From Chaos to Opportunity: Decoding Cancer Heterogeneity for Enhanced Treatment Strategies

 ,
,  ,
,  , ,
, , 
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Cancer Heterogeneity: Definitions
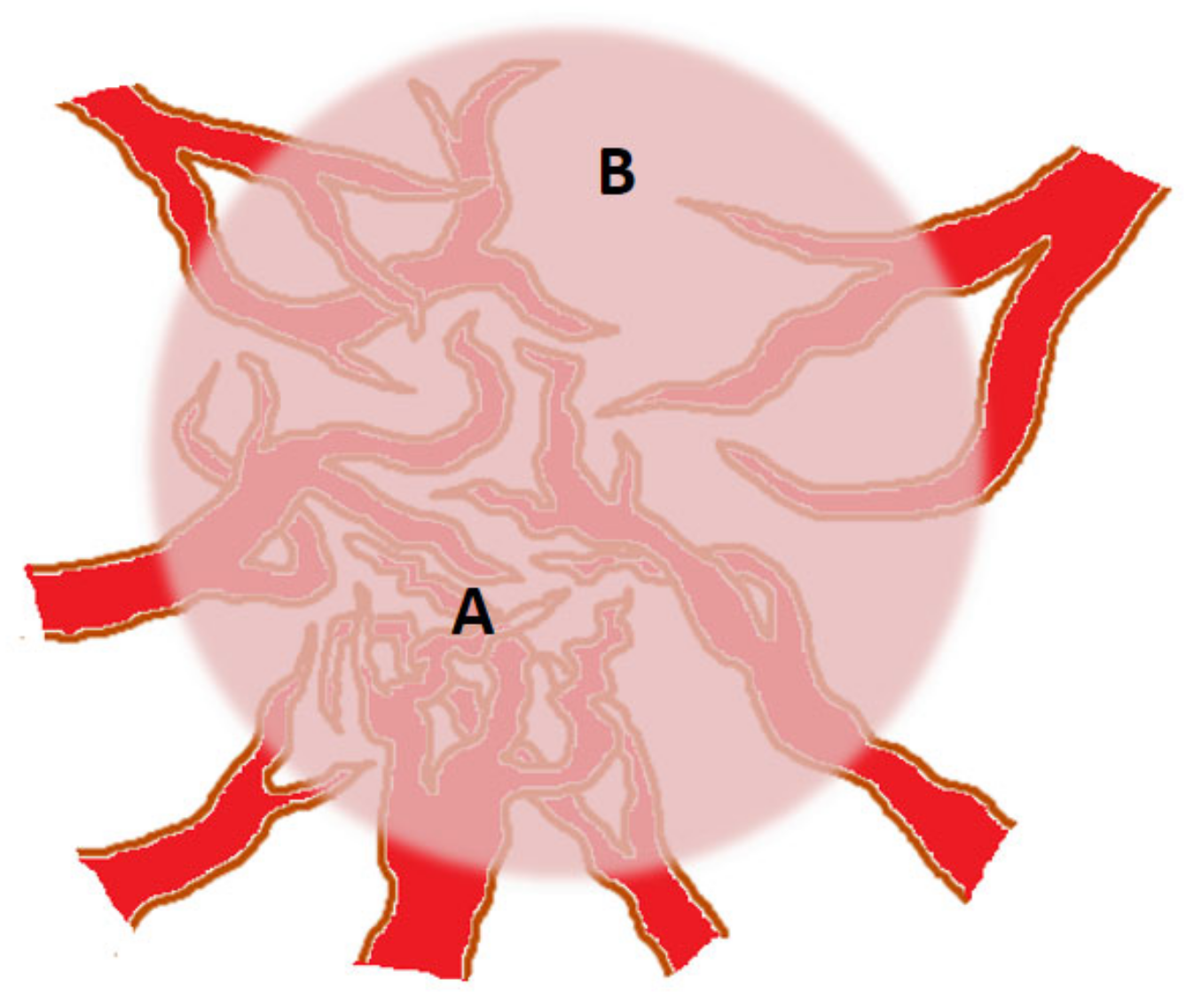
3. Angiogenic Heterogeneity of Tumors
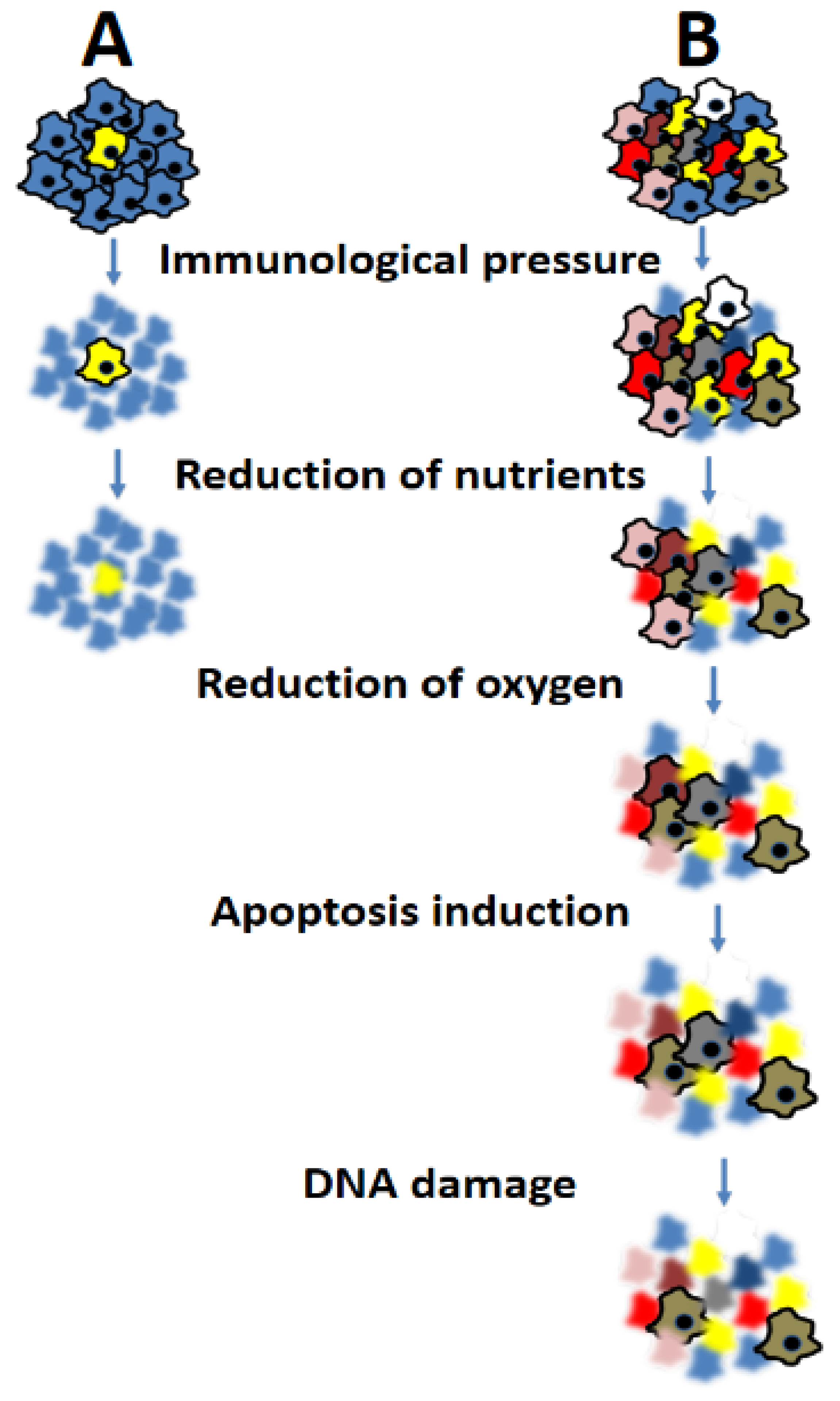
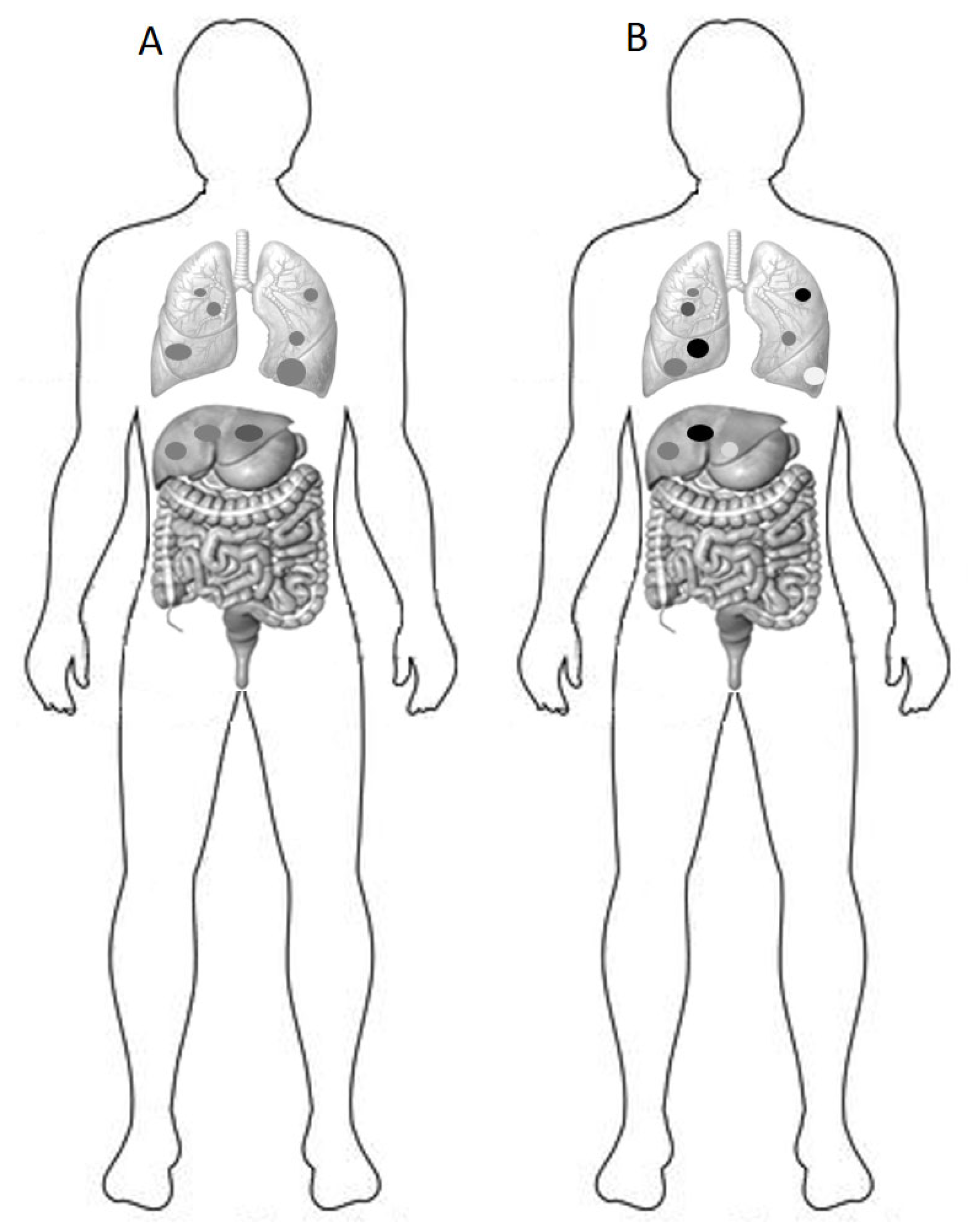
4. Consequences of Heterogeneity on Prognosis and Response to Therapy
5. Mathematical Models Used to Quantify Cancer Heterogeneity
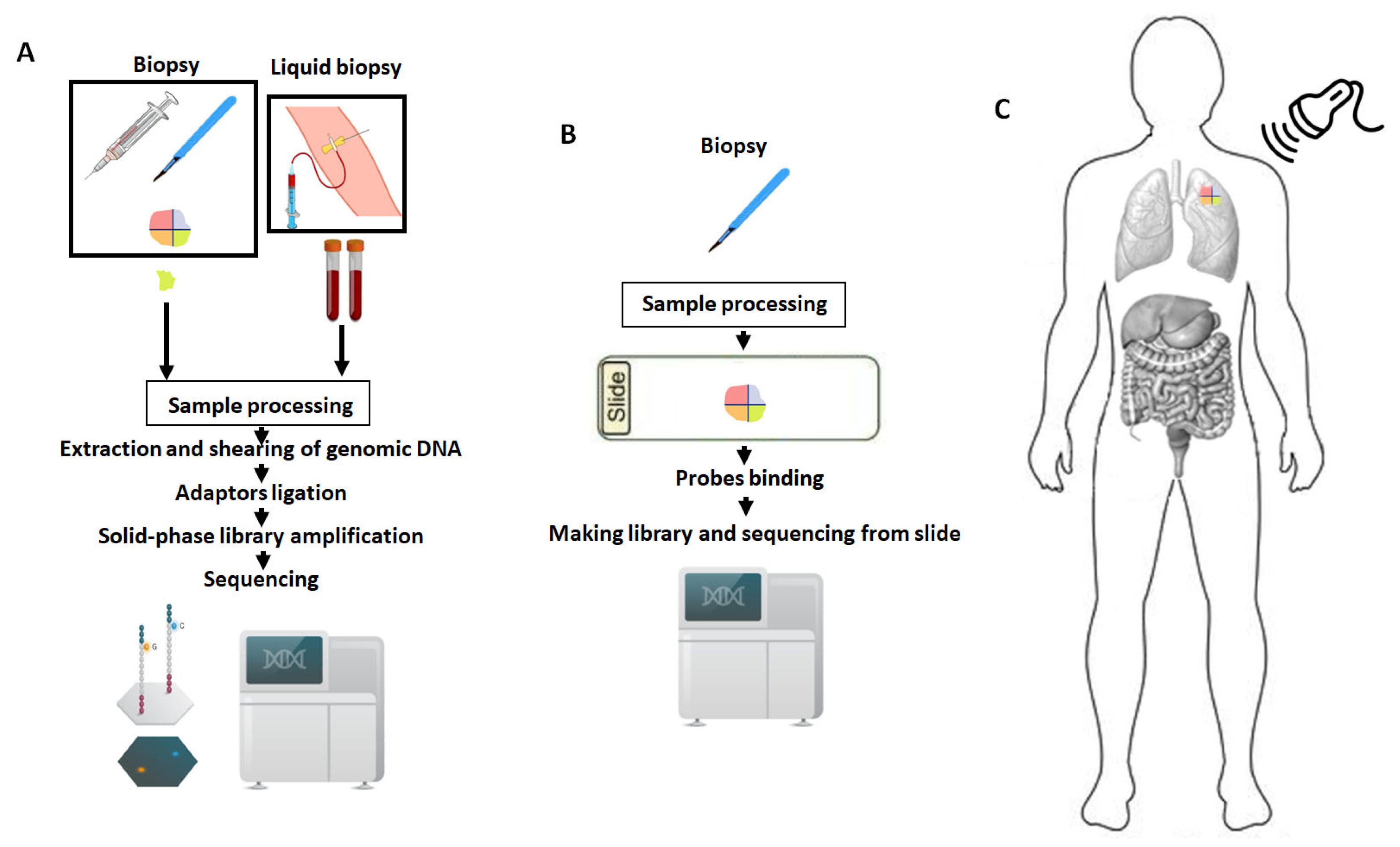
6. Measuring Tumor Heterogeneity: Challenges and Perspectives
7. Exploring Cancer Heterogeneity: Challenges and Prospects in Model Systems
8. Treating Genomic Heterogeneity: Theoretical Pathways for Cure
9. Potential Strategies for Advancing Precision Treatment of Cancer Heterogeneity
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.C.; Bodmer, W.F. Genomic landscape of colorectal carcinogenesis. J. Cancer Res. Clin. Oncol. 2022, 148, 533–545. [Google Scholar] [CrossRef]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021, 18, 79–92. [Google Scholar] [CrossRef]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Ye, K.J.; Liu, G.; Ye, C.J. Genetic and epigenetic heterogeneity in cancer: A genome-centric perspective. J. Cell Physiol. 2009, 220, 538–547. [Google Scholar] [CrossRef]
- Shue, Y.T.; Lim, J.S.; Sage, J. Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Transl. Lung Cancer Res. 2018, 7, 21–31. [Google Scholar] [CrossRef]
- Villanueva, L.; Álvarez-Errico, D.; Esteller, M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Breast Cancer: A Molecularly Heterogenous Disease Needing Subtype-Specific Treatments. Med. Sci. 2020, 8, 18. [Google Scholar] [CrossRef]
- Jacquemin, V.; Antoine, M.; Dom, G.; Detours, V.; Maenhaut, C.; Dumont, J.E. Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers 2022, 14, 280. [Google Scholar] [CrossRef]
- Maniatis, S.; Petrescu, J.; Phatnani, H. Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev. 2021, 66, 70–77. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Cao, Y.; Langer, R.; Ferrara, N. Targeting angiogenesis in oncology, ophthalmology and beyond. Nat. Rev. Drug. Discov. 2023, 22, 476–495. [Google Scholar] [CrossRef]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752S. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Yee, P.P.; Li, W. Tumor necrosis: A synergistic consequence of metabolic stress and inflammation. Bioessays 2021, 43, e2100029. [Google Scholar] [CrossRef]
- Gee, M.S.; Procopio, W.N.; Makonnen, S.; Feldman, M.D.; Yeilding, N.M.; Lee, W.M. Tumor vessel development and maturation impose limits on the effectiveness of anti-vascular therapy. Am. J. Pathol. 2003, 162, 183–193. [Google Scholar] [CrossRef]
