parks, S.K.; Mazure, N.M.; Counillon, L.; Pouysségur, J. Hypoxia promotes tumor cell survival in acidic conditions by preserving ATP levels. J. Cell. Physiol. 2013, 228, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Patiar, S.; Supuran, C.T.; Harris, A.L.; Vaughan-Jones, R.D. The Role of Carbonic Anhydrase 9 in Regulating Extracellular and Intracellular pH in Three-dimensional Tumor Cell Growths. J. Biol. Chem. 2009, 284, 20299–20310. [Google Scholar] [CrossRef] [PubMed]
- Grabmaier, K.; de Weijert, M.C.; Verhaegh, G.W.; Schalken, J.A.; Oosterwijk, E. Strict regulation of CAIXG250/MN by HIF-1α in clear cell renal cell carcinoma. Oncogene 2004, 23, 5624–5631. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.V.; Kuzmin, I.; Wei, M.H.; Pack, S.; Geil, L.; Johnson, B.E.; Stanbridge, E.J.; Lerman, M.I. Down-regulation of transmembrane carbonic anhydrases in renal cell carcinoma cell lines by wild-type von Hippel-Lindau transgenes. Proc. Natl. Acad. Sci. USA 1998, 95, 12596–12601. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Hilvo, M.; Fiore, A.D.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Oosterwijk, E.; Tu, C.; Shiverick, K.T.; Silverman, D.N.; Frost, S.C. Expression and Activity of Carbonic Anhydrase IX Is Associated With Metabolic Dysfunction in MDA-MB-231 Breast Cancer Cells. Cancer Investig. 2009, 27, 613–623. [Google Scholar] [CrossRef]
- Parkkila, A.K.; Herva, R.; Parkkila, S.; Rajaniemi, H. Immunohistochemical demonstration of human carbonic anhydrase isoenzyme II in brain tumours. Histochem. J. 1995, 27, 974–982. [Google Scholar] [CrossRef]
- Nordfors, K.; Haapasalo, J.; Korja, M.; Niemelä, A.; Laine, J.; Parkkila, A.K.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; et al. The tumour-associated carbonic anhydrases CA II, CA IX and CA XII in a group of medulloblastomas and supratentorial primitive neuroectodermal tumours: An association of CA IX with poor prognosis. BMC Cancer 2010, 10, 148. [Google Scholar] [CrossRef]
- Parkkila, S.; Lasota, J.; Fletcher, J.A.; bin Ou, W.; Kivelä, A.J.; Nuorva, K.; Parkkila, A.K.; Ollikainen, J.; Sly, W.S.; Waheed, A.; et al. Carbonic anhydrase II. A novel biomarker for gastrointestinal stromal tumors. Mod. Pathol. 2010, 23, 743–750. [Google Scholar] [CrossRef]
- Pan, P.; Leppilampi, M.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Parkkila, S. Carbonic anhydrase gene expression in CA II-deficient (Car2−/−) and CA IX-deficient (Car9−/−) mice. J. Physiol. 2006, 571, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouysségur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Arranz, J.L.; Wahl, M.L.; Orive, G.; Reshkin, S.J. Proton Transport Inhibitors as Potentially Selective Anticancer Drugs. Anticancer Res. 2009, 29, 2127–2136. [Google Scholar] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Wan, P.; Grinstein, S.; Tannock, I.F. Cytotoxicity of compounds that interfere with the regulation of intracellular pH: A potential new class of anticancer drugs. Cancer Res. 1987, 47, 1497–1504. [Google Scholar] [PubMed]
- Yamagata, M.; Tannock, I. The chronic administration of drugs that inhibit the regulation of intracellular pH: In vitro and anti-tumour effects. Br. J. Cancer 1996, 73, 1328–1334. [Google Scholar] [CrossRef]
- Montcourrier, P.; Silver, I.; Farnoud, R.; Bird, I.; Rochefort, H. Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin. Exp. Metastasis 1997, 15, 382–392. [Google Scholar] [CrossRef]
- Grillon, E.; Farion, R.; Fablet, K.; Waard, M.D.; Tse, C.M.; Donowitz, M.; Rémy, C.; Coles, J.A. The Spatial Organization of Proton and Lactate Transport in a Rat Brain Tumor. PLoS ONE 2011, 6, e17416. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Singleton, P.A.; Diedrich, F.; Stern, R.; Gilad, E. CD44 Interaction with Na+-H+ Exchanger (NHE1) Creates Acidic Microenvironments Leading to Hyaluronidase-2 and Cathepsin B Activation and Breast Tumor Cell Invasion. J. Biol. Chem. 2004, 279, 26991–27007. [Google Scholar] [CrossRef] [PubMed]
