
Immunopeptidomics of Salmonella enterica Serovar Typhimurium-Infected Pig Macrophages Genotyped for Class II Molecules

 , , , and
, , , and
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Genotyping SLA-DRB1 and SLA-DQB1
2.3. Macrophage Differentiation and In Vitro Infection
2.4. Isolating SLA-Class II-Peptide Immunocomplexes
2.5. Mass Spectrometry Analysis
2.6. LC-MS/MS Data Analysis
2.7. Immunoinformatics Analysis
3. Results
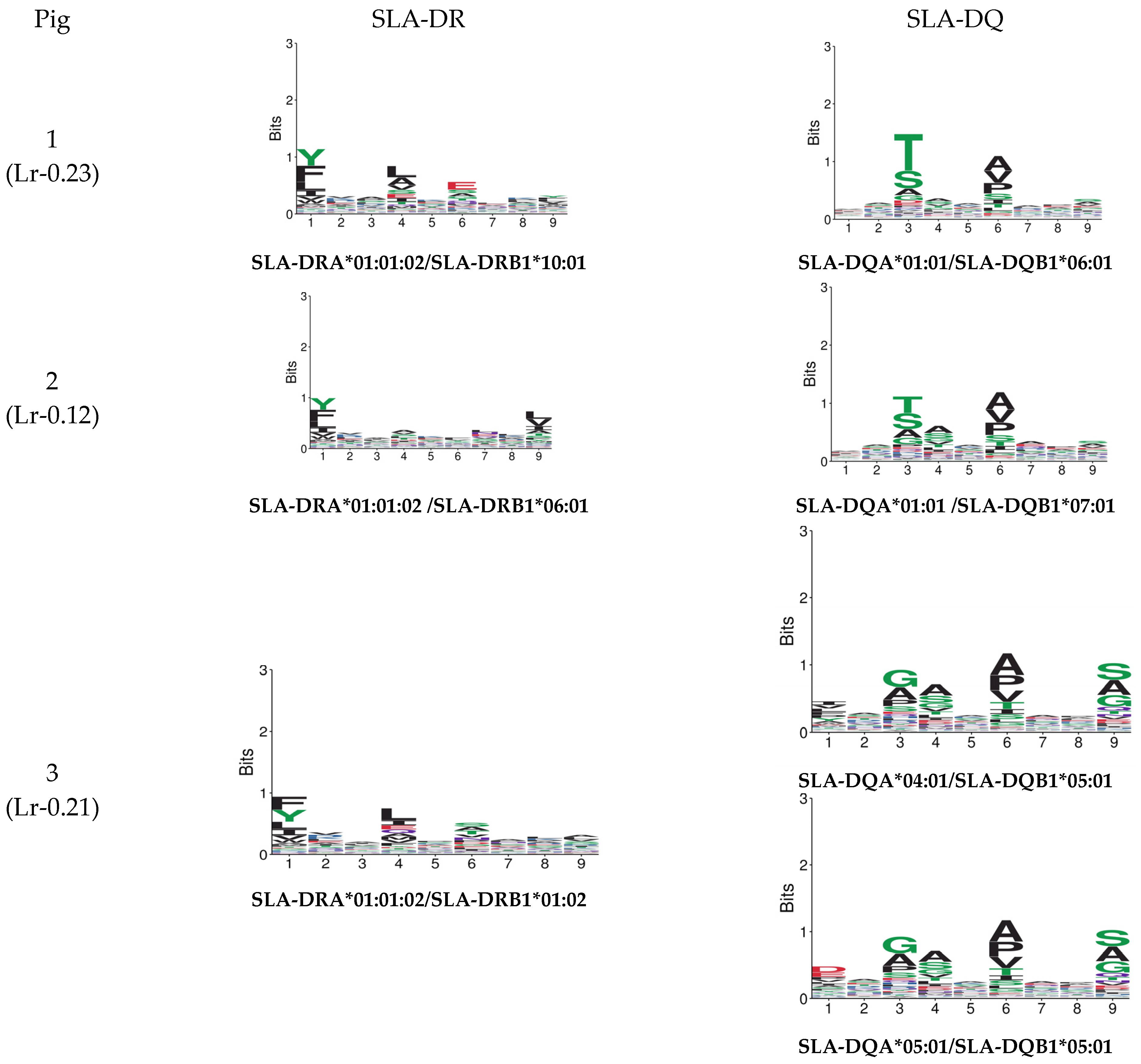
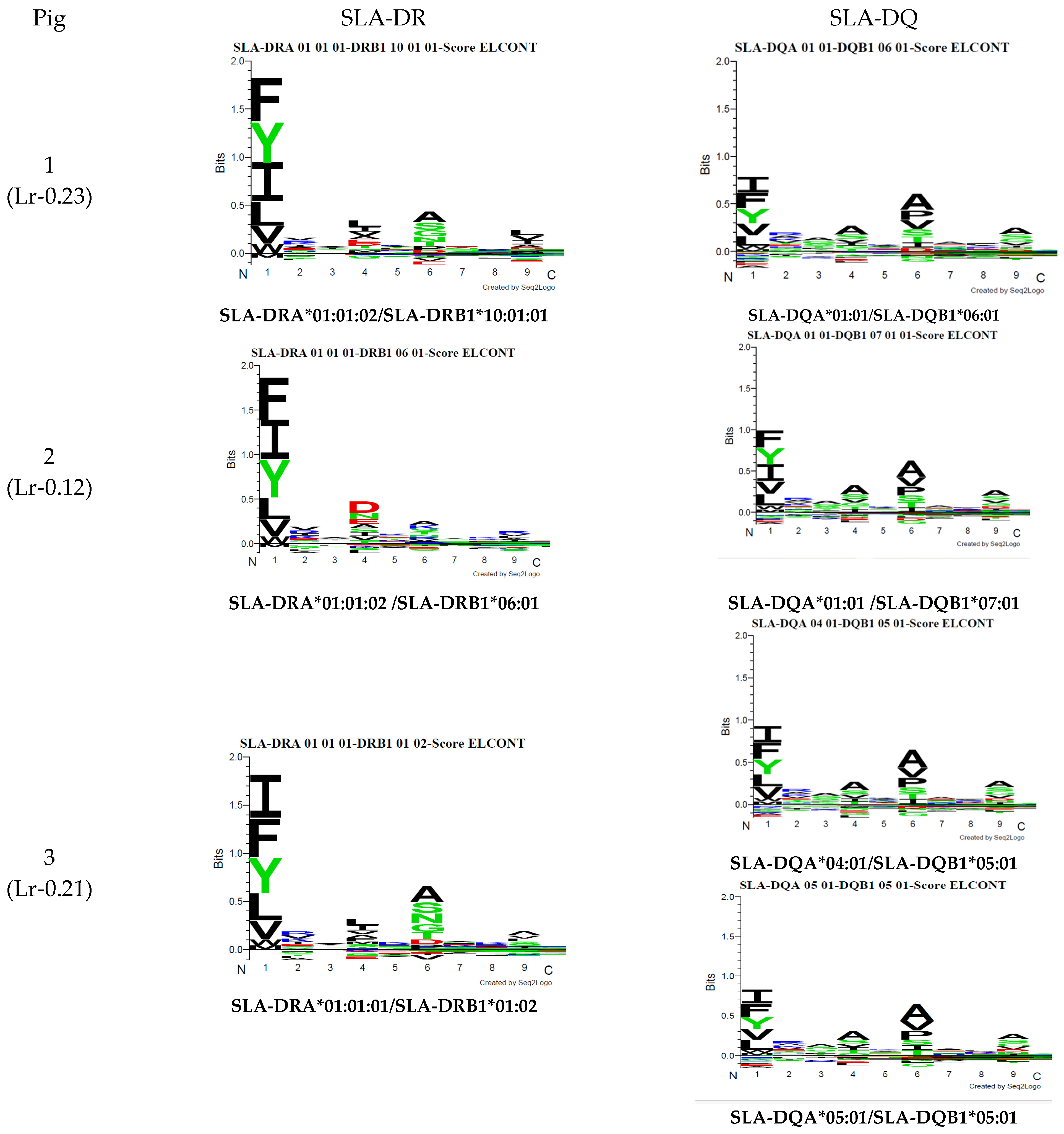
3.1. Haplotype Identification
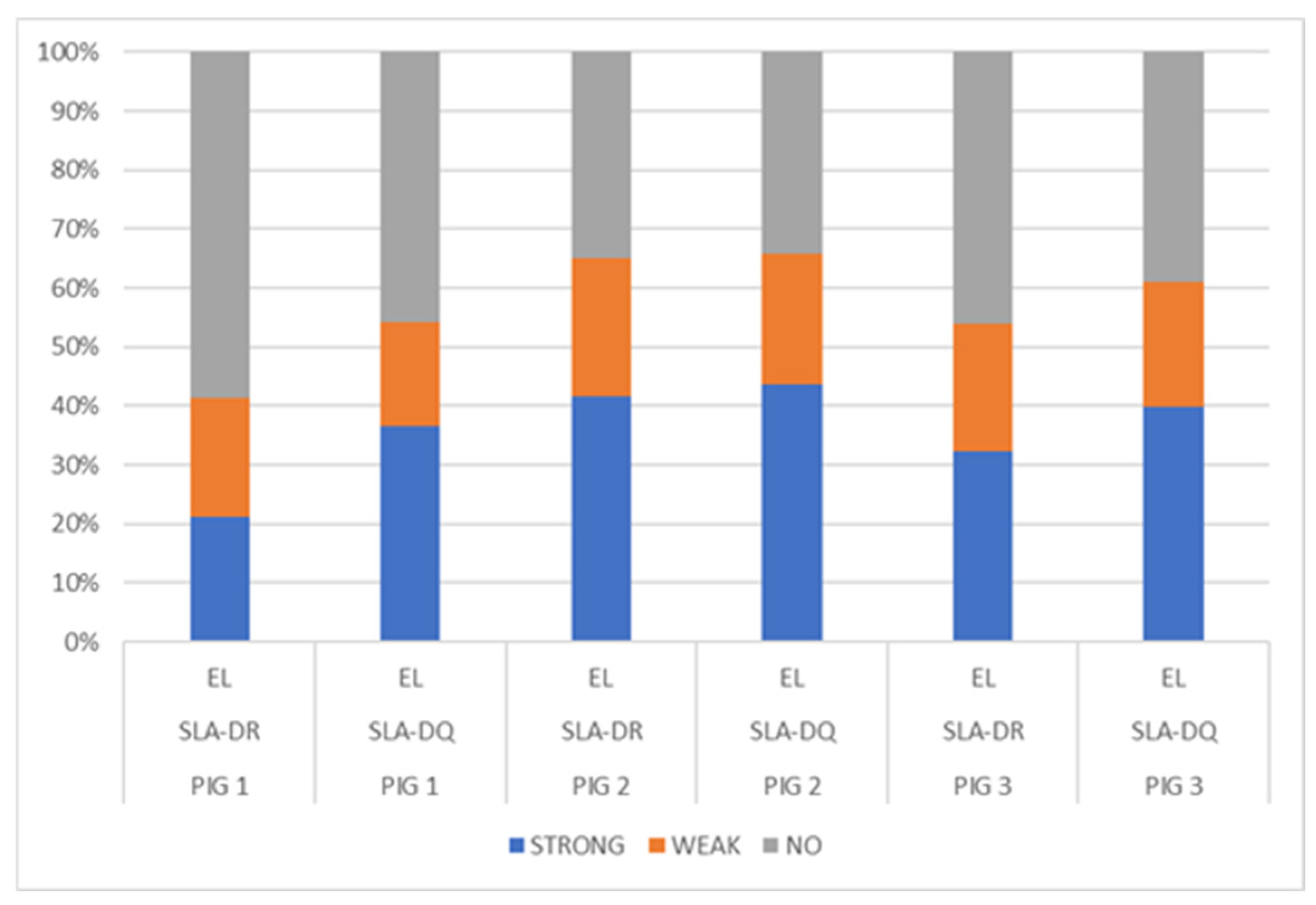
3.2. SLA Class II Ligand Ligandome Data Analysis and Binding Affinity
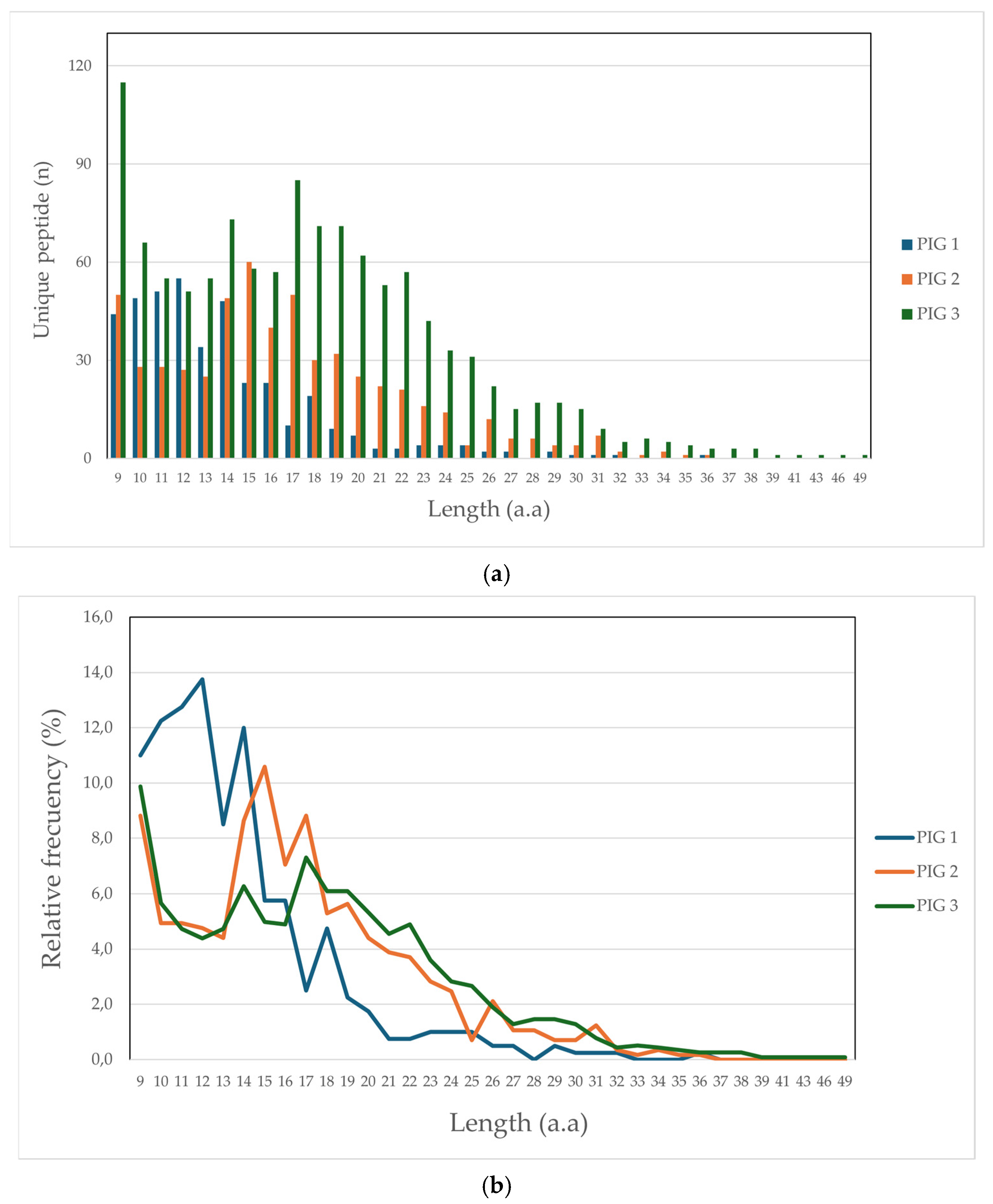
3.2.1. Peptide Length Distribution
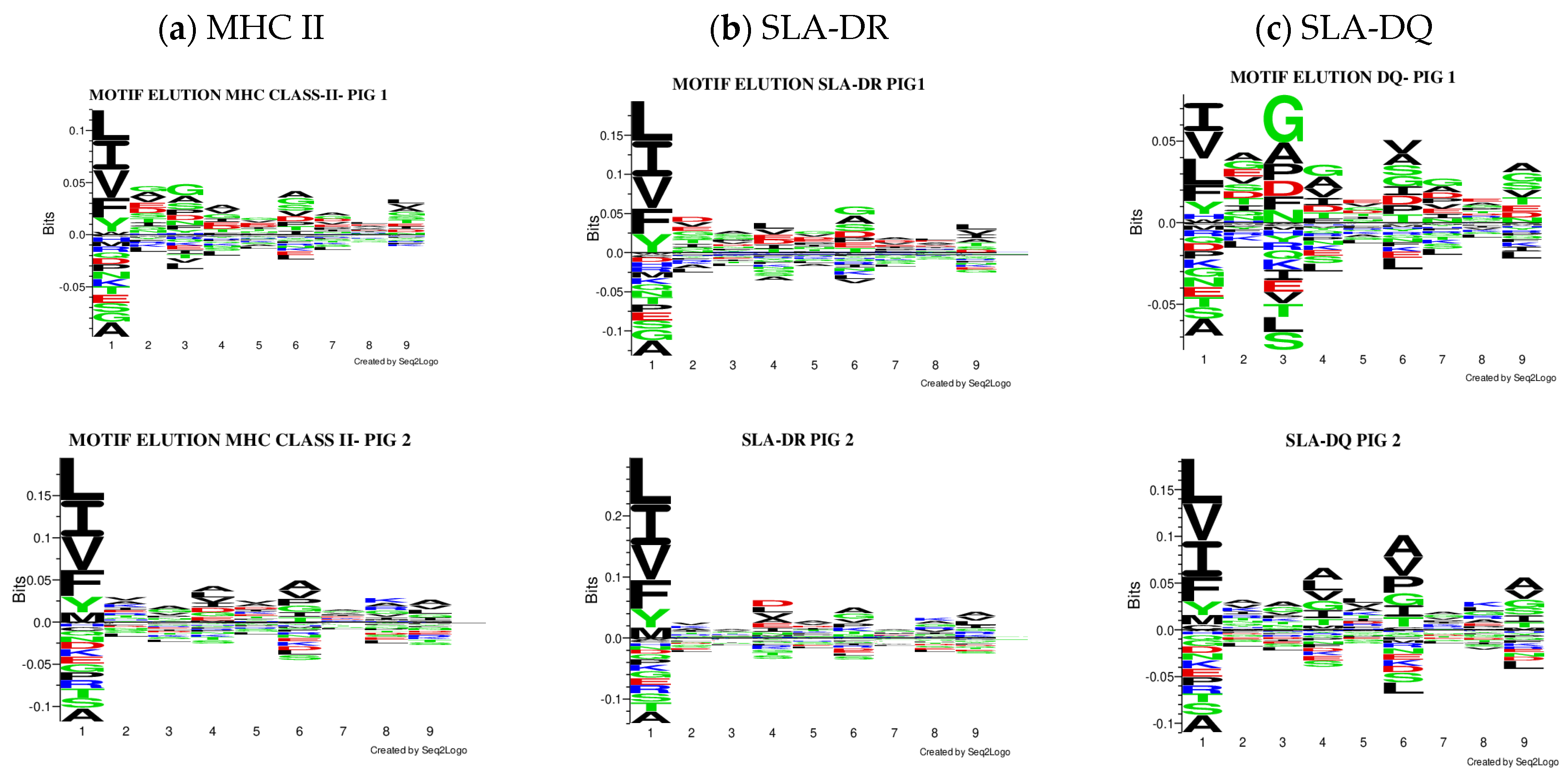
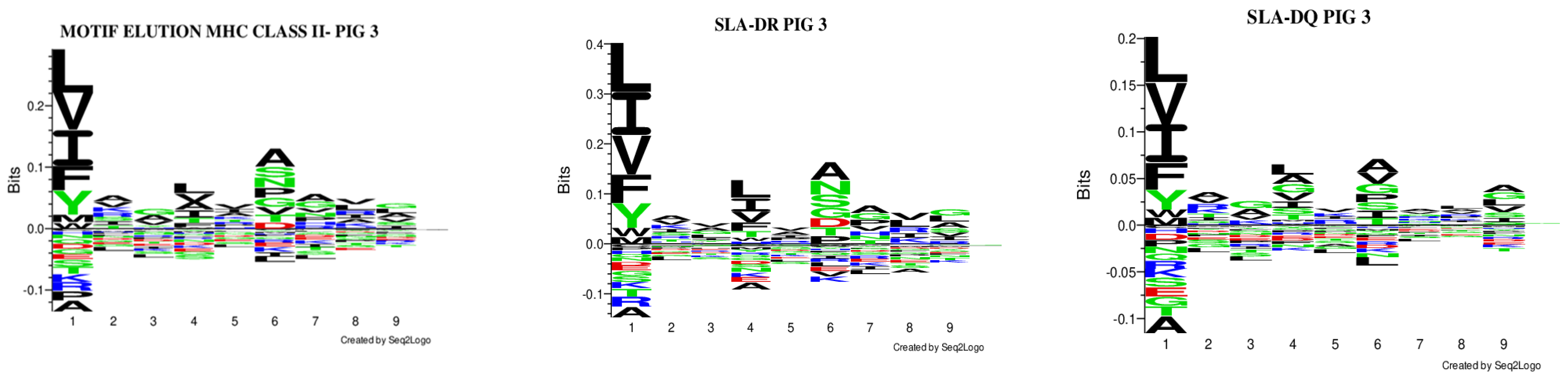
3.2.2. Motif Deconvolution
