
Phenotypic and Genomic Characterization of a Sulfate-Reducing Bacterium Pseudodesulfovibrio methanolicus sp. nov. Isolated from a Petroleum Reservoir in Russia

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Isolation and Cultivation
2.2. Morphological, Physiological, and Chemotaxonomic Characterization
2.3. 16S rRNA Gene and Genome Sequencing and Annotation
2.4. Bioinformatic Analysis
2.5. Nucleotide Sequence Accession Numbers
3. Results and Discussion
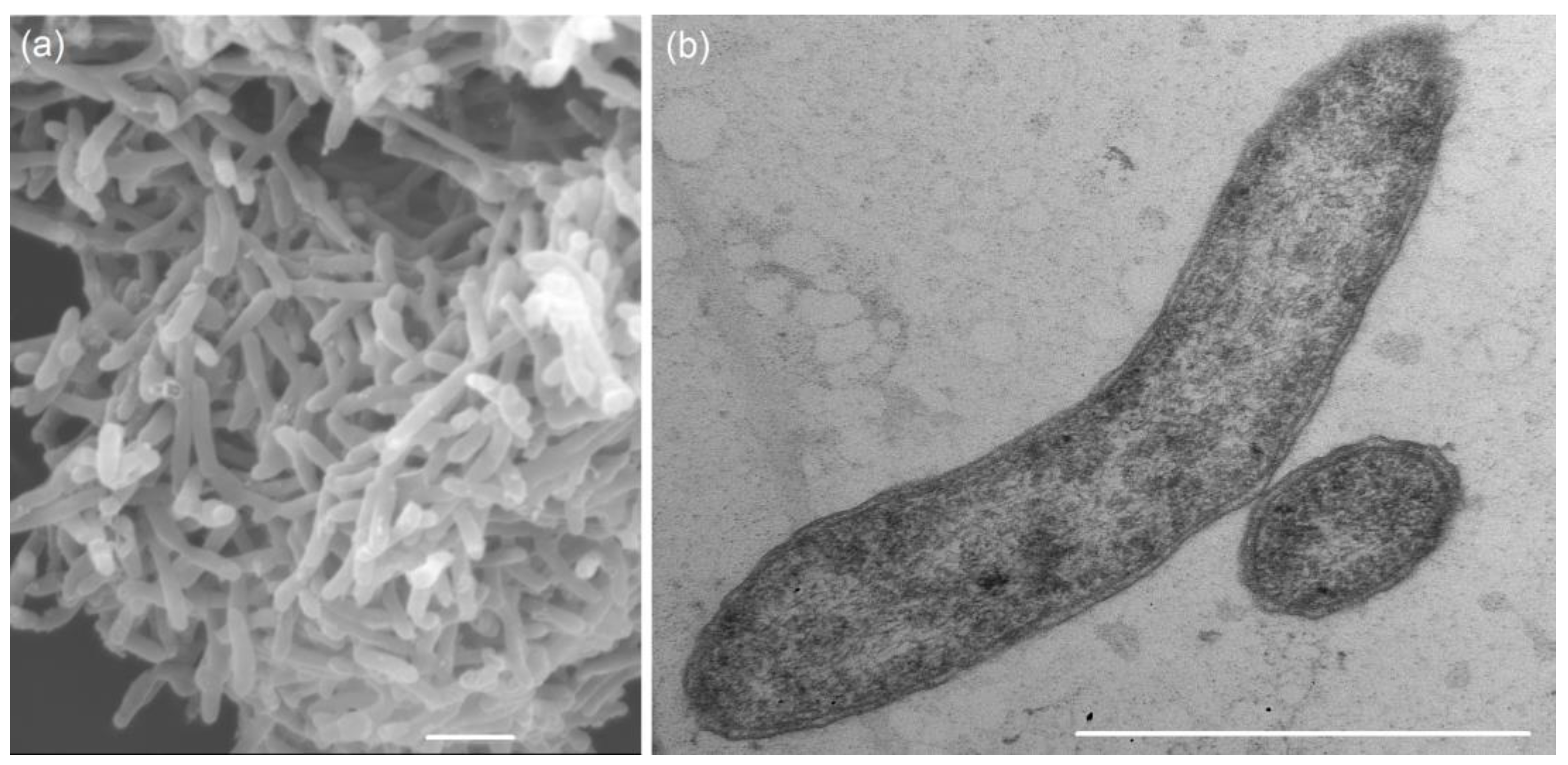
3.1. Phenotypic Characteristics of Strain 5S69T
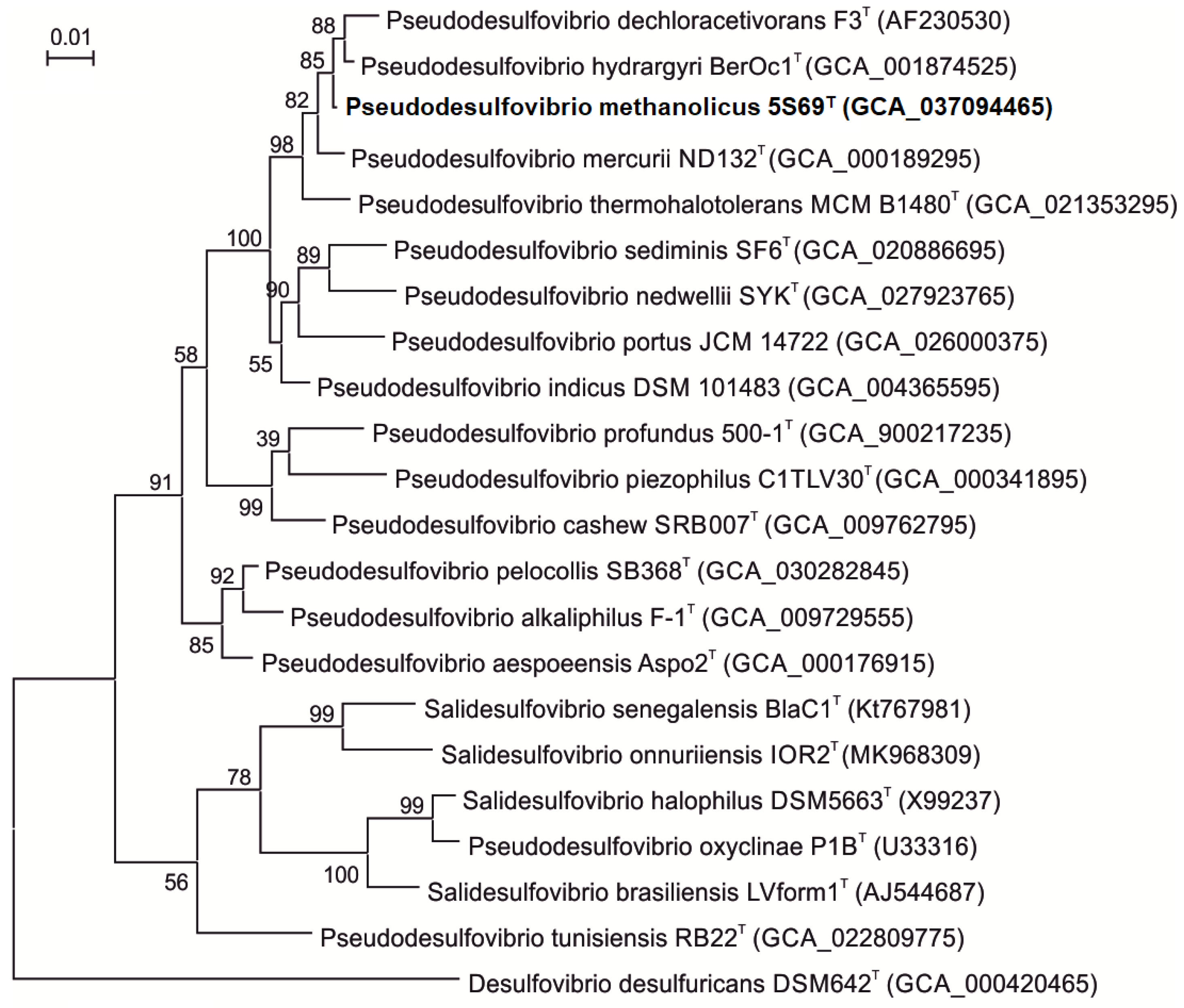
3.2. Phylogenetic Analyses of the 16S rRNA Gene Sequences
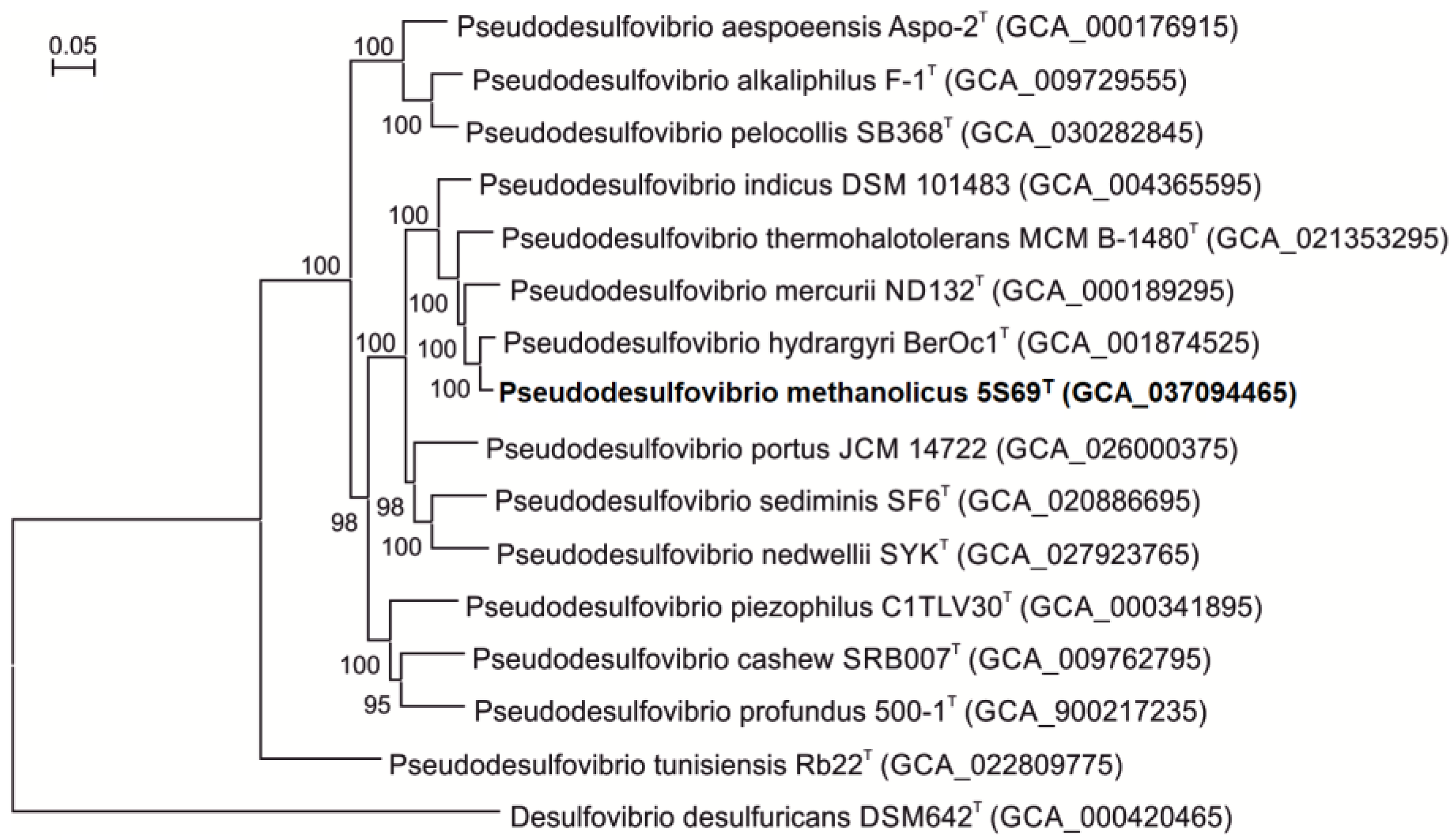
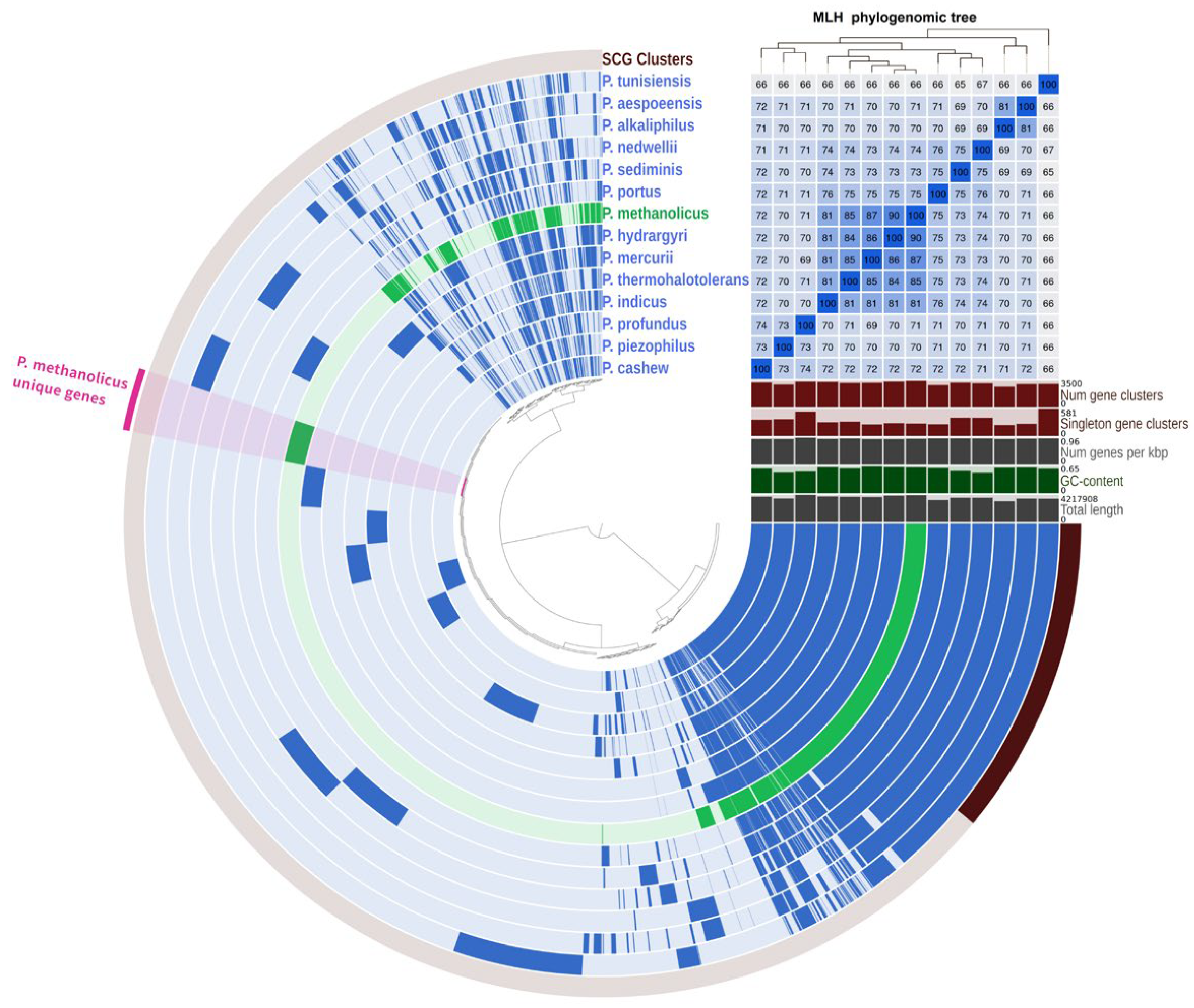
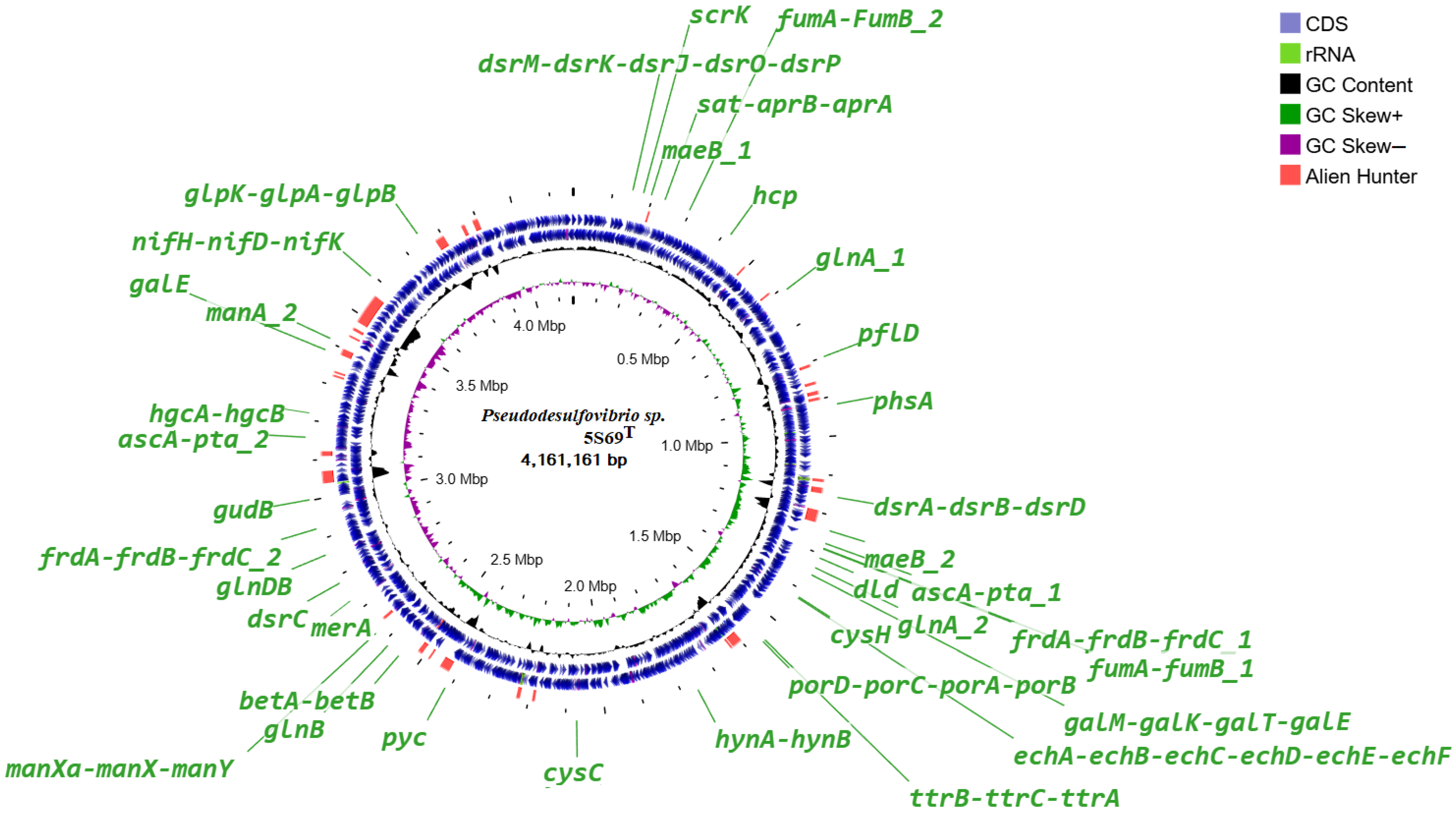
3.3. Genome Features and Phylogeny
3.4. Genome Analysis and Functional Annotation
3.4.1. Carbohydrate Metabolism and Oxidation of Organic Compounds
3.4.2. Hydrogen Utilization and CO2 Fixation
3.4.3. Glycerol and Alcohols Utilization
3.4.4. Pyruvate Fermentation
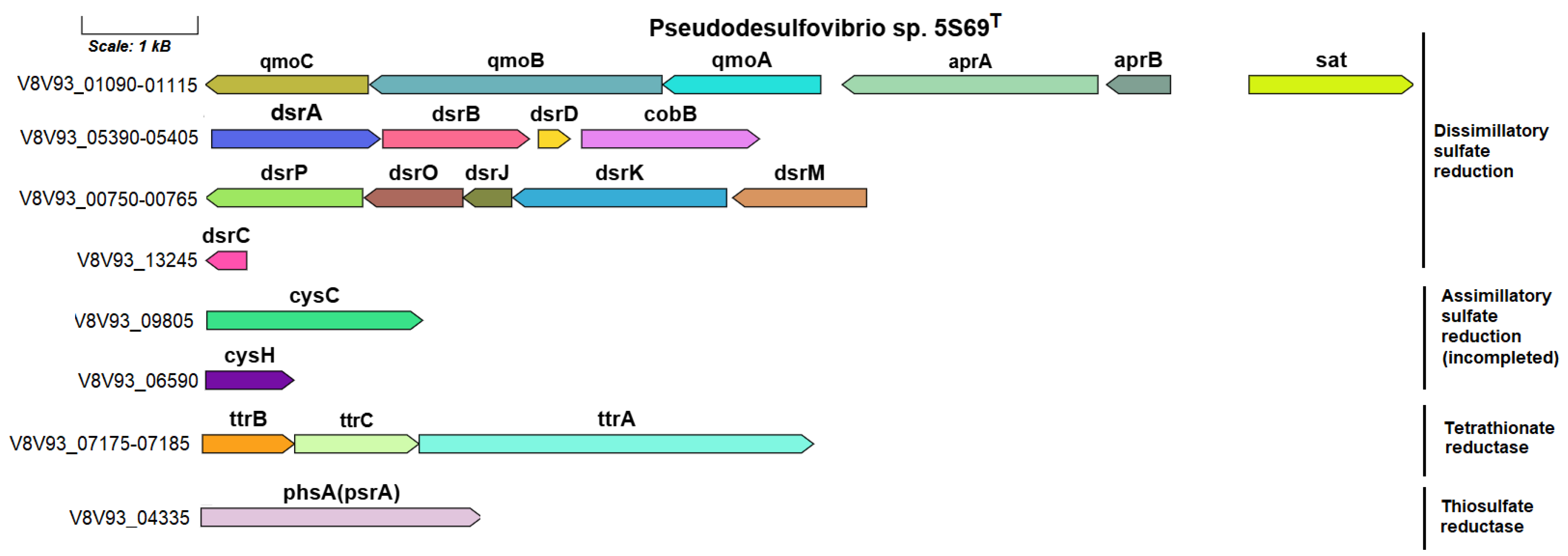
3.4.5. Sulfur Metabolism
3.4.6. Nitrogen Metabolism
3.4.7. Oxidative and Osmotic Stress Response and Heavy Metal Resistance
3.4.8. Hydrogenase Genes
3.4.9. Mercury Methylation Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magot, M.; Ollivier, B.; Patel, B.K.C. Microbiology of petroleum reservoirs. Antonie Van Leeuwenhoek 2000, 77, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.; Elshahed, M.S.; McInerney, M.J. Microbial processes in oil fields: Culprits, problems and opportunities. Adv. Appl. Microbiol. 2009, 66, 141–251. [Google Scholar] [CrossRef] [PubMed]
- Bastin, E.S. The presence of sulfate-reducing bacteria in oilfield waters. Science 1926, 63, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg-Karagicheva, T.L. Microbiological investigations on the sulfur salt waters off Apsheron. Azerb. Petrol. Econ. 1926, 6–7, 30–39. (In Russian) [Google Scholar]