- Golemati, S.; Cokkinos, D.D. Recent advances in vascular ultrasound imaging technology and their clinical implications. Ultrasonics 2022, 119, 106599. [Google Scholar] [CrossRef]
- Oostendorp, M.; Douma, K.; Hackeng, T.M.; Dirksen, A.; Post, M.J.; van Zandvoort, M.A.; Backes, W.H. Quantitative molecular magnetic resonance imaging of tumor angiogenesis using cNGR-labeled paramagnetic quantum dots. Cancer Res. 2008, 68, 7676–7683. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Cai, S.M.; Zhang, L.; Zhu, Q.L.; Sun, Q.; Jiang, Y.X.; Wang, H.Y.; Li, J.C. Association Between Vascular Index Measured via Superb Microvascular Imaging and Molecular Subtype of Breast Cancer. Front. Oncol. 2022, 12, 861151. [Google Scholar] [CrossRef]
- Bhat, S.M.; Badiger, V.A.; Vasishta, S.; Chakraborty, J.; Prasad, S.; Ghosh, S.; Joshi, M.B. 3D tumor angiogenesis models: Recent advances and challenges. J. Cancer Res. Clin. Oncol. 2021, 147, 3477–3494. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Cao, H.; Cao, J.W.; Zuo, L.; Qu, F.J.; Xu, D.; Zhang, H.; Gong, H.Y.; Chen, J.X.; Ye, J.Q.; et al. Heterogeneity of tumor microenvironment is associated with clinical prognosis of non-clear cell renal cell carcinoma: A single-cell genomics study. Cell Death Dis. 2022, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, W.; Dai, Y.; Bao, M.; Yuan, Z.; He, M.; Qin, Z.; Liao, S.; He, J.; Huang, Q.; et al. Single-Cell Transcriptomics Reveals the Complexity of the Tumor Microenvironment of Treatment-Naive Osteosarcoma. Front. Oncol. 2021, 11, 709210, Erratum in Front. Oncol. 2022, 12, 1077067. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Oh, K.; Moffitt, R.; Thompson, P.; Li, J.; Liu, J.; Sasson, A.; Georgakis, G.; Kim, J.; Choi, M.; et al. Comparative single-cell RNA sequencing (scRNA-seq) reveals liver metastasis-specific targets in a patient with small intestinal neuroendocrine cancer. Cold Spring. Harb. Mol. Case Stud. 2020, 6, a004978. [Google Scholar] [CrossRef]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e6. [Google Scholar] [CrossRef]
- Ojha, T.; Pathak, V.; Shi, Y.; Hennink, W.E.; Moonen, C.T.W.; Storm, G.; Kiessling, F.; Lammers, T. Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv. Drug Deliv. Rev. 2017, 119, 44–60. [Google Scholar] [CrossRef]
- Au, J.L.; Yeung, B.Z.; Wientjes, M.G.; Lu, Z.; Wientjes, M.G. Delivery of cancer therapeutics to extracellular and intracellular targets: Determinants, barriers, challenges and opportunities. Adv. Drug Deliv. Rev. 2016, 97, 280–301. [Google Scholar] [CrossRef]