3.2.3. Peptide Clustering Based on Shared Sequence Features
3.2.4. SLA-Binding Prediction: Deconvolution Motif and Annotation
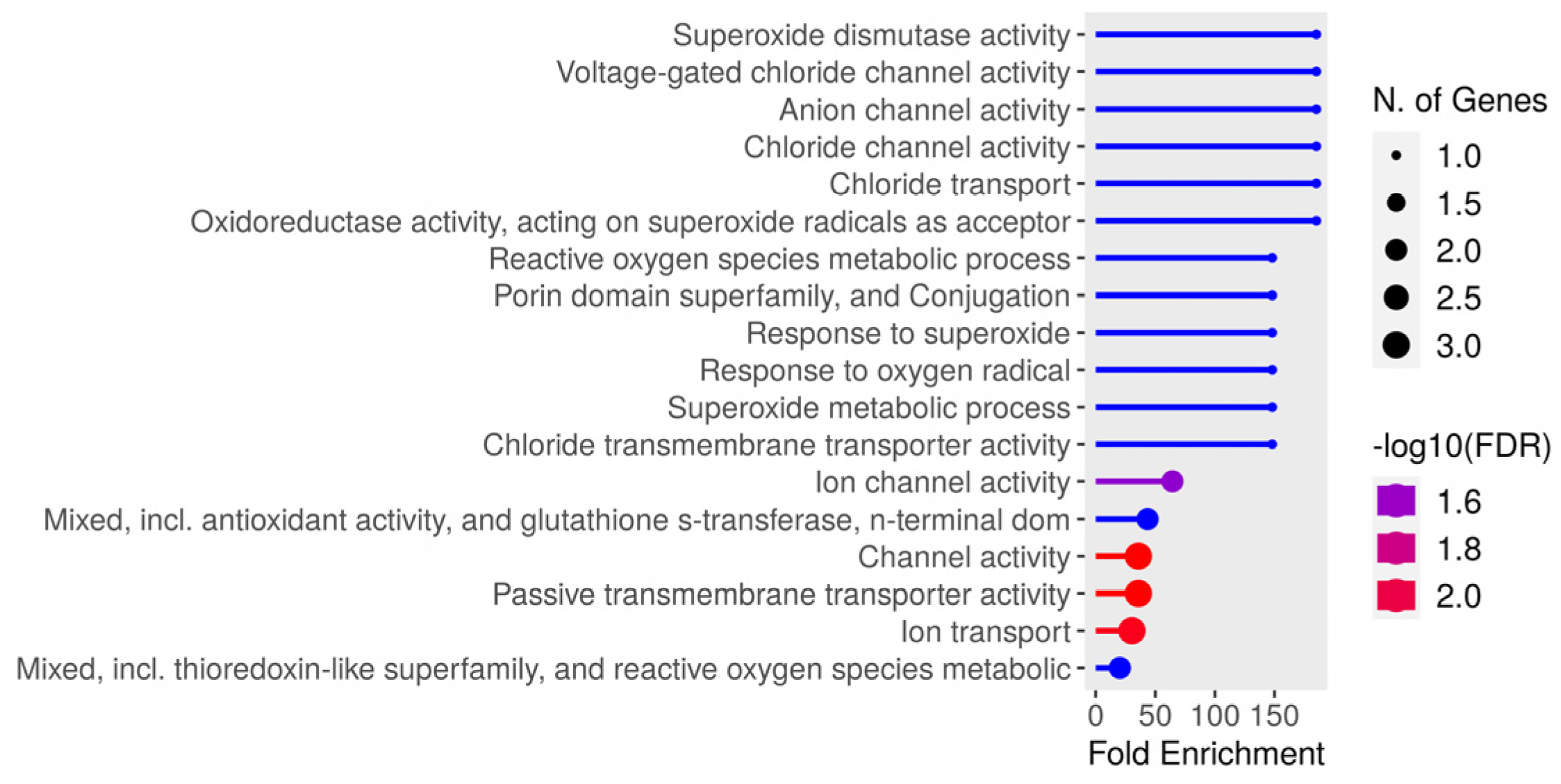
3.3. Salmonella typhimurium Infection Related Peptidome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drozdz, M.; Malaszczuk, M.; Paluch, E.; Pawlak, A. Zoonotic potential and prevalence of Salmonella serovars isolated from pets. Infect. Ecol. Epidemiol. 2021, 11, 1975530. [Google Scholar] [CrossRef]
- Ferrari, R.G.; Rosario, D.K.A.; Cunha-Neto, A.; Mano, S.B.; Figueiredo, E.E.S.; Conte-Junior, C.A. Worldwide Epidemiology of Salmonella Serovars in Animal-Based Foods: A Meta-analysis. Appl. Environ. Microbiol. 2019, 85, e00591-19. [Google Scholar] [CrossRef] [PubMed]
- Shaji, S.; Selvaraj, R.K.; Shanmugasundaram, R. Salmonella Infection in Poultry: A Review on the Pathogen and Control Strategies. Microorganisms 2023, 11, 2814. [Google Scholar] [CrossRef] [PubMed]
