
Integration of Omics Data and Network Models to Unveil Negative Aspects of SARS-CoV-2, from Pathogenic Mechanisms to Drug Repurposing




Abstract
:Simple Summary
Abstract
1. Introduction
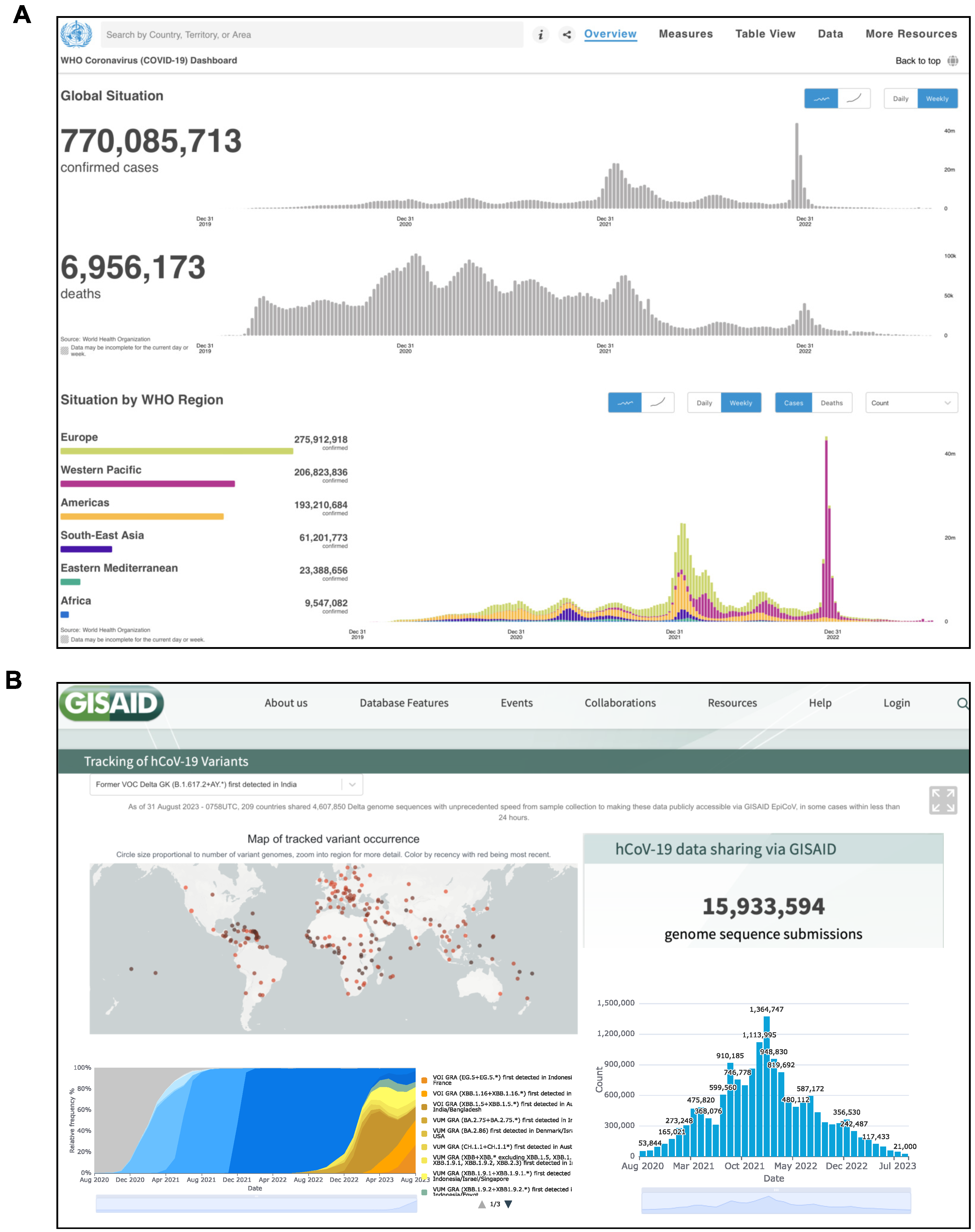
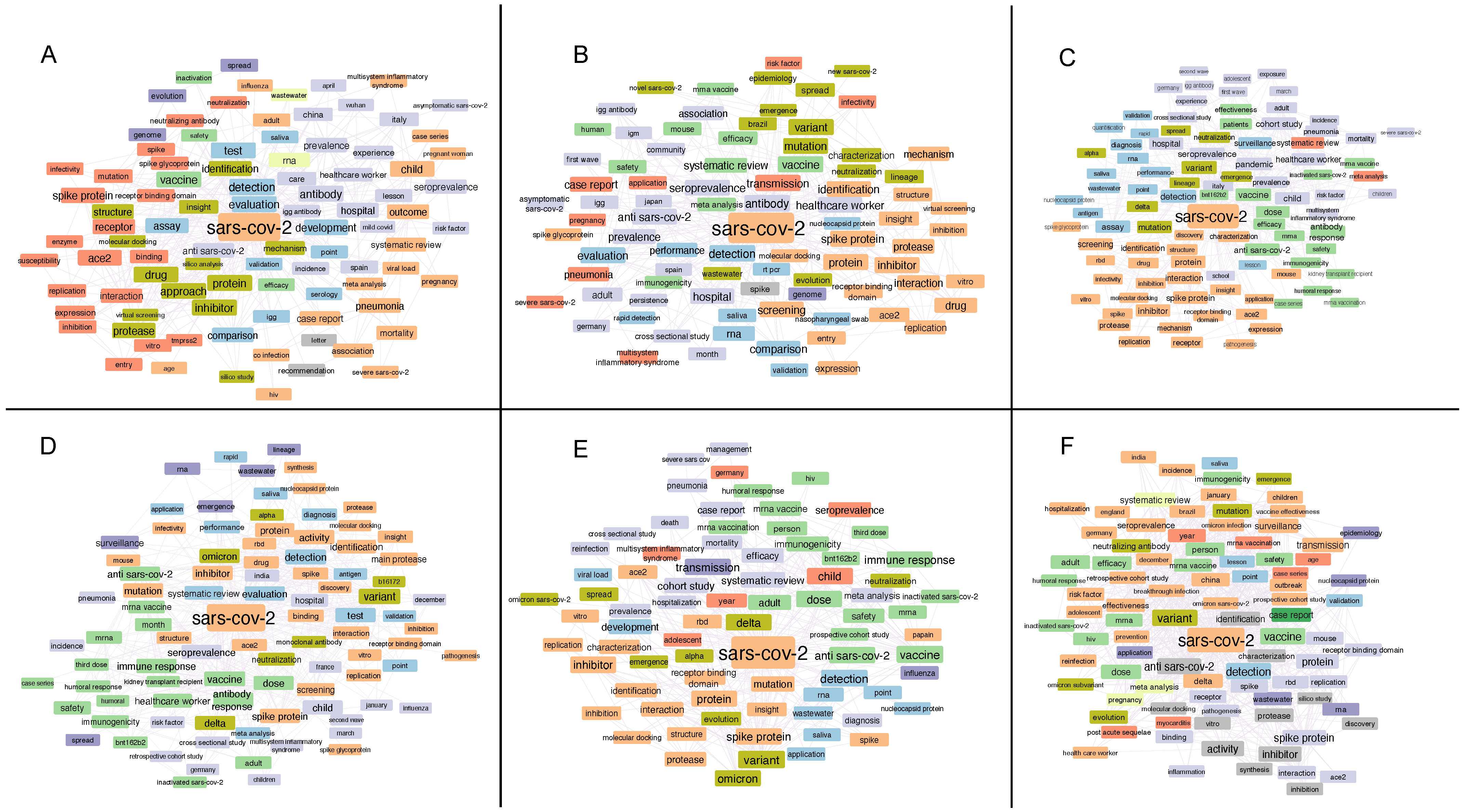
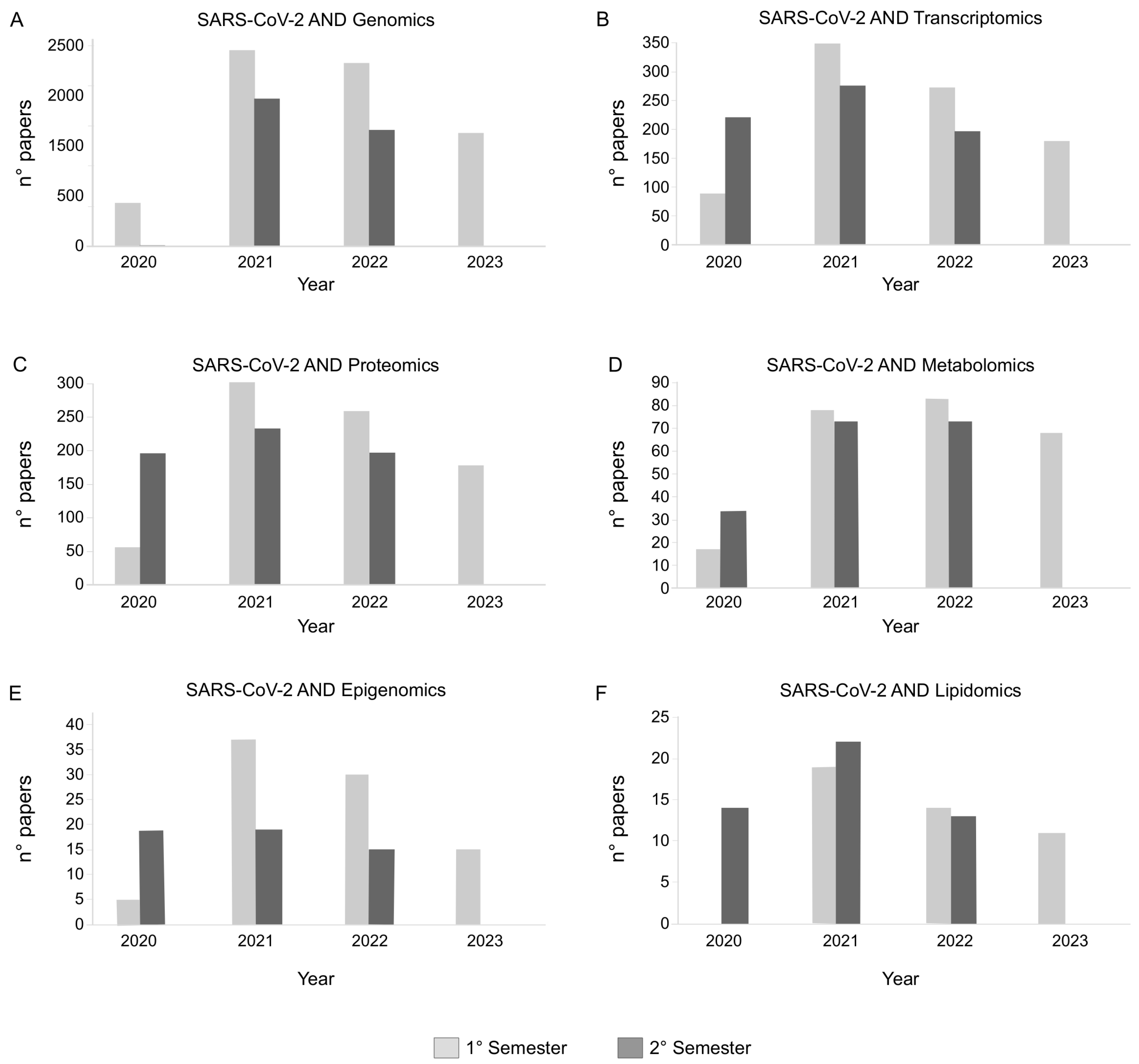
2. Word Associations in Titles of Manuscripts Focused on SARS-CoV-2 Investigation
3. Human Samples and Remodeling of Omics Profiles Following SARS-CoV-2 Infection
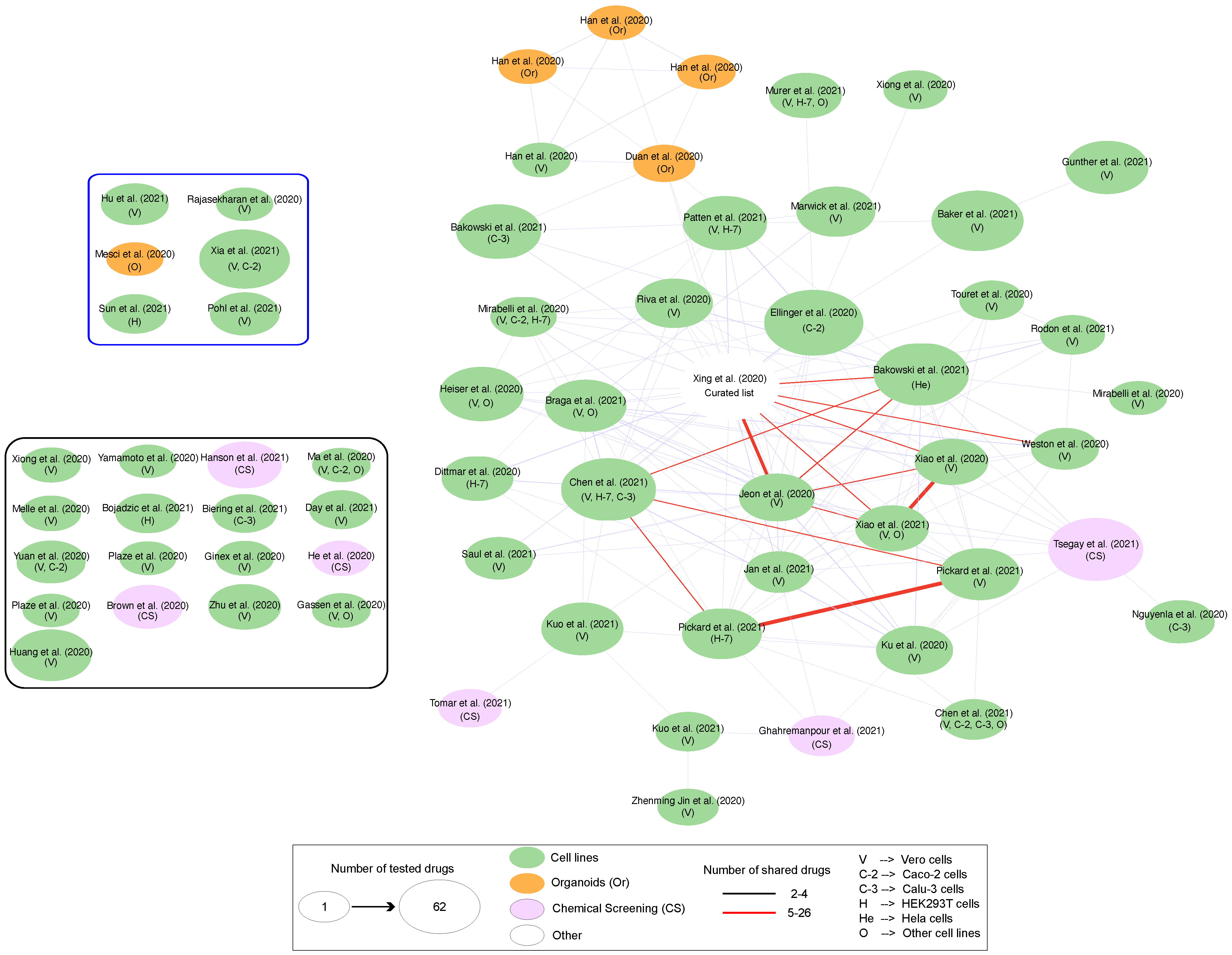
4. Omics Data-Derived Molecular Network Strategies to Explore SARS-CoV-2-Induced Organ/Tissue Damage, Identify Drug Targets and Reposition Existing Drugs
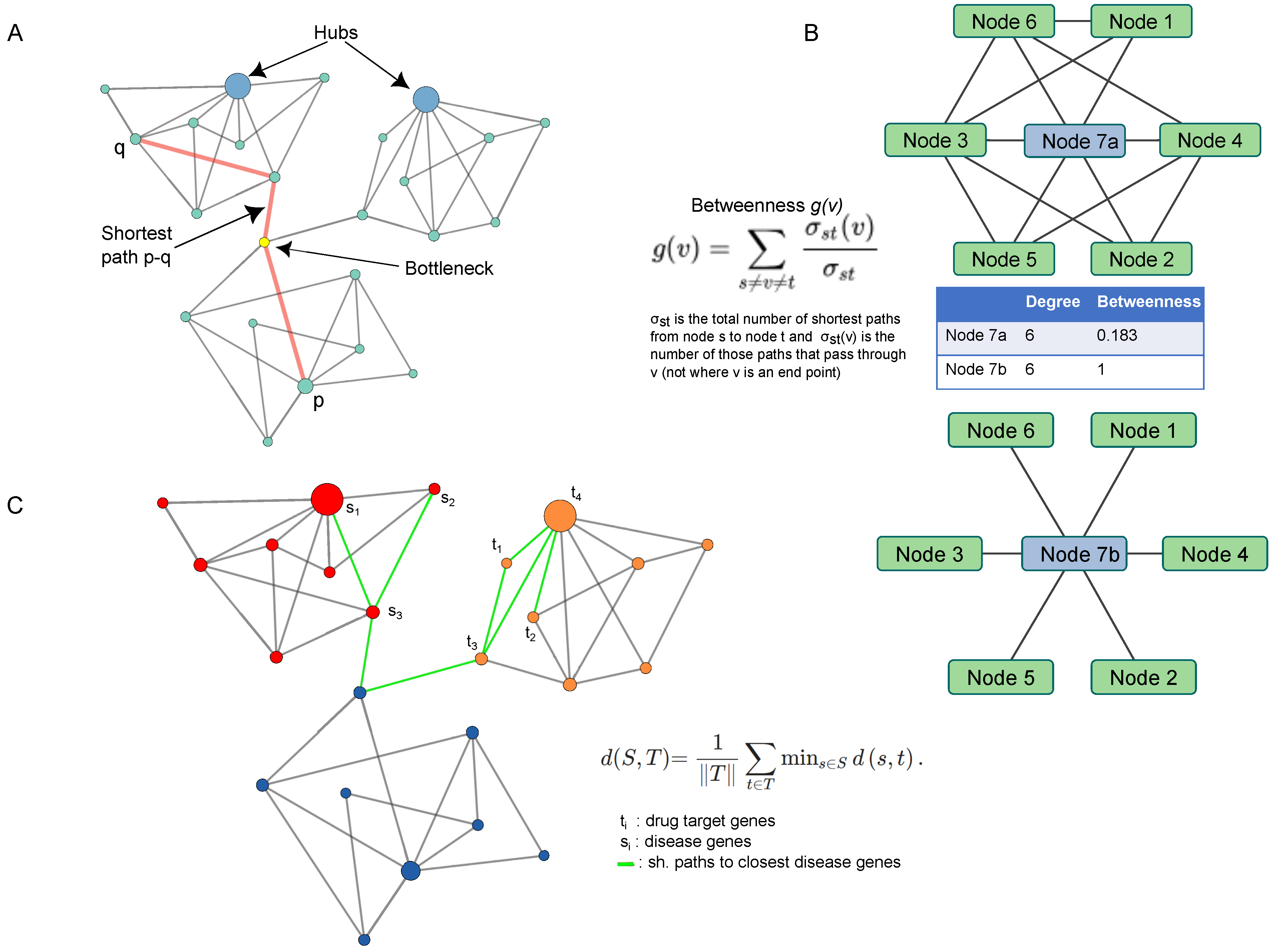
5. Network Topology: From Hubs to Proximity Measure by Way of Shortest Paths
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| COVID-19 | Coronavirus Disease 2019 |
| PPI | Protein–Protein Interaction |
| WHO | World Health Organization |
| MIS-C | Multi-Inflammatory Syndrome |
| EV | Extracellular Vesicles |
| miRNA | microRNA |
| exRNA | Extracellular RNA |
| UMAP | Uniform Manifold Approximation and Projection |
| PRM | Parallel Reaction Monitoring |
| Calu3 | Human Lung Adenocarcinoma Cell Line |
| HL-mECs | Lung Microvascular Endothelial Cells |
| MB610 and MB61222 | Master Mutant and Minor Quasi-Species |
| Caco2 | Human Colorectal Adenocarcinoma Cells |
| MODS | Multiple Organ Dysfunction Syndrome |
| DEGs | Differentially Expressed Genes |
| MD | Molecular Dynamics |
| TM | Traditional Medicine |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| HIV | Human Immunodeficiency Virus |
| FDA | Food and Drug Administration |
| CV | Cardiovascular Disease |
| AUC | Area Under the Curve |
| VeroE6 | Monkey African Green Kidney Cell Line |
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Comish, P.; Kang, R. The hallmarks of COVID-19 disease. PLoS Pathog. 2020, 16, e1008536. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Du, M.; Cai, G.; Chen, F.; Christiani, D.C.; Zhang, Z.; Wang, M. Multiomics Evaluation of Gastrointestinal and Other Clinical Characteristics of COVID-19. Gastroenterology 2020, 158, 2298–2301.e7. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Q.; Ma, L.; Wu, D.; Gao, J.; Chen, G.; Li, H. Systematic profiling of ACE2 expression in diverse physiological and pathological conditions for COVID-19/SARS-CoV-2. J. Cell. Mol. Med. 2020, 24, 9478–9482. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Koch, J.; Uckeley, Z.M.; Doldan, P.; Stanifer, M.; Boulant, S.; Lozach, P.Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821. [Google Scholar] [CrossRef]
- Pizzato, M.; Baraldi, C.; Boscato Sopetto, G.; Finozzi, D.; Gentile, C.; Gentile, M.D.; Marconi, R.; Paladino, D.; Raoss, A.; Riedmiller, I.; et al. SARS-CoV-2 and the Host Cell: A Tale of Interactions. Front. Virol. 2022, 1, 815388. [Google Scholar] [CrossRef]
- Nie, X.; Qian, L.; Sun, R.; Huang, B.; Dong, X.; Xiao, Q.; Zhang, Q.; Lu, T.; Yue, L.; Chen, S.; et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell 2021, 184, 775–791.e14. [Google Scholar] [CrossRef]
- Wadman, M.; Couzin-Frankel, J.; Kaiser, J.; Matacic, C. A rampage through the body. Science 2020, 368, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Rao, R.S.P.; Wilson, R.S.; Punyamurtula, U.; Salvato, F.; Petersen, M.; Ahmed, M.K.; Abid, M.R.; Verburgt, J.C.; Kihara, D.; et al. Mass spectrometry-based proteomic platforms for better understanding of SARS-CoV-2 induced pathogenesis and potential diagnostic approaches. Proteomics 2021, 21, e2000279. [Google Scholar] [CrossRef] [PubMed]
- Cowley, L.A.; Afrad, M.H.; Rahman, S.I.A.; Mamun, M.M.A.; Chin, T.; Mahmud, A.; Rahman, M.Z.; Billah, M.M.; Khan, M.H.; Sultana, S.; et al. Genomics, social media and mobile phone data enable mapping of SARS-CoV-2 lineages to inform health policy in Bangladesh. Nat. Microbiol. 2021, 6, 1271–1278. [Google Scholar] [CrossRef]
- Li, G.; Hilgenfeld, R.; Whitley, R.; Clercq, E.D. Therapeutic strategies for COVID-19: Progress and lessons learned. Nat. Rev. 2023, 22, 449–475. [Google Scholar] [CrossRef]