- Jain, R.K. Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev. 2012, 64, 353–365. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yasuoka, H.; Tsujimoto, M.; Imabun, S.; Nakahara, M.; Nakao, K.; Nakamura, M.; Mori, I.; Kakudo, K. Lymph vessel density correlates with nodal status, VEGF-C expression, and prognosis in breast cancer. Breast Cancer Res. Treat. 2005, 91, 125–132. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Chen, W.L.; Zhang, F.; Wei, X.L.; Zeng, D.; Liang, Y.K.; Wu, J.D.; Zhang, L.Y.; Guo, C.P.; Zeng, H.C.; et al. Over-expression of both VEGF-C and Twist predicts poor prognosis in human breast cancer. Clin. Transl. Oncol. 2019, 21, 1250–1259. [Google Scholar] [CrossRef]
- Niklińska, W.; Burzykowski, T.; Chyczewski, L.; Nikliński, J. Expression of vascular endothelial growth factor (VEGF) in non-small cell lung cancer (NSCLC): Association with p53 gene mutation and prognosis. Lung Cancer 2001, 34, S59–S64. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhu, J.; Liu, D.Y.; Li, H.Y.; Xu, N.; Hou, M. Over-expression of survivin and VEGF in small-cell lung cancer may predict the poorer prognosis. Med. Oncol. 2014, 31, 775. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Sobral, D.; Martins, M.; Kaplan, S.; Golkaram, M.; Salmans, M.; Khan, N.; Vijayaraghavan, R.; Casimiro, S.; Fernandes, A.; Borralho, P.; et al. Genetic and microenvironmental intra-tumor heterogeneity impacts colorectal cancer evolution and metastatic development. Commun. Biol. 2022, 5, 937. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Comito, G.; Chiarugi, P. Microenvironment and tumor cell plasticity: An easy way out. Cancer Lett. 2013, 341, 80–96. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Reiman, J.M.; Kmieciak, M.; Manjili, M.H.; Knutson, K.L. Tumor immunoediting and immunosculpting pathways to cancer progression. Semin. Cancer Biol. 2007, 17, 275–287. [Google Scholar] [CrossRef]
- Cheruku, S.; Rao, V.; Pandey, R.; Rao Chamallamudi, M.; Velayutham, R.; Kumar, N. Tumor-associated macrophages employ immunoediting mechanisms in colorectal tumor progression: Current research in Macrophage repolarization immunotherapy. Int. Immunopharmacol. 2023, 116, 109569. [Google Scholar] [CrossRef]
- Chhabra, Y.; Weeraratna, A.T. Fibroblasts in cancer: Unity in heterogeneity. Cell 2023, 186, 1580–1609. [Google Scholar] [CrossRef]
- Vokurka, M.; Lacina, L.; Brábek, J.; Kolář, M.; Ng, Y.Z.; Smetana, K., Jr. Cancer-Associated Fibroblasts Influence the Biological Properties of Malignant Tumours via Paracrine Secretion and Exosome Production. Int. J. Mol. Sci. 2022, 23, 964. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Temko, D.; Maliga, Z.; Moreira, A.L.; Sei, E.; Minussi, D.C.; Dean, J.; Lee, C.; Xu, Q.; Hochart, G.; et al. Spatial intra-tumor heterogeneity is associated with survival of lung adenocarcinoma patients. Cell Genom. 