- Nazina, T.N.; Rozanova, E.P. Thermophilic sulfate-reducing bacteria from oil strata. Microbiology 1978, 47, 142–148. [Google Scholar]
- Gieg, L.M.; Davidova, I.A.; Duncan, K.E.; Suflita, J.M. Methanogenesis, sulfate reduction and crude oil biodegradation in hot Alaskan oil fields. Environ. Microbiol. 2010, 12, 3074–3086. [Google Scholar] [CrossRef]
- Gieg, L.M.; Jack, T.R.; Foght, J.M. Biological souring and mitigation in oil reservoirs. Appl. Microbiol. Biotechnol. 2011, 92, 263–282. [Google Scholar] [CrossRef]
- Nazina, T.N.; Shestakova, N.M.; Ivoilov, V.S.; Kostrukova, N.K.; Belyaev, S.S.; Ivanov, M.V. Radiotracer assay of microbial processes in petroleum reservoirs. Adv. Biotechnol. Microbiol. 2017, 2, 555591. [Google Scholar] [CrossRef]
- Guan, J.; Zhang, B.L.; Mbadinga, S.M.; Liu, J.F.; Gu, J.D.; Mu, B.Z. Functional genes (dsr) approach reveals similar sulphidogenic prokaryotes diversity but different structure in saline waters from corroding high temperature petroleum reservoirs. Appl. Microbiol. Biotechnol. 2014, 98, 1871–1882. [Google Scholar] [CrossRef]
- Liang, R.; Grizzle, R.S.; Duncan, K.E.; Mcinerney, M.J.; Suflita, J.M. Roles of thermophilic thiosulfate-reducing bacteria and methanogenic archaea in the biocorrosion of oil pipelines. Front. Microbiol. 2014, 5, 89. [Google Scholar] [CrossRef]
- Vigneron, A.; Alsop, E.B.; Chambers, B.; Lomans, B.P.; Head, I.M.; Tsesmetzis, N. Complementary microorganisms in highly corrosive biofilms from an offshore oil production facility. Appl. Environ. Microbiol. 2016, 82, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Voordouw, G. Control of microbial sulfide production with biocides and nitrate in oil reservoir simulating bioreactors. Front. Microbiol. 2015, 6, 1387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-Y.; Hu, B.; Dolfing, J.; Li, Y.; Tang, Y.-Q.; Jiang, Y.; Chi, C.-Q.; Xing, J.; Nie, Y.; Wu, X.