- Wang, X.; Xu, G.; Liu, X.; Liu, Y.; Zhang, S.; Zhang, Z. Multiomics: Unraveling the panoramic landscapes of SARS-CoV-2 infection. Cell. Mol. Immunol. 2021, 18, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Agamah, F.E.; Agamah, F.E.; Bayjanov, J.R.; Niehues, A.; Njoku, K.F.; Skelton, M.; Mazandu, G.K.; Mazandu, G.K.; Mazandu, G.K.; Ederveen, T.H.A.; et al. Computational approaches for network-based integrative multi-omics analysis. Front. Mol. Biosci. 2022, 9, 1214. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Cantwell, A.M.; Singh, H.; Platt, M.; Yu, Y.; Lin, Y.H.; Ikeno, Y.; Hubbard, G.; Xiang, Y.; Gonzalez-Juarbe, N.; Dube, P.H. Kinetic Multi-omic Analysis of Responses to SARS-CoV-2 Infection in a Model of Severe COVID-19. J. Virol. 2021, 95, E01010-21. [Google Scholar] [CrossRef] [PubMed]
- Morselli Gysi, D.; do Valle, Í.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Ghiassian, S.D.; Patten, J.J.; Davey, R.A.; Loscalzo, J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2025581118. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Mehra, R.; Kallianpur, A.; Culver, D.A.; Gack, M.U.; Farha, S.; Zein, J.; Comhair, S.; et al. A network medicine approach to investigation and population-based validation of disease manifestations and drug repurposing for COVID-19. PLoS Biol. 2020, 18, e3000970. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhu, J.; Liu, D.; Sun, Y.; Wu, C. An integrative multiomics analysis identifies putative causal genes for COVID-19 severity. Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 2076–2086. [Google Scholar] [CrossRef]
- Selvaraj, G.; Kaliamurthi, S.; Peslherbe, G.H.; Wei, D.Q. Identifying potential drug targets and candidate drugs for COVID-19: Biological networks and structural modeling approaches. F1000Research 2021, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Han, T.; Lao, Z.; Li, G.; Xiao, J.; Liu, X. Phillyrin for COVID-19 and Influenza Co-infection: A Potential Therapeutic Strategy Targeting Host Based on Bioinformatics Analysis. Front. Pharmacol. 2021, 12, 754241. [Google Scholar] [CrossRef] [PubMed]
- El-Aarag, S.A.; Mahmoud, A.; ElHefnawi, M. Identifying potential novel insights for COVID-19 pathogenesis and therapeutics using an integrated bioinformatics analysis of host transcriptome. Int. J. Biol. Macromol. 2022, 194, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, T.; Liu, S.; Liu, J.; Fang, S.; Tan, S.; Zeng, Y.; Zhang, B.; Li, W. Dissecting the molecular mechanism of cepharanthine against COVID-19, based on a network pharmacology strategy combined with RNA-sequencing analysis, molecular docking, and molecular dynamics simulation. Comput. Biol. Med. 2022, 151, 106298. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, S.; Wang, P.; Chen, X.; Bi, J.; Cheng, L.; Zhang, X. A comprehensive review of the analysis and integration of omics data for SARS-CoV-2 and COVID-19. Brief. Bioinform. 2022, 23, bbab446. [Google Scholar] [CrossRef]
- Vella, D.; Zoppis, I.; Mauri, G.; Mauri, P.; Di Silvestre, D. From protein-protein interactions to protein co-expression networks: A new perspective to evaluate large-scale proteomic data. EURASIP J. Bioinform. Syst. Biol. 2017, 2017, 6. [Google Scholar] [CrossRef]
- Suvarna, K.; Salkar, A.; Palanivel, V.; Bankar, R.; Banerjee, N.; Gayathri J Pai, M.; Srivastava, A.; Singh, A.; Khatri, H.; Agrawal, S.; et al. A Multi-omics Longitudinal Study Reveals Alteration of the Leukocyte Activation Pathway in COVID-19 Patients. J. Proteome Res. 2021, 20, 4667–4680. [Google Scholar] [CrossRef]
- Druzak, S.; Iffrig, E.; Roberts, B.R.; Zhang, T.; Fibben, K.S.; Sakurai, Y.; Verkerke, H.P.; Rostad, C.A.; Chahroudi, A.; Schneider, F.; et al. Multiplatform analyses reveal distinct drivers of systemic pathogenesis in adult versus pediatric severe acute COVID-19. Nat. Commun. 2023, 14, 1638. [Google Scholar] [CrossRef]
- Bi, X.; Liu, W.; Ding, X.; Liang, S.; Zheng, Y.; Zhu, X.; Quan, S.; Yi, X.; Xiang, N.; Du, J.; et al. Proteomic and metabolomic profiling of urine uncovers immune responses in patients with COVID-19. Cell Rep. 2022, 38, 110271. [Google Scholar] [CrossRef]
- Lam, S.M.; Zhang, C.; Wang, Z.; Ni, Z.; Zhang, S.; Yang, S.; Huang, X.; Mo, L.; Li, J.; Lee, B.; et al. A multi-omics investigation of the composition and function of extracellular vesicles along the temporal trajectory of COVID-19. Nat. Metab. 2021, 3, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, B.; Barratt, I.; Townsend, R.; Kalafat, E.; van der Meulen, J.; Gurol-Urganci, I.; O’Brien, P.; Morris, E.; Draycott, T.; Thangaratinam, S.; et al. Effects of the COVID-19 pandemic on maternal and perinatal outcomes: A systematic review and meta-analysis. Lancet Glob. Health 2021, 9, e759–e772. [Google Scholar] [CrossRef]