- Soliani, L.; Rugna, G.; Prosperi, A.; Chiapponi, C.; Luppi, A. Salmonella Infection in Pigs: Disease, Prevalence, and a Link between Swine and Human Health. Pathogens 2023, 12, 1267. [Google Scholar] [CrossRef]
- Gomes-Neves, E.; Antunes, P.; Tavares, A.; Themudo, P.; Cardoso, M.F.; Gartner, F.; Costa, J.M.; Peixe, L. Salmonella cross-contamination in swine abattoirs in Portugal: Carcasses, meat and meat handlers. Int. J. Food Microbiol. 2012, 157, 82–87. [Google Scholar] [CrossRef]
- Patterson, S.K.; Kim, H.B.; Borewicz, K.; Isaacson, R.E. Towards an understanding of Salmonella enterica serovar Typhimurium persistence in swine. Anim. Health Res. Rev. 2016, 17, 159–168. [Google Scholar] [CrossRef]
- Cabral, C.; Panzenhagen, P.; Frensel, K.; Rodrigues, G.; Merces, A.; Rodrigues, D.; Franco, R.; Conte-Junior, C.A. Genetic diversity and multidrug-resistance among Salmonella Typhimurium isolated from swine carcasses and slaughterhouses in Rio de Janeiro, Brazil. Vet. Ital. 2020, 56, 245–250. [Google Scholar] [CrossRef]
- Lamichhane, B.; Mawad, A.M.M.; Saleh, M.; Kelley, W.G.; Harrington, P.J., 2nd; Lovestad, C.W.; Amezcua, J.; Sarhan, M.M.; El Zowalaty, M.E.; Ramadan, H.; et al. Salmonellosis: An Overview of Epidemiology, Pathogenesis, and Innovative Approaches to Mitigate the Antimicrobial Resistant Infections. Antibiotics 2024, 13, 76. [Google Scholar] [CrossRef]
- WHO. Immunization, Vaccines and Biologicals—Nontyphoidal Salmonella Disease 2022. Available online: https://www.who.int/teams/immunization-vaccines-and-biologicals/diseases/nontyphoidal-salmonella-disease (accessed on 16 July 2024).