2022, 2, 100165. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell Lung Cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Leslie, P.L.; Chao, Y.L.; Tsai, Y.H.; Ghosh, S.K.; Porrello, A.; Van Swearingen, A.E.D.; Harrison, E.B.; Cooley, B.C.; Parker, J.S.; Carey, L.A.; et al. Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat. Commun. 2019, 10, 4192. [Google Scholar] [CrossRef]
- Rath, S.; Chakraborty, D.; Pradhan, J.; Imran Khan, M.; Dandapat, J. Epigenomic interplay in tumor heterogeneity: Potential of epidrugs as adjunct therapy. Cytokine 2022, 157, 155967. [Google Scholar] [CrossRef]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef]
- Guillamot, M.; Cimmino, L.; Aifantis, I. The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer 2016, 2, 70–83. [Google Scholar] [CrossRef]
- Schmitz, J.; Schwab, J.; Schwenck, J.; Chen, Q.; Quintanilla-Martinez, L.; Hahn, M.; Wietek, B.; Schwenzer, N.; Staebler, A.; Kohlhofer, U.; et al. Decoding Intratumoral Heterogeneity of Breast Cancer by Multiparametric In Vivo Imaging: A Translational Study. Cancer Res. 2016, 76, 5512–5522. [Google Scholar] [CrossRef]
- Liu, J.; Si, Y.; Zhou, Z.; Yang, X.; Li, C.; Qian, L.; Feng, L.J.; Zhang, M.; Zhang, S.X.; Liu, J.; et al. The prognostic value of 18F-FDG PET/CT intra-tumoural metabolic heterogeneity in pretreatment neuroblastoma patients. Cancer Imaging 2022, 22, 32. [Google Scholar] [CrossRef]
- Ceriani, L.; Gritti, G.; Cascione, L.; Pirosa, M.C.; Polino, A.; Ruberto, T.; Stathis, A.; Bruno, A.; Moccia, A.A.; Giovanella, L.; et al. SAKK38/07 study: Integration of baseline metabolic heterogeneity and metabolic tumor volume in DLBCL prognostic model. Blood Adv. 2020, 4, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Hendlisz, A.; Golfinopoulos, V.; Garcia, C.; Covas, A.; Emonts, P.; Ameye, L.; Paesmans, M.; Deleporte, A.; Machiels, G.; Toussaint, E.; et al. Serial FDG-PET/CT for early outcome prediction in patients with metastatic colorectal cancer undergoing chemotherapy. Ann. Oncol. 2012, 23, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Jung, J.H.; Son, S.H.; Kim, C.Y.; Hong, C.M.; Oh, J.R.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Prognostic Significance of Intratumoral Metabolic Heterogeneity on 18F-FDG PET/CT in Pathological N0 Non-Small Cell Lung Cancer. Clin. Nucl. Med. 2015, 40, 708–714. [Google Scholar] [CrossRef]