- Martin, M.A.; Martin, M.A.; Keith, M.; Keith, M.; Pace, R.M.; Williams, J.E.; Ley, S.H.; Barbosa-Leiker, C.; Caffé, B.; Smith, C.B.; et al. SARS-CoV-2 specific antibody trajectories in mothers and infants over two months following maternal infection. Front. Immunol. 2022, 13, 1015002. [Google Scholar] [CrossRef] [PubMed]
- Ezechukwu, H.C.; Ezechukwu, H.C.; Shi, J.; Shi, J.; Fowora, M.A.; Fowora, M.A.; Diya, C.A.; Elfaki, F.; Adegboye, O.A.; Adegboye, O.A. Fetoplacental transmission and placental response to SARS-CoV-2: Evidence from the literature. Front. Med. 2022, 9, 962937. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.; Zhang, Y.; Ai, J.; Yang, B.; Cui, M.; Liao, Q.; Chen, H.; Bai, H.; Shang, D.; et al. Differential immune responses in pregnant patients recovered from COVID-19. Signal Transduct. Target. Ther. 2021, 6, 289. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Tartaro, D.L.; Gibellini, L.; Paolini, A.; Quong, A.; Petes, C.; Awong, G.; Douglas, S.; Lin, D.; Nieto, J.; et al. Endogenous control of inflammation characterizes pregnant women with asymptomatic or paucisymptomatic SARS-CoV-2 infection. Nat. Commun. 2021, 12, 4677. [Google Scholar] [CrossRef]
- Gomez-Lopez, N.; Romero, R.; Escobar, M.F.; Carvajal, J.A.; Echavarria, M.P.; Albornoz, L.L.; Nasner, D.; Miller, D.; Gallo, D.M.; Galaz, J.; et al. Pregnancy-specific responses to COVID-19 are revealed by high-throughput proteomics of human plasma. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Chen, Y.M.; Zheng, Y.; Yu, Y.; Wang, Y.; Huang, Q.; Qian, F.; Sun, L.; Song, Z.G.; Chen, Z.; Feng, J.; et al. Blood molecular markers associated with COVID-19 immunopathology and multi-organ damage. EMBO J. 2020, 39, e105896. [Google Scholar] [CrossRef]
- Xu, G.; Wu, Y.; Xiao, T.; Qi, F.; Fan, L.; Zhang, S.; Zhou, J.; He, Y.; Gao, X.; Zeng, H.; et al. Multiomics approach reveals the ubiquitination-specific processes hijacked by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 312. [Google Scholar] [CrossRef]
- Caccuri, F.; Bugatti, A.; Zani, A.; De Palma, A.; Di Silvestre, D.; Manocha, E.; Filippini, F.; Messali, S.; Chiodelli, P.; Campisi, G.; et al. SARS-CoV-2 Infection Remodels the Phenotype and Promotes Angiogenesis of Primary Human Lung Endothelial Cells. Microorganisms 2021, 9, 1438. [Google Scholar] [CrossRef] [PubMed]
- Caccuri, F.; Messali, S.; Bortolotti, D.; Silvestre, D.D.; Palma, A.D.; Cattaneo, C.; Bertelli, A.; Zani, A.; Milanesi, M.; Giovanetti, M.; et al. Competition for dominance within replicating quasispecies during prolonged SARS-CoV-2 infection in an immunocompromised host. Virus Evol. 2022, 8, veac042. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.M.; Subbannayya, Y.; Kim, H.; Hagen, L.; Górna, M.W.; Nieminen, A.I.; Bjørås, M.; Espevik, T.; Kainov, D.; Kandasamy, R.K. Multi-OMICs landscape of SARS-CoV-2-induced host responses in human lung epithelial cells. iScience 2023, 26, 105895. [Google Scholar] [CrossRef]
- Zhao, S.; Zhu, W.; Xue, S.; Han, D. Testicular defense systems: Immune privilege and innate immunity. Cell. Mol. Immunol. 2014, 11, 428–437. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Pathak, E.; Atri, N.; Mishra, R. Single-Cell Transcriptome Analysis Reveals the Role of Pancreatic Secretome in COVID-19 Associated Multi-organ Dysfunctions. Interdiscip. Sci. Comput. Life Sci. 2022, 14, 863–878. [Google Scholar] [CrossRef]
- Lim, S.; Zhang, M.; Chang, T.L. ACE2-Independent Alternative Receptors for SARS-CoV-2. Viruses 2022, 14, 2535. [Google Scholar] [CrossRef]
- Rahimi, N. C-type Lectin CD209L/L-SIGN and CD209/DC-SIGN: Cell Adhesion Molecules Turned to Pathogen Recognition Receptors. Biology 2020, 10, 1. [Google Scholar] [CrossRef]
- Vial, C.; Calderón, J.F.; Klein, A.D. NPC1 as a Modulator of Disease Severity and Viral Entry of SARSCoV-2. Curr. Mol. Med. 2021, 21, 2–4. [Google Scholar] [CrossRef]