- Rozhin, J.; Sameni, M.; Ziegler, G.; Sloane, B.F. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res. 1994, 54, 6517–6525. [Google Scholar] [PubMed]
- Yang, X.; Wang, D.; Dong, W.; Song, Z.; Dou, K. Inhibition of Na+/H+ exchanger 1 by 5-(N-ethyl-N-isopropyl) amiloride reduces hypoxia-induced hepatocellular carcinoma invasion and motility. Cancer Lett. 2010, 295, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Lambert, C.A.; Colige, A.C.; Mineur, P.; Noël, A.; Frankenne, F.; Foidart, J.M.; Baba, M.; Hata, R.I.; Miyazaki, K.; et al. Acidic Extracellular pH Induces Matrix Metalloproteinase-9 Expression in Mouse Metastatic Melanoma Cells through the Phospholipase D-Mitogen-activated Protein Kinase Signaling. J. Biol. Chem. 2005, 280, 10938–10944. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Robey, I.F.; Nesbit, L.A. Investigating Mechanisms of Alkalinization for Reducing Primary Breast Tumor Invasion. BioMed Res. Int. 2013, 2013, 485196. [Google Scholar] [CrossRef]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2010, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; Milito, A.D.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of Microenvironment Acidity Reverses Anergy in Human and Murine Tumor-Infiltrating T Lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Heiden, M.G.V. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Loos, J. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. Exp. Cell Res. 1973, 77, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Marquardt, C.; Foker, J. Aerobic glycolysis during lymphocyte proliferation. Nature 1976, 261, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Hedeskov, C.J. Early effects of phytohaemagglutinin on glucose metabolism of normal human lymphocytes. Biochem. J. 1968, 110, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Razungles, J.; Cavaillès, V.; Jalaguier, S.; Teyssier, C. L’effet Warburg. Med. Sci. (Paris) 2013, 29, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxidants Redox Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef]
- Wu, H.; Ying, M.; Hu, X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget 2016, 7, 40621–40629. [Google Scholar] [CrossRef]
- Jacquet, P.; Stéphanou, A. A reduced model of cell metabolism to revisit the glycolysis-OXPHOS relationship in the deregulated tumor microenvironment. J. Theor. Biol. 2023, 562, 111434. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wu, H.; Dai, C.; Pan, Q.; Ding, Z.; Hu, D.; Ji, B.; Luo, Y.; Hu, X. Beyond Warburg effect—Dual metabolic nature of cancer cells. Sci. Rep. 2014, 4, 4927. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, P.; Stéphanou, A. Searching for the Metabolic Signature of Cancer: A Review from Warburg’s Time to Now. Biomolecules 2022, 12, 1412. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Nagaki, T.; Fukuda, I.; Okumura, K. Clastogenicity of low pH to various cultured mammalian cells. Fundam. Mol. Mech. Mutagen. 1992, 268, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Li, T.K.; Yang, J.M.; Liu, L.F. Acidic pH induces topoisomerase II-mediated DNA damage. Proc. Natl. Acad. Sci. USA 2003, 100, 5205–5210. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, T.; Wang, X.; Takahashi, A.; Ohnishi, K.; Saito, H.; Song, C.W.; Ohnishi, T. p53-dependent induction of WAF1 by a low-pH culture condition in human glioblastoma cells. Cancer Res. 1997, 57, 3910–3913. [Google Scholar]
- Williams, A.C.; Collard, T.J.; Paraskeva, C. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: Implications for clonal selection during colorectal carcinogenesis. Oncogene 1999, 18, 3199–3204. [Google Scholar] [CrossRef] [PubMed]