-L. Thermodynamically favorable reactions shape the archaeal community affecting bacterial community assembly in oil reservoirs. Sci. Total Environ. 2021, 781, 146506. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, K.J.; Sierra-Garcia, I.N.; Zafra, G.; de Oliveira, V.M. Genome-resolved meta-analysis of the microbiome in oil reservoirs worldwide. Microorganisms 2021, 9, 1812. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, D.S.; Semenova, E.M.; Grouzdev, D.S.; Bidzhieva, S.K.; Babich, T.L.; Loiko, N.G.; Ershov, A.P.; Kadnikov, V.V.; Beletsky, A.V.; Mardanov, A.V.; et al. Sulfidogenic microbial communities of the Uzen high-temperature oil field in Kazakhstan. Microorganisms 2021, 9, 1818. [Google Scholar] [CrossRef]
- Ivanov, M.V.; Belyaev, S.S. Microbial activity in waterflooded oil fields and its possible regulation. In Proceedings of the 1982 International Conference on Microbial Enhancement of Oil Recovery, Alton, OK, USA, 16–21 May 1982; pp. 48–57. [Google Scholar]
- Belyaev, S.S.; Laurinavichus, K.S.; Obraztsova, A.Y.; Gorlatov, S.N.; Ivanov, M.V. Microbiological processes in the near-bottom zone of injection wells of oil fields. Microbiology 1982, 51, 997–1001. [Google Scholar]
- Belyaev, S.S.; Borzenkov, I.A.; Glumov, I.F.; Ibatullin, R.R.; Milekhina, E.I.; Ivanov, M.V. Activation of the geochemical activity of stratal microflora as basis of a biotechnology for enhancement of oil recovery. Microbiology 1998, 67, 708–714. [Google Scholar]
- Belyaev, S.S.; Borzenkov, I.A. Microbial transformation of low-molecular-weight carbon compounds in the deep subsurface. In Biogeochemistry of Global Change; Chapman & Hall: London, OH, USA, 1993; pp. 825–838. [Google Scholar]
- Ziganshina, E.E.; Mohammed, W.S.; Ziganshin, A.M. Microbial diversity of the produced waters from the oilfields in the Republic of Tatarstan (Russian Federation): Participation in Biocorrosion. Appl. Sci. 2023, 13, 12984. [Google Scholar] [CrossRef]