- Potts, M.; Fletcher-Etherington, A.; Nightingale, K.; Mescia, F.; Bergamaschi, L.; Calero-Nieto, F.J.; Antrobus, R.; Williamson, J.; Cambridge Institute of Therapeutic Immunology and Infectious Disease-National Institute of Health Research (CITIID-NIHR) COVID BioResource Collaboration; Parsons, H.; et al. Proteomic analysis of circulating immune cells identifies cellular phenotypes associated with COVID-19 severity. Cell Rep. 2023, 42, 112613. [Google Scholar] [CrossRef]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cusick, M.E.; Barabási, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, Y.; Gupta, S.; Paramo, M.I.; Hou, Y.; Mao, C.; Luo, Y.; Judd, J.; Wierbowski, S.; Bertolotti, M.; et al. A comprehensive SARS-CoV-2-human protein-protein interactome reveals COVID-19 pathobiology and potential host therapeutic targets. Nat. Biotechnol. 2023, 41, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Guney, E.; Menche, J.; Vidal, M.; Barábasi, A.L. Network-based in silico drug efficacy screening. Nat. Commun. 2016, 7, 10331. [Google Scholar] [CrossRef]
- Das, J.K.; Chakraborty, S.; Roy, S. A scheme for inferring viral-host associations based on codon usage patterns identifies the most affected signaling pathways during COVID-19. J. Biomed. Inform. 2021, 118, 103801. [Google Scholar] [CrossRef]
- Schmidt, N.; Lareau, C.A.; Keshishian, H.; Ganskih, S.; Schneider, C.; Hennig, T.; Melanson, R.; Werner, S.; Wei, Y.; Zimmer, M.; et al. The SARS-CoV-2 RNA-protein interactome in infected human cells. Nat. Microbiol. 2021, 6, 339–353. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, Y.; Wang, S.; Yang, X.; Zhou, K.; Xu, C.; Zhang, X.; Fan, J.; Hou, D.; Li, X.; et al. NPASS database update 2023: Quantitative natural product activity and species source database for biomedical research. Nucleic Acids Res. 2023, 51, D621–D628. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Ru, J.; Li, P.; Wang, J.; Zhou, W.; Li, B.; Huang, C.; Li, P.; Guo, Z.; Tao, W.; Yang, Y.; et al. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminform. 2014, 6, 13. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef]
- Xu, H.Y.; Zhang, Y.Q.; Liu, Z.M.; Chen, T.; Lv, C.Y.; Tang, S.H.; Zhang, X.B.; Zhang, W.; Li, Z.Y.; Zhou, R.R.; et al. ETCM: An encyclopaedia of traditional Chinese medicine. Nucleic Acids Res. 2019, 47, D976–D982. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Szklarczyk, D.; Franceschini, A.; Campillos, M.; von Mering, C.; Jensen, L.J.; Beyer, A.; Bork, P. STITCH 2: An interaction network database for small molecules and proteins. Nucleic Acids Res. 2010, 38, D552–D556. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Chaudhuri, A.G.; Tiwary, B.K.; Chakrabarti, N. Matrix metallopeptidase 9 as a host protein target of chloroquine and melatonin for immunoregulation in COVID-19: A network-based meta-analysis. Meta-Analysis 2020, 257, 118096. [Google Scholar] [CrossRef] [PubMed]
- Vastrad, B.; Vastrad, C.; Tengli, A. Identification of potential mRNA panels for severe acute respiratory syndrome coronavirus 2 (COVID-19) diagnosis and treatment using microarray dataset and bioinformatics methods. 3 Biotech 2020, 10, 422. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Tang, T.; Hu, M. Identification of Differentially Expressed Genes in COVID-19 and Integrated Bioinformatics Analysis of Signaling Pathways. Genet. Res. 2021, 2021, 2728757. [Google Scholar] [CrossRef]
- Ceylan, H. A bioinformatics approach for identifying potential molecular mechanisms and key genes involved in COVID-19 associated cardiac remodeling. Gene Rep. 2021, 24, 101246. [Google Scholar] [CrossRef]
- Li, C.X.; Chen, J.; Lv, S.K.; Li, J.H.; Li, L.L.; Hu, X. Whole-Transcriptome RNA Sequencing Reveals Significant Differentially Expressed mRNAs, miRNAs, and lncRNAs and Related Regulating Biological Pathways in the Peripheral Blood of COVID-19 Patients. Mediat. Inflamm. 2021, 2021, 6635925. [Google Scholar] [CrossRef]
- Jiang, S.T.; Liu, Y.G.; Zhang, L.; Sang, X.T.; Xu, Y.Y.; Lu, X. Systems biology approach reveals a common molecular basis for COVID-19 and non-alcoholic fatty liver disease (NAFLD). Eur. J. Med. Res. 2022, 27, 251. [Google Scholar] [CrossRef]
- Fang, K.; Liang, G.; Zhuang, Z.; Fang, Y.; Dong, Y.; Liang, C.; Chen, X.; Guo, X. Screening the hub genes and analyzing the mechanisms in discharged COVID-19 patients retesting positive through bioinformatics analysis. J. Clin. Lab. Anal. 2022, 36, e24495. [Google Scholar] [CrossRef]