- MacLennan, C.A.; Martin, L.B.; Micoli, F. Vaccines against invasive Salmonella disease: Current status and future directions. Hum. Vaccin. Immunother. 2014, 10, 1478–1493. [Google Scholar] [CrossRef]
- Tizard, I.R. Porcine vaccines. Vaccines Vet. 2021, 17, 225–242.e1. [Google Scholar]
- de la Cruz, M.L.; Conrado, I.; Nault, A.; Perez, A.; Dominguez, L.; Alvarez, J. Vaccination as a control strategy against Salmonella infection in pigs: A systematic review and meta-analysis of the literature. Res. Vet. Sci. 2017, 114, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.L.; Mechtler, K. Immunopeptidomics in the Era of Single-Cell Proteomics. Biology 2023, 12, 1514. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.E.; Bassani-Sternberg, M. The impact of immunopeptidomics: From basic research to clinical implementation. Semin. Immunol. 2023, 66, 101727. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chu, S.; Tan, S.; Yin, X.; Jiang, Y.; Dai, X.; Gong, X.; Fang, X.; Tian, D. Towards Higher Sensitivity of Mass Spectrometry: A Perspective from the Mass Analyzers. Front Chem. 2021, 9, 813359. [Google Scholar] [CrossRef]
- Stopfer, L.E.; D’Souza, A.D.; White, F.M. 1,2,3, MHC: A review of mass-spectrometry-based immunopeptidomics methods for relative and absolute quantification of pMHCs. Immunooncol Technol. 2021, 11, 100042. [Google Scholar] [CrossRef]
- Hunt, D.F.; Henderson, R.A.; Shabanowitz, J.; Sakaguchi, K.; Michel, H.; Sevilir, N.; Cox, A.L.; Appella, E.; Engelhard, V.H. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 1992, 255, 1261–1263. [Google Scholar] [CrossRef]
- Flad, T.; Spengler, B.; Kalbacher, H.; Brossart, P.; Baier, D.; Kaufmann, R.; Bold, P.; Metzger, S.; Bluggel, M.; Meyer, H.E.; et al. Direct identification of major histocompatibility complex class I-bound tumor-associated peptide antigens of a renal carcinoma cell line by a novel mass spectrometric method. Cancer Res. 1998, 58, 5803–5811. [Google Scholar]
- Nelde, A.; Kowalewski, D.J.; Stevanovic, S. Purification and Identification of Naturally Presented MHC Class I and II Ligands. Methods Mol. Biol. 2019, 1988, 123–136. [Google Scholar] [CrossRef]
- Racle, J.; Gfeller, D. How to Predict Binding Specificity and Ligands for New MHC-II Alleles with MixMHC2pred. Methods Mol. Biol. 2024, 2809, 215–235. [Google Scholar] [CrossRef]
- Karunakaran, K.P.; Yu, H.; Jiang, X.; Chan, Q.W.T.; Sigola, L.; Millis, L.A.; Chen, J.; Tang, P.; Foster, L.J.; Brunham, R.C. Immunoproteomic discovery of Mycobacterium bovis antigens, including the surface lipoprotein Mpt83 as a T cell antigen useful for vaccine development. Vaccine 2024, 42, 126266. [Google Scholar] [CrossRef]
- Fisch, A.; Reynisson, B.; Benedictus, L.; Nicastri, A.; Vasoya, D.; Morrison, I.; Buus, S.; Ferreira, B.R.; Kinney Ferreira de Miranda Santos, I.; Ternette, N.; et al. Integral Use of Immunopeptidomics and Immunoinformatics for the Characterization of Antigen Presentation and Rational Identification of BoLA-DR-Presented Peptides and Epitopes. J. Immunol. 2021, 206, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Motz, M.; Stas, M.R.; Hammer, S.E.; Duckova, T.; Fontaine, F.; Kiesler, A.; Seitz, K.; Ladinig, A.; Muller, A.C.; Riedel, C.; et al. Identification of MHC-I-Presented Porcine Respiratory and Reproductive Syndrome Virus (PRRSV) Peptides Reveals Immunogenic Epitopes within Several Non-Structural Proteins Recognized by CD8(+) T Cells. Viruses 2022, 14, 1891. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Liu, Y.; Racle, J. Contemplating immunopeptidomes to better predict them. Semin. Immunol. 2023, 66, 101708. [Google Scholar] [CrossRef] [PubMed]
- Leddy, O.K.; White, F.M.; Bryson, B.D. Leveraging Immunopeptidomics to Study and Combat Infectious Disease. mSystems 2021, 6, e0031021. [Google Scholar] [CrossRef]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef]
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res. 2020, 19, 2304–2315. [Google Scholar] [CrossRef]
- Wei, X.; Li, S.; Wang, S.; Feng, G.; Xie, X.; Li, Z.; Zhang, N. Peptidomes and Structures Illustrate How SLA-I Micropolymorphism Influences the Preference of Binding Peptide Length. Front. Immunol. 2022, 13, 820881. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Delamarre, L.; Jhunjhunwala, S.; Modrusan, Z.; Phung, Q.T.; Elias, J.E.; Lill, J.R. Building proteomic tool boxes to monitor MHC class I and class II peptides. Proteomics 2017, 17, 1600061. [Google Scholar] [CrossRef]
- Hammer, S.E.; Ho, C.S.; Ando, A.; Rogel-Gaillard, C.; Charles, M.; Tector, M.; Tector, A.J.; Lunney, J.K. Importance of the Major Histocompatibility Complex (Swine Leukocyte Antigen) in Swine Health and Biomedical Research. Annu. Rev. Anim. Biosci. 2020, 8, 171–198. [Google Scholar] [CrossRef]
