
Recognition of Aedes aegypti Mosquito Saliva Protein LTRIN by the Human Receptor LTβR for Controlling the Immune Response
 ,
,  and
and 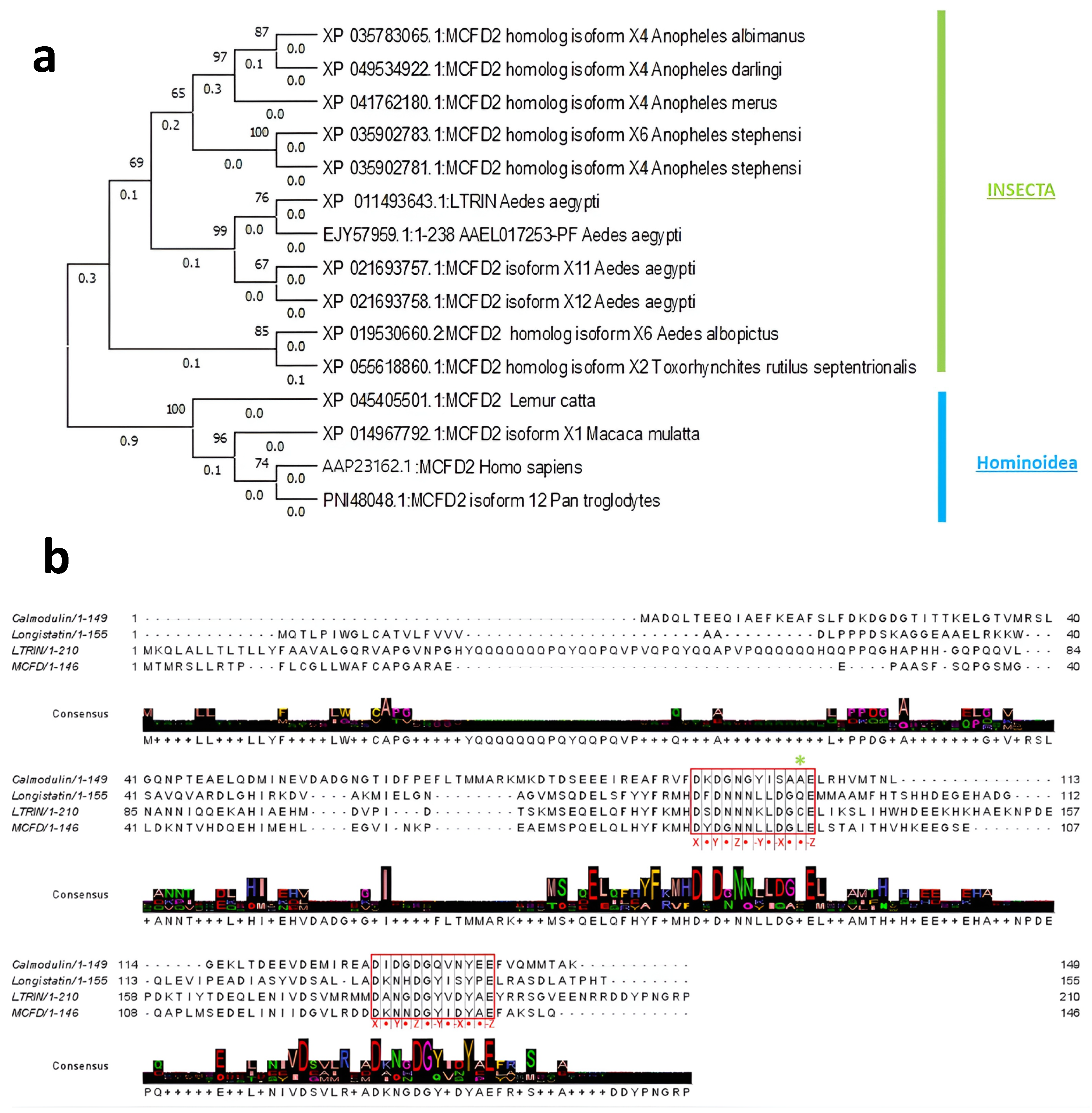{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Maximum Likelihood Phylogenetic Tree
2.2. Protein Expression and Purification
2.3. Protein Refolding of LTβR
2.4. Sedimentation Velocity-Analytical Ultracentrifugation of ΔLTRIN
2.5. Dynamic Light Scattering of ΔLTRIN
2.6. Ruthenium Red Assay of ΔLTRIN
2.7. Isothermal Titration Calorimetry of ΔLTRIN with Different Divalent Ions
2.8. Gel Mobility Shift Assay of ΔLTRIN
2.9. Circular Dichroism of ΔLTRIN and its Mutants
2.10. ELISA
2.11. Hydrogen-Deuterium Exchange Mass Spectrometry
3. Results
3.1. Evolutionary Relationship of LTRIN with Insecta and Hominoidea
3.2. LTRIN Exists as a Dimer
3.3. LTRIN Has EF-Hand Motifs and Binds to Ca2+ and Other Divalent Ions
3.4. LTRIN Has a Stable Alpha-Helix Dominant Structure over a Wide Range of Temperatures
3.5. Disulfide Bonds in LTβR
3.6. The Interactions between LTRIN and LTβR Are Not Affected by the Glycosylation of LTβR or Divalent Ions
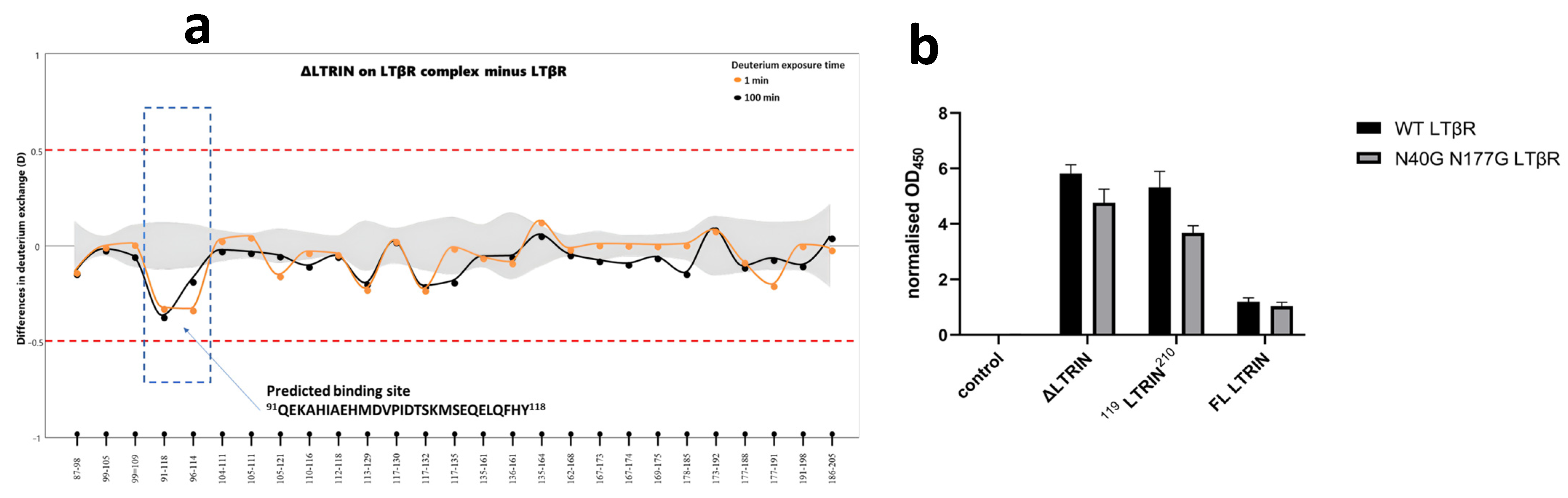
3.7. Mapping the Interaction Sites of ΔLTRIN with LTβR
3.8. The Second EF-Hand of ΔLTRIN Plays an Important Role in Interacting with LTβR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caraballo, H.; King, K. Emergency department management of mosquito-borne illness: Malaria, dengue, and West Nile virus. Emerg. Med. Pract. 2014, 16, 1–23. [Google Scholar] [PubMed]
- Franklinos, L.H.V.; Jones, K.E.; Redding, D.W.; Abubakar, I. The effect of global change on mosquito-borne disease. Lancet Infect. Dis. 2019, 19, e302–e312. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Federman, H.G.; Sievert, D.; Gleeson, J.G. The Neurobiology of Modern Viral Scourges: ZIKV and COVID-19. Neuroscientist 2021, 28, 438–452. [Google Scholar] [CrossRef]
- Sun, P.; Nie, K.; Zhu, Y.; Liu, Y.; Wu, P.; Liu, Z.; Du, S.; Fan, H.; Chen, C.-H.; Zhang, R.; et al. A mosquito salivary protein promotes flavivirus transmission by activation of autophagy. Nat. Commun. 2020, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Rocklöv, J.; Dubrow, R. Climate change: An enduring challenge for vector-borne disease prevention and control. Nat. Immunol. 2020, 21, 479–483. [Google Scholar] [CrossRef]
- Guerrero, D.; Cantaert, T.; Missé, D. Aedes Mosquito Salivary Components and Their Effect on the Immune Response to Arboviruses. Front. Cell. Infect. Microbiol. 2020, 10, 407. [Google Scholar] [CrossRef]