- Meier, A.; Veeraraghavan, H.; Nougaret, S.; Lakhman, Y.; Sosa, R.; Soslow, R.A.; Sutton, E.J.; Hricak, H.; Sala, E.; Vargas, H.A. Association between CT-texture-derived tumor heterogeneity, outcomes, and BRCA mutation status in patients with high-grade serous ovarian cancer. Abdom. Radiol. 2019, 44, 2040–2047. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, G.; Hong, J.H.; Lin, Y.P.; Chen, F.H.; Ng, S.H.; Wang, C.C. Diffusion radiomics analysis of intratumoral heterogeneity in a murine prostate cancer model following radiotherapy: Pixelwise correlation with histology. J. Magn. Reson. Imaging 2017, 46, 483–489. [Google Scholar] [CrossRef]
- Li, Q.; Dormer, J.; Daryani, P.; Chen, D.; Zhang, Z.; Fei, B. Radiomics Analysis of MRI for Predicting Molecular Subtypes of Breast Cancer in Young Women. Proc. SPIE Int. Soc. Opt. Eng. 2019, 10950, 1095044. [Google Scholar] [CrossRef]
- Granata, V.; Fusco, R.; Barretta, M.L.; Picone, C.; Avallone, A.; Belli, A.; Patrone, R.; Ferrante, M.; Cozzi, D.; Grassi, R.; et al. Radiomics in hepatic metastasis by colorectal cancer. Infect. Agent Cancer 2021, 16, 39. [Google Scholar] [CrossRef]
- Basu, S.; Dong, Y.; Kumar, R.; Jeter, C.; Tang, D.G. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin. Cancer Biol. 2022, 78, 90–103. [Google Scholar] [CrossRef]
- Ganguli, P.; Sarkar, R.R. Exploring immuno-regulatory mechanisms in the tumor microenvironment: Model and design of protocols for cancer remission. PLoS ONE 2018, 13, e0203030. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Myeloid-derived suppressor cells in cancer: Recent progress and prospects. Immunol. Cell Biol. 2013, 91, 493–502. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Roberts, C.M.; Cardenas, C.; Tedja, R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11, 1083. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.N.; Wu, Y.M.; Mishler, J.; Hall, S.; Mannan, R.; Wang, L.; Ning, Y.; Zhou, J.; Hopkins, A.C.; Estill, J.C.; et al. The genetic heterogeneity and drug resistance mechanisms of relapsed refractory multiple myeloma. Nat. Commun. 2022, 13, 3750. [Google Scholar] [CrossRef]
- Sexton, R.E.; Hallak, M.N.A.; Uddin, M.H.; Diab, M.; Azmi, A.S. Gastric Cancer Heterogeneity and Clinical Outcomes. Technol. Cancer Res. Treat. 2020, 19, 1533033820935477. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Zhang, S.; Yu, S.; Ma, F.; Wang, B.; Zhang, C.; Sun, J.; Mao, X.; Wei, L. Cellular heterogeneity landscape in laryngeal squamous cell carcinoma. Int. J. Cancer 2020, 147, 2879–2890. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.X.; He, M.H.; Dai, Z.H.; Yu, J.; Wang, J.G.; Li, X.C.; Jiang, B.B.; Ke, Z.F.; Su, T.H.; Peng, Z.W.; et al. Genomic and transcriptional heterogeneity of multifocal hepatocellular carcinoma. Ann. Oncol. 2019, 30, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Mota, A.; Oltra, S.S.; Selenica, P.; Moiola, C.P.; Casas-Arozamena, C.; López-Gil, C.; Diaz, E.; Gatius, S.; Ruiz-Miro, M.; Calvo, A.; et al. Intratumor genetic heterogeneity and clonal evolution to decode endometrial cancer progression. Oncogene 2022, 41, 1835–1850. [Google Scholar] [CrossRef]