- Lighten, J.; Papadopulos, A.S.T.; Mohammed, R.S.; Ward, B.J.; Paterson, I.G.; Baillie, L.; Bradbury, I.R.; Hendry, A.P.; Bentzen, P.; van Oosterhout, C. Evolutionary genetics of immunological supertypes reveals two faces of the Red Queen. Nat. Commun. 2017, 8, 1294. [Google Scholar] [CrossRef]
- Reche, P.A.; Reinherz, E.L. Definition of MHC supertypes through clustering of MHC peptide-binding repertoires. Methods Mol. Biol. 2007, 409, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Pan, C.; Cheng, P.; Wang, J.; Zhao, G.; Wu, X. Peptide-Based Vaccines for Tuberculosis. Front. Immunol. 2022, 13, 830497. [Google Scholar] [CrossRef] [PubMed]
- Kheirvari, M.; Liu, H.; Tumban, E. Virus-like Particle Vaccines and Platforms for Vaccine Development. Viruses 2023, 15, 1109. [Google Scholar] [CrossRef]
- Celis-Giraldo, C.T.; Bohorquez, M.D.; Camargo, M.; Suarez, C.F.; Camargo, A.; Rodriguez-Obediente, K.; Martinez, A.; Lucero, C.E.; Hernandez, B.; Manzano-Roman, R.; et al. A comparative analysis of SLA-DRB1 genetic diversity in Colombian (creoles and commercial line) and worldwide swine populations. Sci. Rep. 2021, 11, 4340. [Google Scholar] [CrossRef]
- Ando, A.; Shigenari, A.; Ota, M.; Sada, M.; Kawata, H.; Azuma, F.; Kojima-Shibata, C.; Nakajoh, M.; Suzuki, K.; Uenishi, H.; et al. SLA-DRB1 and -DQB1 genotyping by the PCR-SSOP-Luminex method. Tissue Antigens 2011, 78, 49–55. [Google Scholar] [CrossRef]
- Luetkemeier, E.S.; Malhi, R.S.; Beever, J.E.; Schook, L.B. Diversification of porcine MHC class II genes: Evidence for selective advantage. Immunogenetics 2009, 61, 119–129. [Google Scholar] [CrossRef]
- Le, M.; Choi, H.; Choi, M.K.; Cho, H.; Kim, J.H.; Seo, H.G.; Cha, S.Y.; Seo, K.; Dadi, H.; Park, C. Development of a simultaneous high resolution typing method for three SLA class II genes, SLA-DQA, SLA-DQB1, and SLA-DRB1 and the analysis of SLA class II haplotypes. Gene 2015, 564, 228–232. [Google Scholar] [CrossRef]
- Celis-Giraldo, C.; Ordonez, D.; Diaz-Arevalo, D.; Bohorquez, M.D.; Ibarrola, N.; Suarez, C.F.; Rodriguez, K.; Yepes, Y.; Rodriguez, A.; Avendano, C.; et al. Identifying major histocompatibility complex class II-DR molecules in bovine and swine peripheral blood monocyte-derived macrophages using mAb-L243. Vaccine 2024, 42, 3445–3454. [Google Scholar] [CrossRef]
- Alvarez, B.; Barra, C.; Nielsen, M.; Andreatta, M. Computational Tools for the Identification and Interpretation of Sequence Motifs in Immunopeptidomes. Proteomics 2018, 18, e1700252. [Google Scholar] [CrossRef]
- Munday, P.R.; Fehring, J.; Revote, J.; Pandey, K.; Shahbazy, M.; Scull, K.E.; Ramarathinam, S.H.; Faridi, P.; Croft, N.P.; Braun, A.; et al. Immunolyser: A web-based computational pipeline for analysing and mining immunopeptidomic data. Comput. Struct. Biotechnol. J. 2023, 21, 1678–1687. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Peters, B.; Sette, A.; Justesen, S.; Buus, S.; Lund, O. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput. Biol. 2008, 4, e1000107. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Thomsen, M.C.; Nielsen, M. Seq2Logo: A method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 2012, 40, W281–W287. [Google Scholar] [CrossRef] [PubMed]
- Gimsa, U.; Ho, C.S.; Hammer, S.E. Preferred SLA class I/class II haplotype combinations in German Landrace pigs. Immunogenetics 2017, 69, 39–47. [Google Scholar] [CrossRef]
- Andreatta, M.; Lund, O.; Nielsen, M. Simultaneous alignment and clustering of peptide data using a Gibbs sampling approach. Bioinformatics 2013, 29, 8–14. [Google Scholar] [CrossRef]
- Sears, K.T.; Galen, J.E.; Tennant, S.M. Advances in the development of Salmonella-based vaccine strategies for protection against Salmonellosis in humans. J. Appl. Microbiol. 2021, 131, 2640–2658. [Google Scholar] [CrossRef]
- Kapetanovic, R.; Fairbairn, L.; Downing, A.; Beraldi, D.; Sester, D.P.; Freeman, T.C.; Tuggle, C.K.; Archibald, A.L.; Hume, D.A. The impact of breed and tissue compartment on the response of pig macrophages to lipopolysaccharide. BMC Genom. 2013, 14, 581. [Google Scholar] [CrossRef]
- Nilsson, J.B.; Kaabinejadian, S.; Yari, H.; Kester, M.G.D.; van Balen, P.; Hildebrand, W.H.; Nielsen, M. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning. Sci. Adv. 2023, 9, eadj6367. [Google Scholar] [CrossRef]
- Strazar, M.; Park, J.; Abelin, J.G.; Taylor, H.B.; Pedersen, T.K.; Plichta, D.R.; Brown, E.M.; Eraslan, B.; Hung, Y.M.; Ortiz, K.; et al. HLA-II immunopeptidome profiling and deep learning reveal features of antigenicity to inform antigen discovery. Immunity 2023, 56, 1681–1698.e1613. [Google Scholar] [CrossRef]
- Ho, C.S.; Lunney, J.K.; Ando, A.; Rogel-Gaillard, C.; Lee, J.H.; Schook, L.B.; Smith, D.M. Nomenclature for factors of the SLA system, update 2008. Tissue Antigens 2009, 73, 307–315. [Google Scholar] [CrossRef]