- Valenzuela-Leon, P.C.; Shrivastava, G.; Martin-Martin, I.; Cardenas, J.C.; Londono-Renteria, B.; Calvo, E. Multiple Salivary Proteins from Aedes aegypti Mosquito Bind to the Zika Virus Envelope Protein. Viruses 2022, 14, 221. [Google Scholar] [CrossRef]
- Isawa, H.; Yuda, M.; Orito, Y.; Chinzei, Y. A mosquito salivary protein inhibits activation of the plasma contact system by binding to factor XII and high molecular weight kininogen. J. Biol. Chem. 2002, 277, 27651–27658. [Google Scholar] [CrossRef]
- Hastings, A.K.; Uraki, R.; Gaitsch, H.; Dhaliwal, K.; Stanley, S.; Sproch, H.; Williamson, E.; MacNeil, T.; Marin-Lopez, A.; Hwang, J.; et al. Aedes aegypti NeSt1 Protein Enhances Zika Virus Pathogenesis by Activating Neutrophils. J. Virol. 2019, 93, e00395-19. [Google Scholar] [CrossRef]
- Marin-Lopez, A.; Wang, Y.; Jiang, J.; Ledizet, M.; Fikrig, E. AgBR1 and NeSt1 antisera protect mice from Aedes aegypti-borne Zika infection. Vaccine 2021, 39, 1675–1679. [Google Scholar] [CrossRef]
- Pielnaa, P.; Al-Saadawe, M.; Saro, A.; Dama, M.F.; Zhou, M.; Huang, Y.; Huang, J.; Xia, Z. Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development. Virology 2020, 543, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Higgs, S.; Horne, K.M.; Vanlandingham, D.L. Flavivirus-mosquito interactions. Viruses 2014, 6, 4703–4730. [Google Scholar] [CrossRef]
- Rodríguez de la Rosa, R.P.; Cano-Torres, J.O.; Rosales, S.; Kleinert, A.P.; Gómez, A.; George, F.; George, J.; Piedad García, M.; Nájera-Cancino, G.; Guerra-de-Blas, P.D.C.; et al. Guillain-Barré syndrome outbreak in Tapachula temporally associated with the Zika virus introduction in Southern Mexico. Epidemiol. Infect. 2022, 150, e181. [Google Scholar] [CrossRef] [PubMed]
- Song, B.H.; Yun, S.I.; Woolley, M.; Lee, Y.M. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 308, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.-Q.; Yang, X.; Wei, Y.; Chen, J.-T.; Wang, X.-J.; Peng, H.-J. A Review on Dengue Vaccine Development. Vaccines 2020, 8, 63. [Google Scholar] [CrossRef]
- Xu, L.; Ma, Z.; Li, Y.; Pang, Z.; Xiao, S. Antibody dependent enhancement: Unavoidable problems in vaccine development. Adv. Immunol. 2021, 151, 99–133. [Google Scholar] [CrossRef]