- Putney, L.K.; Barber, D.L. Na-H Exchange-dependent Increase in Intracellular pH Times G2/M Entry and Transition. J. Biol. Chem. 2003, 278, 44645–44649. [Google Scholar] [CrossRef] [PubMed]
- Smallbone, K.; Maini, P.K.; Gatenby, R.A. Episodic, transient systemic acidosis delays evolution of the malignant phenotype: Possible mechanism for cancer prevention by increased physical activity. BMC Biol. Direct 2010, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, P.C. Intracellular pH. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 1956; pp. 229–277. [Google Scholar] [CrossRef]
- Thomas, R.C. Intracellular pH of snail neurones measured with a new pH-sensitive glass micro-electrode. J. Physiol. 1974, 238, 159–180. [Google Scholar] [CrossRef]
- Lagadic-Gossmann, D.; Chesnais, J.M.; Feuvray, D. Intracellular pH regulation in papillary muscle cells from streptozotocin diabetic rats: An ion-sensitive microelectrode study. Pflügers Archiv.-Eur. J. Physiol. 1988, 412, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.D. Effects of insulin upon ion transport. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 1983, 737, 1–49. [Google Scholar] [CrossRef]
- Ellis, D.; Thomas, R.C. Microelectrode measurement of the intracellular pH of mammalian heart cells. Nature 1976, 262, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Zeuthen, T. Chapter 3 How to Make and Use Double-Barreled Ion-Selective Microelectrodes. In Current Topics in Membranes and Transport; Elsevier: Amsterdam, The Netherlands, 1980; pp. 31–47. [Google Scholar] [CrossRef]
- de Hemptinne, A. Intracellular pH and surface pH in skeletal and cardiac muscle measured with a double-barrelled pH microelectrode. Pflügers Archiv.-Eur. J. Physiol. 1980, 386, 121–126. [Google Scholar] [CrossRef]
- Hagberg, H.; Larsson, S.; Haljamae, H. A new design of double-barrelled microelectrodes for intracellular pH-measurement in vivo. Acta Physiol. Scand. 1983, 118, 149–153. [Google Scholar] [CrossRef]
- Claire, C.; Thomas, R.C. Micro-electrode measurement of the intracellular pH and buffering power of mouse soleus muscle fibres. J. Physiol. 1977, 267, 791–810. [Google Scholar] [CrossRef]
- Heiple, J.M.; Taylor, D.L. Intracellular pH in single motile cells. J. Cell Biol. 1980, 86, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.B.; Richards, J.H. Determination of Intracellular pH by 31P Magnetic Resonance. J. Biol. Chem. 1973, 248, 7276–7278. [Google Scholar] [CrossRef]
- Salhany, J. 31P nuclear magnetic resonance measurement of cardiac pH in perfused guinea-pig hearts. J. Mol. Cell. Cardiol. 1979, 11, 601–610. [Google Scholar] [CrossRef]
- Brindle, K.M.; Rajagopalan, B.; Williams, D.S.; Detre, J.A.; Simplaceanu, E.; Ho, C.; Radda, G.K. 31P NMR measurements of myocardial pH invivo. Biochem. Biophys. Res. Commun. 1988, 151, 70–77. [Google Scholar] [CrossRef]
- Clarke, K.; Stewart, L.C.; Neubauer, S.; Balschi, J.A.; Smith, T.W.; Ingwall, J.S.; Nédélec, J.F.; Humphrey, S.M.; Kléber, A.G.; Springer, C.S. Extracellular volume and transsarcolemmal proton movement during ischemia and reperfusion: A31P NMR spectroscopic study of the isovolumic rat heart. NMR Biomed. 1993, 6, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.A.; Swietach, P.; Atherton, H.J.; Gallagher, F.A.; Lee, P.; Radda, G.K.; Clarke, K.; Tyler, D.J. Measuring intracellular pH in the heart using hyperpolarized carbon dioxide and bicarbonate: A 13C and 31P magnetic resonance spectroscopy study. Cardiovasc. Res. 2009, 86, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R.; Schaffer, S.W.; Ford, C.; Safer, B. Contribution of tissue acidosis to ischemic injury in the perfused rat heart. Circulation 1976, 53 (Suppl. S3), I3-14. [Google Scholar] [PubMed]