- Hammer, S.E.; Duckova, T.; Groiss, S.; Stadler, M.; Jensen-Waern, M.; Golde, W.T.; Gimsa, U.; Saalmueller, A. Comparative analysis of swine leukocyte antigen gene diversity in European farmed pigs. Anim. Genet. 2021, 52, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, K.P.; Yu, H.; Jiang, X.; Chan, Q.; Goldberg, M.F.; Jenkins, M.K.; Foster, L.J.; Brunham, R.C. Identification of MHC-Bound Peptides from Dendritic Cells Infected with Salmonella enterica Strain SL1344: Implications for a Nontyphoidal Salmonella Vaccine. J. Proteome Res. 2017, 16, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; Guillaume, P.; Schmidt, J.; Michaux, J.; Larabi, A.; Lau, K.; Perez, M.A.S.; Croce, G.; Genolet, R.; Coukos, G.; et al. Machine learning predictions of MHC-II specificities reveal alternative binding mode of class II epitopes. Immunity 2023, 56, 1359–1375.e1313. [Google Scholar] [CrossRef]
- Bettencourt, P.; Muller, J.; Nicastri, A.; Cantillon, D.; Madhavan, M.; Charles, P.D.; Fotso, C.B.; Wittenberg, R.; Bull, N.; Pinpathomrat, N.; et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines 2020, 5, 2. [Google Scholar] [CrossRef]
- Andreatta, M.; Alvarez, B.; Nielsen, M. GibbsCluster: Unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 2017, 45, W458–W463. [Google Scholar] [CrossRef]
- Racle, J.; Michaux, J.; Rockinger, G.A.; Arnaud, M.; Bobisse, S.; Chong, C.; Guillaume, P.; Coukos, G.; Harari, A.; Jandus, C.; et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019, 37, 1283–1286. [Google Scholar] [CrossRef]
- Nielsen, M.; Connelley, T.; Ternette, N. Improved Prediction of Bovine Leucocyte Antigens (BoLA) Presented Ligands by Use of Mass-Spectrometry-Determined Ligand and in Vitro Binding Data. J. Proteome Res. 2018, 17, 559–567. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Demmers, L.C.; Wu, W.; Heck, A.J.R. HLA Class II Presentation Is Specifically Altered at Elevated Temperatures in the B-Lymphoblastic Cell Line JY. Mol. Cell Proteom. 2021, 20, 100089. [Google Scholar] [CrossRef]
- Andreatta, M.; Schafer-Nielsen, C.; Lund, O.; Buus, S.; Nielsen, M. NNAlign: A web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLoS ONE 2011, 6, e26781. [Google Scholar] [CrossRef] [PubMed]
- Moise, L.; Gutierrez, A.H.; Khan, S.; Tan, S.; Ardito, M.; Martin, W.D.; De Groot, A.S. New Immunoinformatics Tools for Swine: Designing Epitope-Driven Vaccines, Predicting Vaccine Efficacy, and Making Vaccines on Demand. Front. Immunol. 2020, 11, 563362. [Google Scholar] [CrossRef] [PubMed]
- Pishesha, N.; Harmand, T.J.; Ploegh, H.L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 2022, 22, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Monterrubio-Lopez, G.P.; Gonzalez, Y.M.J.A.; Ribas-Aparicio, R.M. Identification of Novel Potential Vaccine Candidates against Tuberculosis Based on Reverse Vaccinology. Biomed. Res. Int. 2015, 483150, 1–6. [Google Scholar] [CrossRef]
- Confer, A.W.; Ayalew, S. The OmpA family of proteins: Roles in bacterial pathogenesis and immunity. Vet. Microbiol. 2013, 163, 207–222. [Google Scholar] [CrossRef]
- Fourie, K.R.; Wilson, H.L. Understanding GroEL and DnaK Stress Response Proteins as Antigens for Bacterial Diseases. Vaccines 2020, 8, 773. [Google Scholar] [CrossRef]
- Becker, J.P.; Riemer, A.B. The Importance of Being Presented: Target Validation by Immunopeptidomics for Epitope-Specific Immunotherapies. Front. Immunol. 2022, 13, 883989. [Google Scholar] [CrossRef]
- Hamze, M.; Meunier, S.; Karle, A.; Gdoura, A.; Goudet, A.; Szely, N.; Pallardy, M.; Carbonnel, F.; Spindeldreher, S.; Mariette, X.; et al. Characterization of CD4 T Cell Epitopes of Infliximab and Rituximab Identified from Healthy Donors. Front. Immunol. 2017, 8, 500. [Google Scholar] [CrossRef]
- Paul, S.; Karosiene, E.; Dhanda, S.K.; Jurtz, V.; Edwards, L.; Nielsen, M.; Sette, A.; Peters, B. Determination of a Predictive Cleavage Motif for Eluted Major Histocompatibility Complex Class II Ligands. Front. Immunol. 2018, 9, 1795. [Google Scholar] [CrossRef]
- Lichti, C.F.; Vigneron, N.; Clauser, K.R.; Van den Eynde, B.J.; Bassani-Sternberg, M. Navigating Critical Challenges Associated with Immunopeptidomics-Based Detection of Proteasomal Spliced Peptide Candidates. Cancer Immunol. Res. 2022, 10, 275–284. [Google Scholar] [CrossRef]
- de Mey, W.; De Schrijver, P.; Autaers, D.; Pfitzer, L.; Fant, B.; Locy, H.; Esprit, A.; Lybaert, L.; Bogaert, C.; Verdonck, M.; et al. A synthetic DNA template for fast manufacturing of versatile single epitope mRNA. Mol. Ther. Nucleic Acids 2022, 29, 943–954. [Google Scholar] [CrossRef]
- Mayer, R.L.; Verbeke, R.; Asselman, C.; Aernout, I.; Gul, A.; Eggermont, D.; Boucher, K.; Thery, F.; Maia, T.M.; Demol, H.; et al. Immunopeptidomics-based design of mRNA vaccine formulations against Listeria monocytogenes. Nat. Commun. 2022, 13, 6075. [Google Scholar] [CrossRef] [PubMed]
- Yurina, V.; Adianingsih, O.R. Predicting epitopes for vaccine development using bioinformatics tools. Ther. Adv. Vaccines Immunother. 2022, 10, 25151355221100218. [Google Scholar] [CrossRef] [PubMed]