- Manning, J.E.; Oliveira, F.; Coutinho-Abreu, I.V.; Herbert, S.; Meneses, C.; Kamhawi, S.; Baus, H.A.; Han, A.; Czajkowski, L.; Rosas, L.A.; et al. Safety and immunogenicity of a mosquito saliva peptide-based vaccine: A randomised, placebo-controlled, double-blind, phase 1 trial. Lancet 2020, 395, 1998–2007. [Google Scholar] [CrossRef]
- Chuang, Y.M.; Tang, X.D.; Fikrig, E. A Mosquito AgTRIO Monoclonal Antibody Reduces Early Plasmodium Infection of Mice. Infect. Immun. 2022, 90, e0035921. [Google Scholar] [CrossRef]
- Jin, L.; Guo, X.; Shen, C.; Hao, X.; Sun, P.; Li, P.; Xu, T.; Hu, C.; Rose, O.; Zhou, H.; et al. Salivary factor LTRIN from Aedes aegypti facilitates the transmission of Zika virus by interfering with the lymphotoxin-β receptor. Nat. Immunol. 2018, 19, 342–353. [Google Scholar] [CrossRef]
- Sudhamsu, J.; Yin, J.; Chiang, E.Y.; Starovasnik, M.A.; Grogan, J.L.; Hymowitz, S.G. Dimerization of LTβR by LTα1β2 is necessary and sufficient for signal transduction. Proc. Natl. Acad. Sci. USA 2013, 110, 19896–19901. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Miyazaki, M. Refolding Techniques for Recovering Biologically Active Recombinant Proteins from Inclusion Bodies. Biomolecules 2014, 4, 235–251. [Google Scholar] [CrossRef]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- Anisuzzaman, C.W. Methods in Molecular Biology Calcium-Binding Proteins and RAGE From Structural Basics to Clinical Applications Longistatin, an EF-Hand Ca2+-Binding Protein from Vector Tick: Identification, Purification, and Characterization. In Calcium-Binding Proteins and RAGE: Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; pp. 127–146. [Google Scholar] [CrossRef]
- Ozohanics, O.; Ambrus, A. Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life 2020, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Zhang, B. Separate roles of LMAN1 and MCFD2 in ER-to-Golgi trafficking of FV and FVIII. Blood Adv. 2023, 7, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Batoulis, H.; Schmidt, T.H.; Weber, P.; Schloetel, J.-G.; Kandt, C.; Lang, T. Concentration Dependent Ion-Protein Interaction Patterns Underlying Protein Oligomerization Behaviours. Sci. Rep. 2016, 6, 24131. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Prajapati, R.; Chatterjee, S.; Mukherjee, T.K. Concentration-Dependent Reversible Self-Oligomerization of Serum Albumins through Intermolecular β-Sheet Formation. Langmuir 2014, 30, 14894–14904. [Google Scholar] [CrossRef]
- Merten, J.A.; Schultz, K.M.; Klug, C.S. Concentration-dependent oligomerization and oligomeric arrangement of LptA. Protein Sci. 2012, 21, 211–218. [Google Scholar] [CrossRef]