- Ackerman, J.J.; Lowry, M.; Radda, G.K.; Ross, B.D.; Wong, G.G. The role of intrarenal pH in regulation of ammoniagenesis: [31P]NMR studies of the isolated perfused rat kidney. J. Physiol. 1981, 319, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Adam, W.R.; Koretsky, A.P.; Weiner, M.W. Measurement of Renal Intracellular pH by 31P NMR. In Contributions to Nephrology; S.Karger AG: Basel, Switzerland, 1985; pp. 15–21. [Google Scholar] [CrossRef]
- Adam, W.R.; Koretsky, A.P.; Weiner, M.W. 31P-NMR in vivo measurement of renal intracellular pH: Effects of acidosis and K+ depletion in rats. Am. J. Physiol.-Ren. Physiol. 1986, 251, F904–F910. [Google Scholar] [CrossRef] [PubMed]
- Tannen, R.L. Ammonia metabolism. Am. J. Physiol.-Ren. Physiol. 1978, 235, F265–F277. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Ugurbil, K.; Chen, W. Measurement of unidirectional Pi to ATP flux in human visual cortex at 7 T by using 31 P magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 14409–14414. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Zhu, X.H.; Zhang, X.L.; Ugurbil, K.; Chen, W. In vivo31P magnetic resonance spectroscopy of human brain at 7 T: An initial experience. Magn. Reson. Med. 2003, 49, 199–205. [Google Scholar] [CrossRef]
- Leyval, D.; Debay, F.; Engasser, J.M.; Goergen, J.L. Flow cytometry for the intracellular pH measurement of glutamate producing Corynebacterium glutamicum. J. Microbiol. Methods 1997, 29, 121–127. [Google Scholar] [CrossRef]
- Imai, T.; Ohno, T. Measurement of yeast intracellular pH by image processing and the change it undergoes during growth phase. J. Biotechnol. 1995, 38, 165–172. [Google Scholar] [CrossRef]
- Molenaar, D.; Abee, T.; Konings, W.N. Continuous measurement of the cytoplasmic pH in Lactococcus lactis with a fluorescent pH indicator. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1991, 1115, 75–83. [Google Scholar] [CrossRef]
- Brown, D.A.; Garthwaite, J. Intracellular pH and the distribution of weak acids and bases in isolated rat superior cervical ganglia. J. Physiol. 1979, 297, 597–620. [Google Scholar] [CrossRef] [PubMed]
- Padan, E.; Zilberstein, D.; Schuldiner, S. pH homesstasis in bacteria. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 1981, 650, 151–166. [Google Scholar] [CrossRef]
- Waddell, W.J.; Butler, T.C. Calculation of intracellular pH from the distribution of 5,5-Dimethyl-2,4-Oxazolidinedione (DMO). Application to skeletal muscle of the dog. J. Clin. Investig. 1959, 38, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Gores, G.J.; Nieminen, A.L.; Wray, B.E.; Herman, B.; Lemasters, J.J. Intracellular pH during “chemical hypoxia” in cultured rat hepatocytes. Protection by intracellular acidosis against the onset of cell death. J. Clin. Investig. 1989, 83, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Makutonina, A.; Voss, T.G.; Plymale, D.R.; Fermin, C.D.; Norris, C.H.; Vigh, S.; Garry, R.F. Human immunodeficiency virus infection of T-lymphoblastoid cells reduces intracellular pH. J. Virol. 1996, 70, 7049–7055. [Google Scholar] [CrossRef]
- Garry, R.F.; Gottlieb, A.A.; Zuckerman, K.P.; Pace, J.R.; Frank, T.W.; Bostick, D.A. Cell surface effects of human immunodeficiency virus. Biosci. Rep. 1988, 8, 35–48. [Google Scholar] [CrossRef]
- Voss, T.G.; Fermin, C.D.; Levy, J.A.; Vigh, S.; Choi, B.; Garry, R.F. Alteration of intracellular potassium and sodium concentrations correlates with induction of cytopathic effects by human immunodeficiency virus. J. Virol. 1996, 70, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Salvi, A.; Quillan, J.M.; Sadée, W. Monitoring intracellular pH changes in response to osmotic stress and membrane transport activity using 5-chloromethylfluorescein. AAPS PharmSci 2002, 4, 21–28. [Google Scholar] [CrossRef]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef]