- Nazina, T.N.; Shestakova, N.M.; Pavlova, N.K.; Tatarkin, Y.V.; Ivoilov, V.S.; Khisametdinov, M.R.; Sokolova, D.S.; Babich, T.L.; Tourova, T.P.; Poltaraus, A.B.; et al. Functional and phylogenetic microbial diversity in formation waters of a low-temperature carbonate petroleum reservoir. Int. Biodeterior. Biodegrad. 2013, 81, 71–81. [Google Scholar] [CrossRef]
- Nazina, T.; Sokolova, D.; Grouzdev, D.; Semenova, E.; Babich, T.; Bidzhieva, S.; Serdukov, D.; Volkov, D.; Bugaev, K.; Ershov, A.; et al. The potential application of microorganisms for sustainable petroleum recovery from heavy oil reservoirs. Sustainability 2020, 12, 15. [Google Scholar] [CrossRef]
- Kadnikov, V.V.; Ravin, N.V.; Sokolova, D.S.; Semenova, E.M.; Bidzhieva, S.K.; Beletsky, A.V.; Ershov, A.P.; Babich, T.L.; Khisametdinov, M.R.; Mardanov, A.V.; et al. Metagenomic and culture-based analyses of microbial communities from petroleum reservoirs with high-salinity formation water, and their biotechnological botential. Biology 2023, 12, 1300. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Gayet, N.; Zeng, X.; Shao, Z.; Jebbar, M.; Alain, K. Pseudodesulfovibrio indicus gen. nov., sp. nov., a piezophilic sulfate-reducing bacterium from the Indian Ocean and reclassification of four species of the genus Desulfovibrio. Int. J. Syst. Evol. Microbiol. 2016, 66, 3904–3911. [Google Scholar] [CrossRef] [PubMed]
- Galushko, A.; Kuever, J. Pseudodesulfovibrio. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons: New York, NY, USA, 2019; pp. 1–11. [Google Scholar]
- Ranchou-Peyruse, M.; Goni-Urriza, M.; Guignard, M.; Goas, M.; Ranchou-Peyruse, A.; Guyoneaud, R. Pseudodesulfovibrio hydrargyri sp. nov., a mercury-methylating bacterium isolated from a brackish sediment. Int. J. Syst. Evol. Microbiol. 2018, 68, 1461–1466. [Google Scholar] [CrossRef]
- Caumette, P.; Cohen, Y.; Matheron, R. Isolation and characterization of Desulfovibrio halophilus sp. nov., a halophilic sulfate-reducing bacterium isolated from Solar Lake (Sinai). Syst. Appl. Microbiol. 1991, 14, 33–38. [Google Scholar] [CrossRef]