- Rotoli, S.M.; Jones, J.L.; Caradonna, S.J. Cysteine residues contribute to the dimerization and enzymatic activity of human nuclear dUTP nucleotidohydrolase (nDut). Protein Sci. 2018, 27, 1797–1809. [Google Scholar] [CrossRef]
- Wiedemann, C.; Kumar, A.; Lang, A.; Ohlenschläger, O. Cysteines and Disulfide Bonds as Structure-Forming Units: Insights From Different Domains of Life and the Potential for Characterization by NMR. Front. Chem. 2020, 8, 280. [Google Scholar] [CrossRef] [PubMed]
- Anisuzzaman; Islam, M.K.; Miyoshi, T.; Alim, M.A.; Hatta, T.; Yamaji, K.; Matsumoto, Y.; Fujisaki, K.; Tsuji, N. Longistatin, a novel EF-hand protein from the ixodid tick Haemaphysalis longicornis, is required for acquisition of host blood-meals. Int. J. Parasitol. 2010, 40, 721–729. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, M.C.; Ingle, B.L.; Barakat, K.A.; Shrestha, B.; Slavens, K.D.; Cundari, T.R.; Anderson, M.E. The role of strong electrostatic interactions at the dimer interface of human glutathione synthetase. Protein J. 2014, 33, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.L.; Walsh, M.P.; Vogel, H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix–loop–helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. [Google Scholar] [CrossRef]
- Lewit-Bentley, A.; Réty, S. EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol. 2000, 10, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, W.; Kirberger, M.; Lee, H.-W.; Ayalasomayajula, G.; Yang, J.J. Prediction of EF-hand calcium-binding proteins and analysis of bacterial EF-hand proteins. Proteins Struct. Funct. Bioinform. 2006, 65, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Day, I.S.; Reddy, V.S.; Shad Ali, G.; Reddy, A.S. Analysis of EF-hand-containing proteins in Arabidopsis. Genome Biol. 2002, 3, research0056. [Google Scholar] [CrossRef]
- Charuk, J.H.; Pirraglia, C.A.; Reithmeier, R.A. Interaction of ruthenium red with Ca2+-binding proteins. Anal. Biochem. 1990, 188, 123–131. [Google Scholar] [CrossRef]
- Grabarek, Z. Insights into modulation of calcium signaling by magnesium in calmodulin, troponin C and related EF-hand proteins. Biochim. Biophys. Acta 2011, 1813, 913–921. [Google Scholar] [CrossRef]
- Mun, S.A.; Park, J.; Kang, J.Y.; Park, T.; Jin, M.; Yang, J.; Eom, S.H. Structural and biochemical insights into Zn(2+)-bound EF-hand proteins, EFhd1 and EFhd2. IUCrJ 2023, 10, 233–245. [Google Scholar] [CrossRef]