- Tsien, R.Y. A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 1981, 290, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Tafech, A.; Beaujean, C.; Usson, Y.; Stéphanou, A. Generalization of the Ratiometric Method to Extend pH Range Measurements of the BCECF Probe. Biomolecules 2023, 13, 442. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Black, K.C.; Krishnamurty, C.; Baggett, B.K.; Stafford, P.; Rain, M.; Gatenby, R.A.; Gillies, R.J. Acid treatment of melanoma cells selects for invasive phenotypes. Clin. Exp. Metastasis 2008, 25, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Gillies, R.D.; Gatenby, R.A. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin. Cancer Biol. 2008, 18, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Fernández, V.M. An electrochemical cell for reduction of biochemicals: Its application to the study of the effect of pH and redox potential on the activity of hydrogenases. Anal. Biochem. 1983, 130, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Lemma, S.; Cortini, M.; Pellegrini, P.; Perut, F.; Zini, N.; Kusuzaki, K.; Chano, T.; Grisendi, G.; Dominici, M.; et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget 2016, 7, 63408–63423. [Google Scholar] [CrossRef]
- Thews, O.; Gassner, B.; Kelleher, D.K.; Schwerd, G.; Gekle, M. Impact of Extracellular Acidity on the Activity of P-glycoprotein and the Cytotoxicity of Chemotherapeutic Drugs. Neoplasia 2006, 8, 143–152. [Google Scholar] [CrossRef]
- Cheng, G.M.Y.; To, K.K.W. Adverse Cell Culture Conditions Mimicking the Tumor Microenvironment Upregulate ABCG2 to Mediate Multidrug Resistance and a More Malignant Phenotype. ISRN Oncol. 2012, 2012, 746025. [Google Scholar] [CrossRef]
- Fan, S.; Niu, Y.; Tan, N.; Wu, Z.; Wang, Y.; You, H.; Ke, R.; Song, J.; Shen, Q.; Wang, W.; et al. LASS2 enhances chemosensitivity of breast cancer by counteracting acidic tumor microenvironment through inhibiting activity of V-ATPase proton pump. Oncogene 2012, 32, 1682–1690. [Google Scholar] [CrossRef]
- Visioli, F.; Wang, Y.; Alam, G.N.; Ning, Y.; Rados, P.V.; Nör, J.E.; Polverini, P.J. Glucose-Regulated Protein 78 (Grp78) Confers Chemoresistance to Tumor Endothelial Cells under Acidic Stress. PLoS ONE 2014, 9, e101053. [Google Scholar] [CrossRef]
- Tarling, E.J.; de Aguiar Vallim, T.Q.; Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 2013, 24, 342–350. [Google Scholar] [CrossRef]
- Gillet, J.; Remacle, J. Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2007, 1775, 237–262. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome Release and Low pH Belong to a Framework of Resistance of Human Melanoma Cells to Cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [PubMed]
- Genovese, I.; Ilari, A.; Assaraf, Y.G.; Fazi, F.; Colotti, G. Not only P-glycoprotein: Amplification of the ABCB1- containing chromosome region 7q21 confers multidrug resistance upon cancer cells by coordinated overexpression of an assortment of resistance-related proteins. Drug Resist. Updat. 2017, 32, 23–46. [Google Scholar] [CrossRef]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. β-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef]
- Ponte-Sucre, A. Availability and applications of ATP-binding cassette (ABC) transporter blockers. Appl. Microbiol. Biotechnol. 2007, 76, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Thews, O.; Riemann, A.; Nowak, M.; Gekle, M. Impact of Hypoxia-Related Tumor Acidosis on Cytotoxicity of Different Chemotherapeutic Drugs In Vitro and In Vivo. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2014; pp. 51–58. [Google Scholar] [CrossRef]
- Mellor, H.R.; Callaghan, R. Accumulation and distribution of doxorubicin in tumour spheroids: The influence of acidity and expression of P-glycoprotein. Cancer Chemother. Pharmacol. 2011, 68, 1179–1190. [Google Scholar] [CrossRef]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.G.; Tsang, L.L.H.; Slack, J.A. NMR and Molecular Modeling Investigation of the Mechanism of Activation of the Antitumor Drug Temozolomide and Its Interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef]