- Waite, D.W.; Chuvochina, M.; Pelikan, C.; Parks, D.H.; Yilmaz, P.; Wagner, M.; Loy, A.; Naganuma, T.; Nakai, R.; Whitman, W.B.; et al. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int. J. Syst. Evol. Microbiol. 2020, 70, 5972–6016. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, C.C.; Soren, A.B.; Gionfriddo, C.M.; Podar, M.; Wall, J.D.; Brown, S.D.; Michener, J.K.; Urriza, M.S.G.; Elias, D.A. Pseudodesulfovibrio mercurii sp. nov., a mercury-methylating bacterium isolated from sediment. Int. J. Syst. Evol. Microbiol. 2019, 71, 004697. [Google Scholar] [CrossRef]
- Ben Ali Gam, Z.; Oueslati, R.; Abdelkafi, S.; Casalot, L.; Tholozan, J.L.; Labat, M. Desulfovibrio tunisiensis sp. nov., a novel weakly halotolerant, sulfate-reducing bacterium isolated from exhaust water of a Tunisian oil refinery. Int. J. Syst. Evol. Microbiol. 2009, 59, 1059–1063. [Google Scholar] [CrossRef]
- Zheng, R.; Wu, S.; Sun, C. Pseudodesulfovibrio cashew sp. nov., a novel deep-sea sulfate-reducing bacterium, linking heavy metal resistance and sulfur cycle. Microorganisms 2021, 9, 429. [Google Scholar] [CrossRef]
- Gaikwad, S.L.; Pore, S.D.; Dhakephalkar, P.K.; Dagar, S.S.; Soni, R.; Kaur, M.P.; Rawat, H.N. Pseudodesulfovibrio thermohalotolerans sp. nov., a novel obligately anaerobic, halotolerant, thermotolerant, and sulfate-reducing bacterium isolated from a western offshore hydrocarbon reservoir in India. Anaerobe 2023, 83, 102780. [Google Scholar] [CrossRef]
- Slobodkina, G.; Merkel, A.; Novikov, A.; Slobodkin, A. Pseudodesulfovibrio pelocollis sp. nov. a sulfate-reducing bacterium isolated from a terrestrial mud volcano. Curr. Microbiol. 2024, 81, 120. [Google Scholar] [CrossRef]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef] [PubMed]
- Genus Pseudodesulfovibrio. Available online: https://lpsn.dsmz.de/genus/pseudodesulfovibrio (accessed on 1 August 2024).