- Andrews, S.S.; Tretton, J. Physical Principles of Circular Dichroism. J. Chem. Educ. 2020, 97, 4370–4376. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Ware, C.F. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol. Rev. 2008, 223, 186–201. [Google Scholar] [CrossRef]
- Dutra, G.; Komuczki, D.; Jungbauer, A.; Satzer, P. Continuous capture of recombinant antibodies by ZnCl2 precipitation without polyethylene glycol. Eng. Life Sci. 2020, 20, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Abou-Abbass, H.; Abou-El-Hassan, H.; Bahmad, H.; Zibara, K.; Zebian, A.; Youssef, R.; Ismail, J.; Zhu, R.; Zhou, S.; Dong, X.; et al. Glycosylation and other PTMs alterations in neurodegenerative diseases: Current status and future role in neurotrauma. Electrophoresis 2016, 37, 1549–1561. [Google Scholar] [CrossRef]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct. Target. Ther. 2021, 6, 396. [Google Scholar] [CrossRef]
- Krautter, F.; Iqbal, A.J. Glycans and Glycan-Binding Proteins as Regulators and Potential Targets in Leukocyte Recruitment. Front. Cell. Dev. Biol. 2021, 9, 624082. [Google Scholar] [CrossRef]
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta 2018, 1865, 507–521. [Google Scholar] [CrossRef]
- Zhang, M.; Abrams, C.; Wang, L.; Gizzi, A.; He, L.; Lin, R.; Chen, Y.; Loll, P.J.; Pascal, J.M.; Zhang, J.F. Structural basis for calmodulin as a dynamic calcium sensor. Structure 2012, 20, 911–923. [Google Scholar] [CrossRef]
- Halling, D.B.; Aracena-Parks, P.; Hamilton, S.L. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci. STKE 2005, 2005, re15. [Google Scholar] [CrossRef]
- Zheng, C.; Liu, H.H.; Zhou, J.; Zhang, B. EF-hand domains of MCFD2 mediate interactions with both LMAN1 and coagulation factor V or VIII. Blood 2010, 115, 1081–1087. [Google Scholar] [CrossRef][Green Version]
- Guy, J.E.; Wigren, E.; Svärd, M.; Härd, T.; Lindqvist, Y. New Insights into Multiple Coagulation Factor Deficiency from the Solution Structure of Human MCFD2. J. Mol. Biol. 2008, 381, 941–955. [Google Scholar] [CrossRef] [PubMed]
- Berggård, T.; Miron, S.; Önnerfjord, P.; Thulin, E.; Åkerfeldt, K.S.; Enghild, J.J.; Akke, M.; Linse, S. Calbindin D28k Exhibits Properties Characteristic of a Ca2+ Sensor. J. Biol. Chem. 2002, 277, 16662–16672. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.W.; Almalki, R.; Roe, S.M.; Wagner, A.; Duman, R.; Atack, J.R. The X-ray structure of human calbindin-D28K: An improved model. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, T.M.; Lee, G.M.; Okon, M.; Reid, R.E.; McIntosh, L.P. Calcium-induced folding of a fragment of calmodulin composed of EF-hands 2 and 3. Protein Sci. 2007, 16, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, Y.; Wong, H.C.; Chen, Y.; Chen, Y.; Wang, S.; Castiblanco, A.; Liu, A.; Yang, J.J. A single EF-hand isolated from STIM1 forms dimer in the absence and presence of Ca2+. FEBS J. 2009, 276, 5589–5597. [Google Scholar] [CrossRef] [PubMed]
- Benoit, J.B.; Lopez-Martinez, G.; Patrick, K.R.; Phillips, Z.P.; Krause, T.B.; Denlinger, D.L. Drinking a hot blood meal elicits a protective heat shock response in mosquitoes. Proc. Natl. Acad. Sci. USA 2011, 108, 8026–8029. [Google Scholar] [CrossRef]
- Ma, J.; Martin, J.D.; Zhang, H.; Auger, K.R.; Ho, T.F.; Kirkpatrick, R.B.; Grooms, M.H.; Johanson, K.O.; Tummino, P.J.; Copeland, R.A.; et al. A second p53 binding site in the central domain of Mdm2 is essential for p53 ubiquitination. Biochemistry 2006, 45, 9238–9245. [Google Scholar] [CrossRef]
- Garg, R.; Juncadella, I.J.; Ramamoorthi, N.; Ashish; Ananthanarayanan, S.K.; Thomas, V.; Rincón, M.; Krueger, J.K.; Fikrig, E.; Yengo, C.M.; et al. Cutting edge: CD4 is the receptor for the tick saliva immunosuppressor, Salp15. J. Immunol. 2006, 177, 6579–6583. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, S.N.; Anthony, I.R.; Gavor, E.; Lim, X.S.; Kini, R.M.; Mok, Y.K.; Sivaraman, J. Recognition of Aedes aegypti Mosquito Saliva Protein LTRIN by the Human Receptor LTβR for Controlling the Immune Response. Biology 2024, 13, 42. https://doi.org/10.3390/biology13010042
Loh SN, Anthony IR, Gavor E, Lim XS, Kini RM, Mok YK, Sivaraman J. Recognition of Aedes aegypti Mosquito Saliva Protein LTRIN by the Human Receptor LTβR for Controlling the Immune Response. Biology. 2024; 13(1):42. https://doi.org/10.3390/biology13010042
Chicago/Turabian StyleLoh, Su Ning, Ian Russell Anthony, Edem Gavor, Xin Shan Lim, R. Manjunatha Kini, Yu Keung Mok, and J. Sivaraman. 2024. "Recognition of Aedes aegypti Mosquito Saliva Protein LTRIN by the Human Receptor LTβR for Controlling the Immune Response" Biology 13, no. 1: 42. https://doi.org/10.3390/biology13010042
APA StyleLoh, S. N., Anthony, I. R., Gavor, E., Lim, X. S., Kini, R. M., Mok, Y. K., & Sivaraman, J. (2024). Recognition of Aedes aegypti Mosquito Saliva Protein LTRIN by the Human Receptor LTβR for Controlling the Immune Response. Biology, 13(1), 42. https://doi.org/10.3390/biology13010042