- Danner, R.; Prochniak, L.M.; Pereckas, M.; Rouse, J.R.; Wahhab, A.; Hackner, L.G.; Lochhead, R.B. Identification of Major Histocompatibility Complex Class II Epitopes From Lyme Autoantigen Apolipoprotein B-100 and Borrelia burgdorferi Mcp4 in Murine Lyme Arthritis. J. Infect. Dis. 2024, 230, S27–S39. [Google Scholar] [CrossRef]
- Dorigatti, E.; Schubert, B. Joint epitope selection and spacer design for string-of-beads vaccines. Bioinformatics 2020, 36, i643–i650. [Google Scholar] [CrossRef]
- Tahir Ul Qamar, M.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Gutierrez, A.H.; Martin, W.D.; Bailey-Kellogg, C.; Terry, F.; Moise, L.; De Groot, A.S. Development and validation of an epitope prediction tool for swine (PigMatrix) based on the pocket profile method. BMC Bioinform. 2015, 16, 290. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pig | SLA-DRB1 1 | SLA-DQB1 2 | SLA-DQA 3 | Haplotype 3 |
|---|---|---|---|---|
| 1 | 10:01:01 | 06:01 | 01:XX | Lr-0.23 |
| 2 | 06:01 | 07:01 | 01:XX | Lr-0.12 |
| 3 | 01:02 | 05:01 | 04:XX/05:XX | Lr-0.21 |
| CORE Position |
|---|
| 1 2 3 4 5 6 7 8 9 |
| VLSAADKANVKAAWGKVGGQA |
| VLSAADKANVKAAWG |
| VLSAADKANVKAAWGKVGG |
| VLSAADKANVKAAWGKVGGQAGA |
| VLSAADKANVK |
| VLSAADKANVKAA |
| VLSAADKANVKAAW |
| VLSAADKANVKAAWGKVG |
| GSYTQAAGSDSAQGSDVSLTKDPRV |
| SYTQAAGSDSDQGSDVSLTKDPRV |
| GSDSAQGSDVSLTKDPRV |
| AGSDSAQGSDVSLTKDPRV |
| SDSAQGSDVSLTKDPRV |
| SYTQAAGSDSAQGSDVSLTKDPRV |
| SDVSLTKDPRV |
| SAQGSDVSLTKDPRV |
| GSDVSLTKDPRV |
| TEDLSSGLGVTKQDL |
| TEDLSSGLGVTKQ |
| TEDLSSGLGVTK |
| TEDLSSGLGVTKQD |
| ID | Eluted Ligand < 5% | Unique Eluted Ligand < 5% | |||
|---|---|---|---|---|---|
| Class II | SLA-DR | SLA-DQ | SLA-DR | SLA-DQ | |
| Pig 1 (Lr-0.23) | 3962 | 2149 | 1813 | 160 | 206 |
| Pig 2 (Lr-0.12) | 11764 | 3991 | 5343 | 287 | 202 |
| Pig 3 (Lr-0.21) | 3420 | 2511 | 3630 | 94 | 493 |
| Protein Accession | Gene Name | Peptide Sequence | Total Peptides |
|---|---|---|---|
| P02936|OMPA_SALTY | Outer membrane protein A (outer membrane major heat-modifiable protein) | APKDNTWYAGAKLGWSQYHDTGFIH | 2 |
| GWSQYHDTG | 2 | ||
| APKDNTWYAGAKLGWSQYHDTGFIHN | 1 | ||
| RFGQQEAAPVVAPAPAPAPEVQ | 1 | ||
| IGTRPDNGLLSVGVSYRFGQQEA | 1 | ||
| P0A1D3|CH60_SALTY | Chaperonin GroEL (EC 5.6.1.7) | VEDALHATRAAVEEGVVAGGGVALIRVASKIADL | 2 |
| GNDARVKMLRGVNVLADAVKVTLGPKGR | 2 | ||
| AAVEEGVVAGGGVALIRVASKIADL | 2 | ||
| ATRAAVEEGVVAGGGVALIRVASKIADL | 2 | ||
| AAVEEGVVAGGGVALIRVASKIADLKGQ | 1 | ||
| P0A1H5|EFTU_SALTY | Elongation factor Tu (EF-Tu) | GQVLAKPGTIKPH | 2 |
| VDHGKTTLTAAITTVLAKTYGGAAR | 1 | ||
| VNVGTIGHVDHGKTTLTA | 1 | ||
| VDHGKTTLTAAITTVLAKTYGGAA | 1 | ||
| P0A1P0|G3P_SALTY | Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | VPTPNVSVVDLTVRLEKAATYEQIK | 1 |
| DNETGYSNKVLDLIAHISK | 1 | ||
| VPTPNVSVVDLTVRLEKAATYEQIKAAVK | 1 | ||
| SNKVLDLIAHISK | 1 | ||
| STGAAKAVGKVLPELNGKLTGMAF | 1 | ||
| P0A2F4|SODF_SALTY | Superoxide dismutase [Fe] | SFELPALPY | 1 |
| P0A6B1|ACP_SALTY | Acyl carrier protein (ACP) | ALEEEFDTEIPDEEAEKIT | 1 |
| EEEFDTEIPDEEAEKITTVQ | 1 | ||
| EEEFDTEIPDEEAEKIT | 1 | ||
| P43019|SODM_SALTY | Superoxide dismutase [Mn] | SYTLPSLPY | 1 |
| Q7CPE2|ATPB_SALTY | ATP synthase subunit beta (EC 7.1.2.2) (ATP synthase F1 sector subunit beta) | YTLAGTEVSALLGRMPSAV | 1 |
| TLAGTEVSALLGRMPSAV | 1 | ||
| PADDLTDPSPA | 1 | ||
| TLAGTEVSALL | 1 | ||
| Q8ZN40|ISCS_SALTY | Cysteine desulfurase (IscS) | MKLPIYLDYSATTPVD | 1 |
| Q8ZRP8|CLCA_SALTY | H(+)/Cl(-) exchange transporter ClcA | GREGPTVQIGGNL | 1 |
| Q8ZP45|Q8ZP45_SALTY | Aldehyde-alcohol dehydrogenase | SVPETTKILIGEVTVVDESEPF | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celis-Giraldo, C.; Suárez, C.F.; Agudelo, W.; Ibarrola, N.; Degano, R.; Díaz, J.; Manzano-Román, R.; Patarroyo, M.A. Immunopeptidomics of Salmonella enterica Serovar Typhimurium-Infected Pig Macrophages Genotyped for Class II Molecules. Biology 2024, 13, 832. https://doi.org/10.3390/biology13100832
Celis-Giraldo C, Suárez CF, Agudelo W, Ibarrola N, Degano R, Díaz J, Manzano-Román R, Patarroyo MA. Immunopeptidomics of Salmonella enterica Serovar Typhimurium-Infected Pig Macrophages Genotyped for Class II Molecules. Biology. 2024; 13(10):832. https://doi.org/10.3390/biology13100832
Chicago/Turabian StyleCelis-Giraldo, Carmen, Carlos F. Suárez, William Agudelo, Nieves Ibarrola, Rosa Degano, Jaime Díaz, Raúl Manzano-Román, and Manuel A. Patarroyo. 2024. "Immunopeptidomics of Salmonella enterica Serovar Typhimurium-Infected Pig Macrophages Genotyped for Class II Molecules" Biology 13, no. 10: 832. https://doi.org/10.3390/biology13100832
APA StyleCelis-Giraldo, C., Suárez, C. F., Agudelo, W., Ibarrola, N., Degano, R., Díaz, J., Manzano-Román, R., & Patarroyo, M. A. (2024). Immunopeptidomics of Salmonella enterica Serovar Typhimurium-Infected Pig Macrophages Genotyped for Class II Molecules. Biology, 13(10), 832. https://doi.org/10.3390/biology13100832