- Kuever, J.; Rainey, F.A.; Widdel, F. Order II. Desulfovibrionales ord. nov. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; Brenner, D.J., Krieg, N.R., Staley, J.T., Garrity, G.M., Eds.; Volume 2 (The Proteobacteria), Part C (The Alpha-, Beta-, Delta-, and Epsilonproteobacteria); Springer: New York, NY, USA, 2005; pp. 925–926. [Google Scholar]
- Park, M.J.; Kim, Y.J.; Park, M.; Yu, J.; Namirimu, T.; Roh, Y.R.; Kwon, K.K. Establishment of genome based criteria for classification of the family Desulfovibrionaceae and proposal of two novel genera, Alkalidesulfovibrio gen. nov. and Salidesulfovibrio gen. nov. Front. Microbiol. 2022, 13, 738205. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.N.; Sokolova, D.S.; Babich, T.L.; Semenova, E.M.; Ershov, A.P.; Bidzhieva, S.K.; Borzenkov, I.A.; Poltaraus, A.B.; Khisametdinov, M.R.; Tourova, T.P. Microorganisms of low-temperature heavy oil reservoirs (Russia) and their possible application for enhanced oil recovery. Microbiology 2017, 86, 773–785. [Google Scholar] [CrossRef]
- Widdel, F.; Bak, F. Gram-negative mesophilic sulphate-reducing bacteria. In The Prokaryotes, 2nd ed.; Balows, A., Trüper, H.G., Dworkin, M., Harder, W., Schleifer, K.-H., Eds.; Springer: New York, NY, USA, 1992; Volume IV, pp. 3352–3378. [Google Scholar]
- Wolin, E.A.; Wolin, M.J.; Wolfe, R.S. Formation of methane by bacterial extracts. J. Biol. Chem. 1963, 238, 2882–2888. [Google Scholar] [CrossRef] [PubMed]
- Pfennig, N.; Lippert, K.D. Über das Vitamin B12-Bedürfnis phototropher Schwefelbakterien. Arch. Mikrobiol. 1966, 55, 245–256. [Google Scholar] [CrossRef]
- Hungate, R.E. A roll tube method for the cultivation of strict anaerobes. In Methods in Microbiology; Norris, J.L., Ribbons, D.W., Eds.; Academic Press: New York, NY, USA, 1969; Volume 3b, pp. 117–132. [Google Scholar]
- Trüper, H.G.; Schlegel, H.G. Sulfur metabolism in Thiorhodaceae. I. Quantitative measurements on growing cells of Chromatium okenii. Antonie Van Leeuwenhoek 1964, 30, 321–323. [Google Scholar] [CrossRef]
- Bidzhieva, S.K.; Sokolova, D.S.; Grouzdev, D.S.; Kostrikina, N.A.; Poltaraus, A.B.; Tourova, T.P.; Shcherbakova, V.A.; Troshina, O.Y.; Nazina, T.N. Sphaerochaeta halotolerans sp. nov., a novel spherical halotolerant spirochete from a Russian heavy oil reservoir, emended description of the genus Sphaerochaeta, reclassification of Sphaerochaeta coccoides to a new genus Parasphaerochaeta gen. nov. as Parasphaerochaeta coccoides comb. nov. and proposal of Sphaerochaetaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 4748–4759. [Google Scholar] [CrossRef]
- Troshina, O.; Oshurkova, V.; Suzina, N.; Machulin, A.; Ariskina, E.; Vinokurova, N.; Kopitsyn, D.; Novikov, A.; Shcherbakova, V. Sphaerochaeta associata sp. nov., a spherical spirochaete isolated from cultures of Methanosarcina mazei JL01. Int. J. Syst. Evol. Microbiol. 2015, 65, 4315–4322. [Google Scholar] [CrossRef]
- Collins, M.D.; Jones, D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [CrossRef]
- Minnikin, D.E.; O’Donnell, A.G.; Goodfellow, M.; Alderson, G.; Athalye, M.; Schaal, A.; Parlett, J.H. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 1984, 2, 233–241. [Google Scholar] [CrossRef]
- Nichols, B.W. Separation of the lipids of photosynthetic tissues: Improvements in analysis by thin-layer chromatography. Biochim. Biophys. Acta 1963, 70, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.M.; Grouzdev, D.S.; Sokolova, D.S.; Tourova, T.P.; Poltaraus, A.B.; Potekhina, N.V.; Shishina, P.N.; Bolshakova, M.A.; Avtukh, A.N.; Ianutsevich, E.A.; et al. Physiological and genomic characterization of Actinotalea subterranea sp. nov. from oil-degrading methanogenic enrichment and reclassification of the family Actinotaleaceae. Microorganisms 2022, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Available online: https://github.com/najoshi/sickle (accessed on 24 July 2024).