- Sinha, S.; Castillo, V.; Espinoza, C.R.; Tindle, C.; Fonseca, A.G.; Dan, J.M.; Katkar, G.D.; Das, S.; Sahoo, D.; Ghosh, P. COVID-19 lung disease shares driver AT2 cytopathic features with Idiopathic pulmonary fibrosis. EBioMedicine 2022, 82, 104185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Q.; Zhang, J.; Xie, H. Exploration of the Potential Link, Hub Genes, and Potential Drugs for Coronavirus Disease 2019 and Lung Cancer Based on Bioinformatics Analysis. J. Oncol. 2022, 2022, 8124673. [Google Scholar] [CrossRef] [PubMed]
- Osuna-Martinez, U.; Aviña-Padilla, K.; Olimon-Andalon, V.; Angulo-Rojo, C.; Guadron-Llanos, A.; Rivas-Ferreira, J.C.; Urrea, F.; Calderon-Zamora, L. In Silico Prediction of Hub Genes Involved in Diabetic Kidney and COVID-19 Related Disease by Differential Gene Expression and Interactome Analysis. Genes 2022, 13, 2412. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Xiao, Y.; Luo, Y. Identifying the Effect of COVID-19 Infection in Multiple Myeloma and Diffuse Large B-Cell Lymphoma Patients Using Bioinformatics and System Biology. Comput. Math. Methods Med. 2022, 2022, 7017317. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Li, X.; Gao, S.; Zhang, Y.; Chen, H.; Zhai, X. Identifying Potential Gene Defect Patterns Related to COVID-19 Based on Pharmacological and Bioinformatics Analysis for Lung Adenocarcinoma. Int. J. Gen. Med. 2022, 15, 4285–4301. [Google Scholar] [CrossRef]
- Zamanian Azodi, M.; Arjmand, B.; Zali, A.; Razzaghi, M. Introducing APOA1 as a key protein in COVID-19 infection: A bioinformatics approach. Gastroenterol. Hepatol. Bed Bench 2020, 13, 367–373. [Google Scholar]
- Zhang, M.; Zhang, X.; Pei, J.; Guo, B.; Zhang, G.; Li, M.; Huang, L. Identification of phytochemical compounds of Fagopyrum dibotrys and their targets by metabolomics, network pharmacology and molecular docking studies. Heliyon 2023, 9, e14029. [Google Scholar] [CrossRef]
- Datta, S.; Sarkar, I.; Sen, G.; Sen, A. Neem and Turmeric in the management of Covid Associated Mucormycosis (CAM) derived through network pharmacology. J. Biomol. Struct. Dyn. 2023, 41, 3281–3294. [Google Scholar] [CrossRef]
- Xu, F.; Gao, J.; Munkhsaikhan, U.; Li, N.; Gu, Q.; Pierre, J.F.; Starlard-Davenport, A.; Towbin, J.A.; Cui, Y.; Purevjav, E.; et al. The Genetic Dissection of Ace2 Expression Variation in the Heart of Murine Genetic Reference Population. Front. Cardiovasc. Med. 2020, 7, 582949. [Google Scholar] [CrossRef]
- Zhou, Y.; Chu, Y.; Shi, J.; Hu, Y. Analysis of the Molecular Mechanism of Huangqi Herb Treating COVID- 19 with Myocardial Injury by Pharmacological Tools, Programming Software and Molecular Docking. Comb. Chem. High Throughput Screen. 2023, 26, 1015–1029. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, Y.Z.; Xie, Y.Z.; Peng, C.W.; Peng, C.W.; Yao, K.N.; Lin, X.Y.; Lin, X.Y.; Zhan, S.F.; Zhuang, H.F.; et al. Modeling Kaempferol as a Potential Pharmacological Agent for COVID-19/PF Co-Occurrence Based on Bioinformatics and System Pharmacological Tools. Front. Pharmacol. 2022, 13, 865097. [Google Scholar] [CrossRef]
- Li, X.; Liang, W.; Yu, C.; Meng, Q.; Zhang, W.; Wu, X.; Xue, J.; Deng, S.; Wang, H. Potential therapeutic strategies for quercetin targeting critical pathological mechanisms associated with colon adenocarcinoma and COVID-19. Front. Pharmacol. 2022, 13, 988153. [Google Scholar] [CrossRef]
- Alzahrani, F.A.; Khan, M.F.; Ahmad, V. Recognition of Differentially Expressed Molecular Signatures and Pathways Associated with COVID-19 Poor Prognosis in Glioblastoma Patients. Int. J. Mol. Sci. 2023, 24, 3562. [Google Scholar] [CrossRef]
- Hossain, M.A.; Rahman, M.H.; Sultana, H.; Ahsan, A.; Rayhan, S.I.; Hasan, M.I.; Sohel, M.; Somadder, P.D.; Moni, M.A. An integrated in-silico Pharmaco-BioInformatics approaches to identify synergistic effects of COVID-19 to HIV patients. Comput. Biol. Med. 2023, 155, 106656. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, J.; Yu, Z.; Gu, X.; Lu, Y.; Ruan, Y.; Wang, T. Exploration of Fuzheng Yugan Mixture on COVID-19 based on network pharmacology and molecular docking. Medicine 2023, 102, e32693. [Google Scholar] [CrossRef]