- Sinha, N.; Sinha, S.; Valero, C.; Schäffer, A.A.; Aldape, K.; Litchfield, K.; Chan, T.A.; Morris, L.G.T.; Ruppin, E. Immune Determinants of the Association between Tumor Mutational Burden and Immunotherapy Response across Cancer Types. Cancer Res. 2022, 82, 2076–2083. [Google Scholar] [CrossRef]
- Motta, R.; Cabezas-Camarero, S.; Torres-Mattos, C.; Riquelme, A.; Calle, A.; Figueroa, A.; Sotelo, M.J. Immunotherapy in microsatellite instability metastatic colorectal cancer: Current status and future perspectives. J. Clin. Transl. Res. 2021, 7, 511–522. [Google Scholar] [PubMed]
- Wolf, Y.; Samuels, Y. Intratumor Heterogeneity and Antitumor Immunity Shape One Another Bidirectionally. Clin. Cancer Res. 2022, 28, 2994–3001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bao, M.; Gao, G.; Cai, Y.; Wu, L.; Lei, L.; Zhao, J.; Ji, X.; Huang, Y.; Su, C. Increased blood-based intratumor heterogeneity (bITH) is associated with unfavorable outcomes of immune checkpoint inhibitors plus chemotherapy in non-small cell Lung Cancer. BMC Med. 2022, 20, 256. [Google Scholar] [CrossRef]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef]
- Altrock, P.M.; Liu, L.L.; Michor, F. The mathematics of cancer: Integrating quantitative models. Nat. Rev. Cancer 2015, 15, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Bearer, E.L.; Lowengrub, J.S.; Frieboes, H.B.; Chuang, Y.L.; Jin, F.; Wise, S.M.; Ferrari, M.; Agus, D.B.; Cristini, V. Multiparameter computational modeling of tumor invasion. Cancer Res. 2009, 69, 4493–4501. [Google Scholar] [CrossRef]
- Kim, J.Y.; Gatenby, R.A. Quantitative Clinical Imaging Methods for Monitoring Intratumoral Evolution. Methods Mol. Biol. 2017, 1513, 61–81. [Google Scholar] [CrossRef]
- Chung, Y.R.; Kim, H.J.; Kim, Y.A.; Chang, M.S.; Hwang, K.T.; Park, S.Y. Diversity index as a novel prognostic factor in breast cancer. Oncotarget 2017, 8, 97114–97126. [Google Scholar] [CrossRef]
- Iwasa, Y.; Michor, F. Evolutionary dynamics of intratumor heterogeneity. PLoS ONE 2011, 6, e17866. [Google Scholar] [CrossRef]
- Stensrud, M.J.; Valberg, M. Inequality in genetic cancer risk suggests bad genes rather than bad luck. Nat. Commun. 2017, 8, 1165. [Google Scholar] [CrossRef][Green Version]
- Martinez, P.; Kimberley, C.; BirkBak, N.J.; Marquard, A.; Szallasi, Z.; Graham, T.A. Quantification of within-sample genetic heterogeneity from SNP-array data. Sci. Rep. 2017, 7, 3248. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Kim, H.J.; Cheng, Y.K.; Gönen, M.; Itzkovitz, S.; Argani, P.; van Oudenaarden, A.; Sukumar, S.; Michor, F.; Polyak, K. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014, 74, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Tsoucas, D.; Yuan, G.C. Assessing Inequality in Transcriptomic Data. Cell Syst. 2018, 6, 149–150. [Google Scholar] [CrossRef]