- Ballesta, A.; Zhou, Q.; Zhang, X.; Lv, H.; Gallo, J. Multiscale Design of Cell-Type-Specific Pharmacokinetic/Pharmacodynamic Models for Personalized Medicine: Application to Temozolomide in Brain Tumors. CPT Pharmacometrics Syst. Pharmacol. 2014, 3, 112. [Google Scholar] [CrossRef]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Stéphanou, A.; Ballesta, A. pH as a potential therapeutic target to improve temozolomide antitumor efficacy: A mechanistic modeling study. Pharmacol. Res. Perspect. 2019, 7, e00454. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2020, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol. Ther. 2000, 85, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Miraglia, E.; Viarisio, D.; Riganti, C.; Costamagna, C.; Ghigo, D.; Bosia, A. Na+/H+ exchanger activity is increased in doxorubicin-resistant human colon cancer cells and its modulation modifies the sensitivity of the cells to doxorubicin. Int. J. Cancer 2005, 115, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Rebillard, A.; Tekpli, X.; Meurette, O.; Sergent, O.; LeMoigne-Muller, G.; Vernhet, L.; Gorria, M.; Chevanne, M.; Christmann, M.; Kaina, B.; et al. Cisplatin-Induced Apoptosis Involves Membrane Fluidification via Inhibition of NHE1 in Human Colon Cancer Cells. Cancer Res. 2007, 67, 7865–7874. [Google Scholar] [CrossRef]
- Spugnini, E.P.; Citro, G.; Fais, S. Proton pump inhibitors as anti vacuolar-ATPases drugs: A novel anticancer strategy. J. Exp. Clin. Cancer Res. 2010, 29, 44. [Google Scholar] [CrossRef]
- Milito, A.D.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2009, 127, 207–219. [Google Scholar] [CrossRef]
- Murakami, T.; Shibuya, I.; Ise, T.; Chen, Z.S.; ichi Akiyama, S.; Nakagawa, M.; Izumi, H.; Nakamura, T.; ichi Matsuo, K.; Yamada, Y.; et al. Elevated expression of vacuolar proton pump genes and cellular ph in cisplatin resistance. Int. J. Cancer 2001, 93, 869–874. [Google Scholar] [CrossRef]
- Torigoe, T.; Izumi, H.; Ishiguchi, H.; Uramoto, H.; Murakami, T.; Ise, T.; Yoshida, Y.; Tanabe, M.; Nomoto, M.; Itoh, H.; et al. Enhanced Expression of the Human Vacuolar H+-ATPase c subunit Gene (ATP6L) in Response to Anticancer Agents. J. Biol. Chem. 2002, 277, 36534–36543. [Google Scholar] [CrossRef] [PubMed]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-κB/IL-8 Pathway that Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Bueno, V.; Binet, I.; Steger, U.; Bundick, R.; Ferguson, D.; Murray, C.; Donald, D.; Wood, K. The Specific Monocarboxylate Transporter (MCT1) Inhibitor, AR-C117977, a Novel Immunosuppressant, Prolongs Allograft Survival in the Mouse. Transplantation 2007, 84, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, H.; Qi, Z.; Pahlman, C.; Veress, B.; Bundick, R.V.; Craggs, R.I.; Holness, E.; Edwards, S.; Murray, C.M.; Ferguson, D.; et al. The Specific Monocarboxylate Transporter-1 (MCT-1) Inhibitor, AR-C117977, Induces Donor-Specific Suppression, Reducing Acute and Chronic Allograft Rejection in the Rat. Transplantation 2007, 84, 1191–1199. [Google Scholar] [CrossRef]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tafech, A.; Stéphanou, A. On the Importance of Acidity in Cancer Cells and Therapy. Biology 2024, 13, 225. https://doi.org/10.3390/biology13040225
Tafech A, Stéphanou A. On the Importance of Acidity in Cancer Cells and Therapy. Biology. 2024; 13(4):225. https://doi.org/10.3390/biology13040225
Chicago/Turabian StyleTafech, Alaa, and Angélique Stéphanou. 2024. "On the Importance of Acidity in Cancer Cells and Therapy" Biology 13, no. 4: 225. https://doi.org/10.3390/biology13040225
APA StyleTafech, A., & Stéphanou, A. (2024). On the Importance of Acidity in Cancer Cells and Therapy. Biology, 13(4), 225. https://doi.org/10.3390/biology13040225