- Xia, S.; Zhong, Z.; Gao, B.; Vong, C.T.; Lin, X.; Cai, J.; Gao, H.; Chan, G.; Li, C. The important herbal pair for the treatment of COVID-19 and its possible mechanisms. Chin. Med. 2021, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, W.; Kui, F.; Gao, F.; Niu, Y.; Li, W.; Zhang, Y.; Guo, Z.; Du, G. The mechanism and active compounds of semen armeniacae amarum treating coronavirus disease 2019 based on network pharmacology and molecular docking. Food Nutr. Res. 2021, 65, 5623. [Google Scholar] [CrossRef]
- Gao, K.; Song, Y.; Song, A. Exploring active ingredients and function mechanisms of Ephedra-bitter almond for prevention and treatment of Corona virus disease 2019 (COVID-19) based on network pharmacology. BioData Min. 2020, 13, 19. [Google Scholar] [CrossRef]
- Han, L.; Wei, X.X.; Zheng, Y.J.; Zhang, L.L.; Wang, X.M.; Yang, H.Y.; Ma, X.; Zhao, L.H.; Tong, X.L. Potential mechanism prediction of Cold-Damp Plague Formula against COVID-19 via network pharmacology analysis and molecular docking. Chin. Med. 2020, 15, 78. [Google Scholar] [CrossRef]
- Hu, H.; Wang, K.; Wang, L.; Du, Y.; Chen, J.; Li, Y.; Fan, C.; Li, N.; Sun, Y.; Tu, S.; et al. He-Jie-Shen-Shi Decoction as an Adjuvant Therapy on Severe Coronavirus Disease 2019: A Retrospective Cohort and Potential Mechanistic Study. Front. Pharmacol. 2021, 12, 700498. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.K.; Adnan, M. Revealing Potential Bioactive Compounds and Mechanisms of Lithospermum erythrorhizon against COVID-19 via Network Pharmacology Study. Curr. Issues Mol. Biol. 2022, 44, 1788–1809. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Scardoni, G.; Tosadori, G.; Faizan, M.; Spoto, F.; Fabbri, F.; Laudanna, C. Biological network analysis with CentiScaPe: Centralities and experimental dataset integration. F1000Research 2014, 3, 139. [Google Scholar] [CrossRef]
- Martínez, V.; Navarro, C.; Cano, C.; Fajardo, W.; Blanco, A. DrugNet: Network-based drug-disease prioritization by integrating heterogeneous data. Artif. Intell. Med. 2015, 63, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Lu, W.; Liu, C.; Fang, J.; Hou, Y.; Handy, D.E.; Wang, R.; Zhao, Y.; Yang, Y.; Huang, J.; et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. 2019, 10, 3476. [Google Scholar] [CrossRef]
- Fiscon, G.; Conte, F.; Farina, L.; Paci, P. SAveRUNNER: A network-based algorithm for drug repurposing and its application to COVID-19. PLoS Comput. Biol. 2021, 17, e1008686. [Google Scholar] [CrossRef]
- Gates, L.E.; Hamed, A.A. The Anatomy of the SARS-CoV-2 Biomedical Literature: Introducing the CovidX Network Algorithm for Drug Repurposing Recommendation. J. Med. Internet Res. 2020, 22, e21169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Peng, S.; Wei, D.Q.; Zhong, W.; Dou, Y.; Xie, X. LUNAR: Drug Screening for Novel Coronavirus Based on Representation Learning Graph Convolutional Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Ghiassian, S.D.; Menche, J.; Barabási, A.L. A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput. Biol. 2015, 11, e1004120. [Google Scholar] [CrossRef]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.L. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Desai, R.J.; Handy, D.E.; Wang, R.; Schneeweiss, S.; Barabási, A.L.; Loscalzo, J. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun. 2018, 9, 2691. [Google Scholar] [CrossRef]
- Zitnik, M.; Sosič, R.; Leskovec, J. Prioritizing network communities. Nat. Commun. 2018, 9, 2544. [Google Scholar] [CrossRef] [PubMed]
- Trojak, R.M.; Lenger, M.; Birner, A.; Maget, A.; Dalkner, N.; Lang, J.N.; Fellendorf, F.T.; Ratzenhofer, M.; Schönthaler, E.M.D.; Fleischmann, E.; et al. Impact of the COVID-19 Pandemic on Productivity of Workers in the Health Sector between Working in a Hospital and from Home. J. Clin. Med. 2023, 12, 5129. [Google Scholar] [CrossRef]
- Diray-Arce, J.; Fourati, S.; Doni Jayavelu, N.; Patel, R.; Maguire, C.; Chang, A.C.; Dandekar, R.; Qi, J.; Lee, B.H.; van Zalm, P.; et al. Multi-omic longitudinal study reveals immune correlates of clinical course among hospitalized COVID-19 patients. Cell Rep. Med. 2023, 4, 101079. [Google Scholar] [CrossRef]