- Kashyap, A.; Rapsomaniki, M.A.; Barros, V.; Fomitcheva-Khartchenko, A.; Martinelli, A.L.; Rodriguez, A.F.; Gabrani, M.; Rosen-Zvi, M.; Kaigala, G. Quantification of tumor heterogeneity: From data acquisition to metric generation. Trends Biotechnol. 2022, 40, 647–676. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Bagheri, N. Agent-Based Models Predict Emergent Behavior of Heterogeneous Cell Populations in Dynamic Microenvironments. Front. Bioeng. Biotechnol. 2020, 8, 249. [Google Scholar] [CrossRef]
- Rai, V.; Nadar, S.R.; Upadhyay, R.K. Nonlinear phenomena in biology and medicine. Comput. Math. Methods Med. 2012, 2012, 183879. [Google Scholar] [CrossRef]
- Buiatti, M.; Longo, G. Randomness and multilevel interactions in biology. Theory Biosci. 2013, 132, 139–158. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef]
- Petegrosso, R.; Li, Z.; Kuang, R. Machine learning and statistical methods for clustering single-cell RNA-sequencing data. Brief Bioinform. 2020, 21, 1209–1223. [Google Scholar] [CrossRef]
- Reiter, J.G.; Baretti, M.; Gerold, J.M.; Makohon-Moore, A.P.; Daud, A.; Iacobuzio-Donahuee, C.A. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 2019, 19, 639–650. [Google Scholar] [CrossRef]
- Lynch, M.; Ackerman, M.S.; Gout, J.F.; Long, H.; Sung, W.; Thomase, W.K. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Kokko, H.; Eyre-Walker, A. Population size and the rate of evolution. Trends Ecol. Evol. 2014, 29, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Scally, A. The mutation rate in human evolution and demographic inference. Curr. Opin. Genet. Dev. 2016, 41, 36–43. [Google Scholar] [CrossRef]
- Thoma, F. Light and dark in chromatin repair: Repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J. 1999, 18, 6585–6598. [Google Scholar] [CrossRef] [PubMed]
- Doig, K.D.; Fellowes, A.; Scott, P.; Fox, S.B. Tumour mutational burden: An overview for pathologists. Pathology 2022, 54, 249–253. [Google Scholar] [CrossRef]
- Kelly, C.M.; Gutierrez Sainz, L.; Chi, P. The management of metastatic GIST: Current standard and investigational therapeutics. J. Hematol. Oncol. 2021, 14, 2. [Google Scholar] [CrossRef]
- Liu, C.; Ye, D.; Yang, H.; Chen, X.; Su, Z.; Li, X.; Ding, M.; Liu, Y. RAS-targeted cancer therapy: Advances in drugging specific mutations. MedComm 2023, 4, e285. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Sun, J.; Pedersen, M.; Bengtsson, S.; Rönnstrand, L. Grb2 mediates negative regulation of stem cell factor receptor/c-Kit signaling by recruitment of Cbl. Exp. Cell Res. 2007, 313, 3935–3942. [Google Scholar] [CrossRef]