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ’omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Harrison, K.J.; de Crécy-Lagard, V.; Zallot, R. Gene Graphics: A genomic neighborhood data visualization web application. Bioinformatics 2018, 34, 1406–1408. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Badziong, W.; Ditter, B.; Thauer, R.K. Acetate and carbon dioxide assimilation by Desulfovibrio vulgaris (Marburg), growing on hydrogen and sulfate as sole energy source. Arch. Microbiol. 1979, 123, 301–305. [Google Scholar] [CrossRef]
- Tang, K.-H.; Tang, Y.J.; Blankenship, R.E. Carbon metabolic pathways in phototrophic bacteria and their broader evolutionary. Front. Microbiol. 2011, 2, 165. [Google Scholar] [CrossRef]
- Santos, S.C.; Liebensteiner, M.G.; van Gelder, A.H.; Dimitrov, M.R.; Almeida, P.F.; Quintella, C.M.; Stams, A.J.M.; Sánchez-Andrea, I. Bacterial glycerol oxidation coupled to sulfate reduction at neutral and acidic pH. J. Gen. Appl. Microbiol. 2018, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goorissen, H.P. Thermophilic Methanol Utilization by Sulfate Reducing Bacteria. Ph.D. Thesis, University of Groningen, Groningen, The Netherlands, 2002. Chapter 3. pp. 55–61. [Google Scholar]
- Sousa, D.Z.; Visser, M.; van Gelder, A.H.; Boeren, S.; Pieterse, M.M.; Pinkse, M.W.H.; Verhaert, P.D.E.M.; Vogt, C.; Franke, S.; Kümmel, S.; et al. The deep-subsurface sulfate reducer Desulfotomaculum kuznetsovii employs two methanol-degrading pathways. Nat. Commun. 2018, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.N.; Tourova, T.P.; Grouzdev, D.S.; Bidzhieva, S.K.; Poltaraus, A.B. A novel view on the taxonomy of sulfate-reducing bacterium ‘Desulfotomaculum salinum’ and a description of a new species Desulfofundulus salinus sp. nov. Microorganisms 2024, 12, 1115. [Google Scholar] [CrossRef] [PubMed]
- Friedeheim, L.; Boeren, S.; Sánchez-Andrea, I.; Stams, A.J.M.; Sousa, D.Z. Alcohol dehydrogenase system acts as the sole pathway for methanol oxidation in Desulfofundulus kuznetsovii strain TPOSR. Antonie Van Leeuwenhoek 2024, 117, 47. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Fonseca, L.; Moura, I.; Moura, J.J.G. Reduction of carbon dioxide by a molybdenum-containing formate dehydrogenase: A kinetic and mechanistic study. J. Am. Chem. Soc. 2016, 138, 8834–8846. [Google Scholar] [CrossRef] [PubMed]
- Lupa, B.; Hendrickson, E.L.; Leigh, J.A.; Whitman, W.B. Formate-dependent H2 production by the mesophilic methanogen Methanococcus maripaludis. Appl. Environ. Microbiol. 2008, 74, 6584–6590. [Google Scholar] [CrossRef]
- Postgate, J.R. The reduction of sulphur compounds by Desulphovibrio desulphuricans. J. Gen. Microbiol. 1951, 5, 725–738. [Google Scholar] [CrossRef]
- Barrett, E.L.; Clark, M.A. Tetrathionate reduction and production of hydrogen sulfide from thiosulfate. Microbiol. Rev. 1987, 51, 192–205. [Google Scholar] [CrossRef]
- Hensel, M.; Hinsley, A.P.; Nikolaus, T.; Sawers, G.; Berks, B.C. The genetic basis of tetrathionate respiration in Salmonella typhimurium. Mol. Microbiol. 1999, 32, 275–287. [Google Scholar] [CrossRef]
- Lumppio, H.L.; Shenvi, N.V.; Summers, A.O.; Voordouw, G.; Kurtz, D.M., Jr. Rubrerythrin and rubredoxin oxidoreductase in Desulfovibrio vulgaris: A novel oxidative stress protection system. J. Bacteriol. 2001, 183, 101–108. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Lu, S.; Lou, H.; Wang, X.; Wang, W. The protective role of potassium in the adaptation of Pseudomonas protegens SN15-2 to hyperosmotic stress. Microbiol. Res. 2024, 289, 127887. [Google Scholar] [CrossRef] [PubMed]
- Patzer, S.I.; Hantke, K. The ZnuABC high-affinity zinc uptake system and its regulator Zur in Escherichia coli. Mol. Microbiol. 1998, 28, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.H.; Moreno-Sánchez, R.; Cervantes, C. Chromate efflux by means of the ChrA chromate resistance protein from Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 7398–7400. [Google Scholar] [CrossRef] [PubMed]
- Hedderich, R. Energy-converting [NiFe] hydrogenases from archaea and extremophiles: Ancestors of complex I. J. Bioenerg. Biomembr. 2004, 36, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Baffert, C.; Kpebe, A.; Avilan, L.; Brugna, M. Hydrogenases and H2 metabolism in sulfate-reducing bacteria of the Desulfovibrio genus. Adv. Microb. Physiol. 2019, 74, 143–189. [Google Scholar] [CrossRef]
- Pereira, P.M.; He, Q.; Valente, F.M.; Xavier, A.V.; Zhou, J.; Pereira, I.A.; Louro, R.O. Energy metabolism in Desulfovibrio vulgaris Hildenborough: Insights from transcriptome analysis. Antonie Van Leeuwenhoek. 2008, 93, 347–362. [Google Scholar] [CrossRef]
- Keller, K.L.; Wall, J.D. Genetics and molecular biology of the electron flow for sulfate respiration in Desulfovibrio. Front. Microbiol. 2011, 2, 135. [Google Scholar] [CrossRef]
- Hatchikian, E.C.; Forget, N.; Bernadac, A.; Alazard, D.; Ollivier, B. Involvement of a single periplasmic hydrogenase for both hydrogen uptake and production in some Desulfovibrio species. Res. Microbiol. 1995, 146, 129–141. [Google Scholar] [CrossRef]
- Caffrey, S.M.; Park, H.S.; Voordouw, J.K.; He, Z.; Zhou, J.; Voordouw, G. Function of periplasmic hydrogenases in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 2007, 189, 6159–6167. [Google Scholar] [CrossRef]
- Schaefer, J.K.; Rocks, S.S.; Zheng, W.; Liang, L.; Gu, B.; Morel, F.M.M. Active transport, substrate specificity, and methylation of Hg(II) in anaerobic bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 8714–8719. [Google Scholar] [CrossRef]
- Parks, J.M.; Johs, A.; Podar, M.; Bridou, R.; Hurt, R.A., Jr.; Smith, S.D.; Tomanicek, S.J.; Qian, Y.; Brown, S.D.; Brandt, C.C.; et al. The genetic basis for bacterial mercury methylation. Science 2013, 339, 1332–1335. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Type strain | 5S69T | J2T | BerOc1T | ND132T | MCM B-1480T |
| Motility | + | + | + | + | + |
| NaCl, min-opt-max, % (w/v) | 0.2–(2–4)–6 | 0.2–2.5–6.0 | 0.2–1.5–4.0 | 0–2–3 | 1–3–6 |
| Temperature, min-opt-max, °C | 15–(23–28)–37 | 9–(30–35)–40 | 25–30–35 | 20–32–37 | 20–37–60 |
| pH, min-opt-max | 4.6–6.5–8.6 | 5.0–(6.5–7.0)–8.0 | –(6.0–7.4)– | 6.8–7.8–8.2 | 6–7–8 |