- Golob, J.L.; Lugogo, N.; Lauring, A.S.; Lok, A.S. SARS-CoV-2 vaccines: A triumph of science and collaboration. J. Clin. Investig. Insight 2021, 6, e149187. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sue, A.C.H.; Goh, W.W.B. Feature selection in clinical proteomics: With great power comes great reproducibility. Drug Discov. Today 2017, 22, 912–918. [Google Scholar] [CrossRef]
- Montanez, G.; Cho, Y.R. Predicting False Positives of Protein-Protein Interaction Data by Semantic Similarity Measures §. Curr. Bioinform. 2013, 8, 339–346. [Google Scholar] [CrossRef]
- Vajda, S.; Emili, A. Mapping global protein contacts. Science 2019, 365, 120–121. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Stein, D.J.; Clarke, D.J.; Kropiwnicki, E.; Jagodnik, K.M.; Bartal, A.; Evangelista, J.E.; Hom, J.; Cheng, M.; Bailey, A.; et al. The COVID-19 Drug and Gene Set Library. Patterns 2020, 1, 100090. [Google Scholar] [CrossRef]
- Pickard, A.; Calverley, B.C.; Chang, J.; Garva, R.; Gago, S.; Lu, Y.; Kadler, K.E. Discovery of re-purposed drugs that slow SARS-CoV-2 replication in human cells. PLoS Pathog. 2021, 17, e1009840. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, C.; Chang, D.; Wang, Y.; Dong, X.; Jiao, T.; Zhao, Z.; Ren, L.; Dela Cruz, C.S.; Sharma, L.; et al. Identification of Potent and Safe Antiviral Therapeutic Candidates Against SARS-CoV-2. Front. Immunol. 2020, 11, 586572. [Google Scholar] [CrossRef] [PubMed]
- Kale-Pradhan, P.B.; Pacitto, R.; Giuliano, C.A.; Johnson, L.B. Evaluation of High-dose versus Standard-dose of Dexamethasone on Mortality among the Mechanically Ventilated COVID-19 Patients. Curr. Drug Saf. 2023, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, T.; Guihur, A.; Rebeaud, M.E.; Mulot, M.; Peiffer-Smadja, N.; Mahamat-Saleh, Y. Effect of hydroxychloroquine with or without azithromycin on the mortality of coronavirus disease 2019 (COVID-19) patients: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2021, 27, 19–27. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database/Link | Description | License | Ref. |
|---|---|---|---|
| DGIdb dgidb.org (accessed on 1 August 2023) | Web resource that provides information on drug–gene interactions and druggable genes from publications, databases and other web-based sources | OSP | [59] |
| TTD db.idrblab.net/ttd/ (accessed on 1 August 2023) | Database that provides information about the known and explored therapeutic protein and nucleic acid targets, the targeted disease, pathway information and the corresponding drug target | Free | [60] |
| NPASS bidd.group/NPASS/index.php (accessed on 1 August 2023) | It integrates species sources of natural products and connects natural products to biological targets via experimentally derived quantitative activity data | Free | [61] |
| SwissADME swissadme.ch (accessed on 1 August 2023) | It allows predictive models for physicochemical properties, pharmacokinetics, ADME parameters, druglikeness and medicinal chemistry friendliness of one or multiple small molecules to support drug discovery | Free | [62] |
| STP swisstargetprediction.ch (accessed on 1 August 2023) | It estimates the most probable macromolecular targets of a small molecule, assumed as bioactive from three different species (Homo sapiens, Mus musculus, Rattus norvegicus) | Free | [63] |
| SEA sea.bkslab.org (accessed on 1 August 2023) | It quantitatively groups and relates proteins based on the chemical similarity of their ligands. It can be used to rapidly search large compound databases and to build cross-target similarity maps | Free | [64] |
| PharmMapper lilab-ecust.cn/pharmmapper/ (accessed on 1 August 2023) | It is a web server that allows the identification of potential small molecule targets using a pharmacophore mapping approach. The server hosts a large repertoire of pharmacophore models annotated from various sources and it finds the best mapping poses of the query molecule against all the pharmacophore models in the database | Free | [65] |
| TCMSP cmsp-e.com/tcmsp.php (accessed on 1 August 2023) | It captures the relationships between drugs, targets and diseases as well as pharmacokinetic properties for natural compounds | Free | [66] |
| PubChem pubchem.ncbi.nlm.nih.gov (accessed on 1 August 2023) | It is an open chemistry database that mainly contains small molecules, but also nucleotides, carbohydrates, lipids, peptides and chemically modified macromolecules. It collects information on chemical structures, identifiers, chemical and physical properties, biological activity, patents, health and safety, toxicity data and many others | OSP | [67] |
| GeneCards genecards.org (accessed on 1 August 2023) | It is a searchable integrative database that provides comprehensive user-friendly information on all annotated and predicted human genes. It automatically integrates gene-centric data from approximately 150 web sources, including genomic, transcriptomic, proteomic, genetic, clinical and functional information | OSP | [68] |
| OMIM omim.org (accessed on 1 August 2023) | It is a comprehensive, authoritative compendium of human genes and genetic phenotypes that is freely available and updated daily. The full-text, referenced overviews in OMIM contain information on all known Mendelian disorders and over 16,000 genes. OMIM focuses on the relationships between phenotypes and genotypes | OSP | [69] |
| ETCM tcmip.cn/ETCM/ (accessed on 1 August 2023) | It includes the most commonly used herbs and formulas of Traditional Chinese Medicine, as well as their ingredients, to explore the relationships or build networks among TCM herbs, formulas, ingredients, gene targets and related pathways or diseases | Free | [70] |
| STRING string-db.org (accessed on 1 August 2023) | It aims to integrate all known and predicted associations between proteins, including both physical interactions and functional associations. To achieve this, STRING collects and scores evidence from a number of sources: (i) automated text mining of the scientific literature, (ii) databases of interaction experiments and annotated complexes/pathways, (iii) computational interaction predictions from co-expression and from conserved genomic context and (iv) systematic transfers of interaction evidence from one organism to another. The upcoming version 11.5 of the resource will contain more than 14,000 organisms | Free | [71] |
| STITCH 2 stitch.embl.de (accessed on 1 August 2023) | It aims to integrate the data dispersed throughout the literature and various databases of biological pathways, drug–target relationships and binding affinities. In STITCH 2, the number of relevant interactions is increased by the incorporation of BindingDB, PharmGKB and the Comparative Toxicogenomics Database. The resulting network can be explored interactively or used as the basis for large-scale analyses. STITCH 2 connects proteins from 630 organisms to over 74,000 different chemicals, including 2200 drugs | Free | [72] |
| CHEMBL https://www.ebi.ac.uk/chembl/ (accessed on 1 August 2023) | ChEMBL is a manually curated database of bioactive molecules with druglike properties. It brings together chemical, bioactivity and genomic data to aid the translation of genomic information into effective new drugs | Free | [73] |
| Pathguide pathguide.org (accessed on 1 August 2023) | It contains information about 702 biological pathway-related resources and molecular interaction-related resources. Databases that are free and those supporting BioPAX, CellML, PSI-MI or SBML standards are indicated | Free |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardo, L.; Lomagno, A.; Mauri, P.L.; Di Silvestre, D. Integration of Omics Data and Network Models to Unveil Negative Aspects of SARS-CoV-2, from Pathogenic Mechanisms to Drug Repurposing. Biology 2023, 12, 1196. https://doi.org/10.3390/biology12091196
Bernardo L, Lomagno A, Mauri PL, Di Silvestre D. Integration of Omics Data and Network Models to Unveil Negative Aspects of SARS-CoV-2, from Pathogenic Mechanisms to Drug Repurposing. Biology. 2023; 12(9):1196. https://doi.org/10.3390/biology12091196
Chicago/Turabian StyleBernardo, Letizia, Andrea Lomagno, Pietro Luigi Mauri, and Dario Di Silvestre. 2023. "Integration of Omics Data and Network Models to Unveil Negative Aspects of SARS-CoV-2, from Pathogenic Mechanisms to Drug Repurposing" Biology 12, no. 9: 1196. https://doi.org/10.3390/biology12091196
APA StyleBernardo, L., Lomagno, A., Mauri, P. L., & Di Silvestre, D. (2023). Integration of Omics Data and Network Models to Unveil Negative Aspects of SARS-CoV-2, from Pathogenic Mechanisms to Drug Repurposing. Biology, 12(9), 1196. https://doi.org/10.3390/biology12091196