| Electron donor with sulfate: | |||||
| H2/CO2 | + * | + | + | + * | ND |
| Formate | + | + | − | + * | + |
| Succinate | + | ND | − | − | + |
| Fumarate | + | − | + | + | + |
| Citrate | W | ND | − | ND | − |
| Malate | + | + | − | − | ND |
| Benzoate | − | ND | − | ND | ND |
| Methanol | + | ND | − | − | ND |
| Ethanol | + | − | W | − | ND |
| Glycerol | + | ND | − | ND | |
| Glucose | − | ND | − | − | + |
| Sucrose | − | ND | − | ND | + |
| Fructose | + | ND | − | ND | + |
| Lactose | − | ND | − | ND | + |
| Galactose | W | ND | − | ND | + |
| Electron acceptor: | |||||
| Sulfite, thiosulfate | + | + | + | + | + |
| Elemental sulfur | + | − | − | ND | − |
| Nitrate | − | W | − | − | + |
| Fermentation of: | |||||
| Lactate | − | + | − | − | + |
| Fumarate | + | − | − | + | + |
| Genome size (Mb) | 4.16 | 3.96 | 4.1 | 3.86 | 3.87 |
| Genomic G + C content (mol %) | 63.0 | 63.5 | 64.0 | 65.2 | 60.39 |
| Major cellular fatty acids | i-C15:0, ai-C15:0, C16:0 | i-C15:0, ai-C15:0, i-C17:1 ω9c, i-C17:0 | C18:0, ai-C15:0, C16:0, C18:1 ω7 | i-C15:0, ai-C15:0, i-C17:0, i-C17:1 ω9c | i-C15:0, ai-C15:0, C16:0, ai-C17:0 |
| Isolation source | Hydrocarbon reservoir | Deep hydrothermal vent | Brackish sediments | Estuarine sediment | Hydrocarbon reservoir |
| Relatedness to strain 5S69T | |||||
| Identity of 16S rRNA (%) | 100 | 98.0 | 99.5 | 98.9 | 98.4 |
| ANI (%) | 100 | 83.7 | 90.0 | 87.7 | 84.6 |
| AAI (%) | 100 | 80.9 | 90.0 | 87.2 | 84.8 |
| dDDH (%) | 100 | 26.2 | 40.3 | 34.5 | 28.5 |
| Parameter | Pseudodesulfovibrio methanolicus sp. nov. |
|---|---|
| Genus name | Pseudodesulfovibrio |
| Species name | Pseudodesulfovibrio methanolicus |
| Species status | sp. nov. |
| Species etymology | me.tha.no’li.cus. N.L. neut. n. methanol, methanol; N.L. masc. adj. methanolicus, pertaining to methanol |
| Designation of the Type Strain | 5S69T |
| Strain Collection Numbers | VKM B-3653T = KCTC 25499T = UQM 41509T |
| Genome accession number | GCF_037094465.1 |
| Genome status | Complete |
| Genome size | 4.16 Mb |
| GC mol% | 63.0 |
| 16S rRNA gene accession nr. | PP792559.1 |
| Description of the new taxon and diagnostic traits | The cells are straight or slightly curved rods, motile due a single flagellum, stained Gram-negative, and have cell wall structure typical of Gram-negative bacteria. Growth is observed in the presence of 0.2–6.0% (w/v) NaCl (optimum, 2.0–4.0% NaCl), at pH 4.6–8.6 (optimum, pH 6.5), and at 15–37 °C (optimum, 23–28 °C) under sulfate-reducing conditions. Strictly anaerobic. Reduces sulfate to sulfide in media with formate, lactate, pyruvate, malate, fumarate, succinate, methanol, ethanol, glycerol, fructose, and yeast extract as carbon and energy sources; weak growth observed on glutamate, citrate, propanol, galactose, and mannose, but does not use acetate, propionate, butyrate, glycine, L-serine, ornithine, glucose, lactose, sucrose, and benzoate. Hydrogen is utilized as electron donor for sulfate reduction in the presence of acetate as a carbon source. Lactate is oxidized with the production of acetate. Fermentative growth is observed with pyruvate, but lactate is not fermented. It uses sulfate, thiosulfate, sulfite, and fumarate as electron acceptors in the presence of lactate, but does not use nitrate. The predominant cellular fatty acids are iso-C15:0, anteiso-C15:0, and C16:0. The major polar lipids are phosphatidylethanolamine, diphosphatidylglycerol, phosphatidylglycerol, glycolipid, and phosphatidylserine. The major respiratory quinone is menaquinone MK-6(H4). The genome size of the type strain is 4.16 Mb with a genomic G + C content of 63.0 mol%. The type strain, 5S69T (VKM B-3653T = KCTC 25499T = UQM 41509T), was isolated from the Vostochno-Anzirskoe oil field, in Yelabuzhsky district, Tatarstan, Russian Federation. The GenBank/EMBL/DDBJ accession number for the 16S rRNA gene sequence is PP792559.1 and the genomic assembly accession number is GCF_037094465.1. |
| Country and region of origin | Russian Federation, Tatarstan, Yelabuzhsky district |
| Date of isolation | 2018 |
| Source of isolation | A mixture of injection fresh river water and production water from the Vostochno-Anzirskoe oil field |
| Sampling date | June 2016 |
| Latitude, Longitude | 55°66′69″ N, 51°49′84.00″ E |
| Depth (meters below sea level) | 1585 |
| Number of strains in study | 1 |
| Information related to the Nagoya Protocol | Not applicable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bidzhieva, S.K.; Tourova, T.P.; Kadnikov, V.V.; Samigullina, S.R.; Sokolova, D.S.; Poltaraus, A.B.; Avtukh, A.N.; Tereshina, V.M.; Beletsky, A.V.; Mardanov, A.V.; et al. Phenotypic and Genomic Characterization of a Sulfate-Reducing Bacterium Pseudodesulfovibrio methanolicus sp. nov. Isolated from a Petroleum Reservoir in Russia. Biology 2024, 13, 800. https://doi.org/10.3390/biology13100800
Bidzhieva SK, Tourova TP, Kadnikov VV, Samigullina SR, Sokolova DS, Poltaraus AB, Avtukh AN, Tereshina VM, Beletsky AV, Mardanov AV, et al. Phenotypic and Genomic Characterization of a Sulfate-Reducing Bacterium Pseudodesulfovibrio methanolicus sp. nov. Isolated from a Petroleum Reservoir in Russia. Biology. 2024; 13(10):800. https://doi.org/10.3390/biology13100800
Chicago/Turabian StyleBidzhieva, Salimat K., Tatyana P. Tourova, Vitaly V. Kadnikov, Salima R. Samigullina, Diyana S. Sokolova, Andrey B. Poltaraus, Alexander N. Avtukh, Vera M. Tereshina, Alexey V. Beletsky, Andrey V. Mardanov, and et al. 2024. "Phenotypic and Genomic Characterization of a Sulfate-Reducing Bacterium Pseudodesulfovibrio methanolicus sp. nov. Isolated from a Petroleum Reservoir in Russia" Biology 13, no. 10: 800. https://doi.org/10.3390/biology13100800
APA StyleBidzhieva, S. K., Tourova, T. P., Kadnikov, V. V., Samigullina, S. R., Sokolova, D. S., Poltaraus, A. B., Avtukh, A. N., Tereshina, V. M., Beletsky, A. V., Mardanov, A. V., & Nazina, T. N. (2024). Phenotypic and Genomic Characterization of a Sulfate-Reducing Bacterium Pseudodesulfovibrio methanolicus sp. nov. Isolated from a Petroleum Reservoir in Russia. Biology, 13(10), 800. https://doi.org/10.3390/biology13100800