- Liang, J.; Wu, Y.L.; Chen, B.J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef]
- Alem, D.; Yang, X.; Beato, F.; Sarcar, B.; Tassielli, A.F.; Dai, R.; Hogenson, T.L.; Park, M.A.; Jiang, K.; Cai, J.; et al. Translational relevance of SOS1 targeting for KRAS-mutant colorectal cancer. Mol. Carcinog. 2023, 62, 1025–1037. [Google Scholar] [CrossRef]
- Tellez-Gabriel, M.; Ory, B.; Lamoureux, F.; Heymann, M.F.; Heymann, D. Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis. Int. J. Mol. Sci. 2016, 17, 2142. [Google Scholar] [CrossRef]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- Ottaiano, A.; Circelli, L.; Lombardi, A.; Scala, S.; Martucci, N.; Galon, J.; Buonanno, M.; Scognamiglio, G.; Botti, G.; Hermitte, F.; et al. Genetic trajectory and immune microenvironment of lung-specific oligometastatic colorectal cancer. Cell Death Dis. 2020, 11, 275. [Google Scholar] [CrossRef]
- Ottaiano, A.; Caraglia, M.; Di Mauro, A.; Botti, G.; Lombardi, A.; Galon, J.; Luce, A.; D’Amore, L.; Perri, F.; Santorsola, M.; et al. Evolution of Mutational Landscape and Tumor Immune-Microenvironment in Liver Oligo-Metastatic Colorectal Cancer. Cancers 2020, 12, 3073. [Google Scholar] [CrossRef] [PubMed]
- Capuozzo, M.; Santorsola, M.; Bocchetti, M.; Perri, F.; Cascella, M.; Granata, V.; Celotto, V.; Gualillo, O.; Cossu, A.M.; Nasti, G.; et al. p53: From Fundamental Biology to Clinical Applications in Cancer. Biology 2022, 11, 1325. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R.; Thavornwatanayong, T.; Venkatesh, M.K.; Chandy, C.; Vachss Dustine, T.; Guzik, H.; Koba, W.; Liu, Q.; Goel, S. Development and Characterization of a Genetic Mouse Model of KRAS Mutated Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 5677. [Google Scholar] [CrossRef] [PubMed]
- Creedon, H.; Balderstone, L.A.; Muir, M.; Balla, J.; Gomez-Cuadrado, L.; Tracey, N.; Loane, J.; Klinowska, T.; Muller, W.J.; Brunton, V.G. Use of a genetically engineered mouse model as a preclinical tool for HER2 breast cancer. Dis. Model Mech. 2016, 9, 131–140. [Google Scholar] [CrossRef]
- Puzio-Kuter, A.M.; Castillo-Martin, M.; Kinkade, C.W.; Wang, X.; Shen, T.H.; Matos, T.; Shen, M.M.; Cordon-Cardo, C.; Abate-Shen, C. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev. 2009, 23, 675–680. [Google Scholar] [CrossRef]
- Zhou, Z.; Flesken-Nikitin, A.; Corney, D.C.; Wang, W.; Goodrich, D.W.; Roy-Burman, P.; Nikitin, A.Y. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006, 66, 7889–7898. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Miyado, M.; Igarashi, M.; Tamano, M.; Kubo, A.; Yamashita, S.; Asahara, H.; Fukami, M.; Takada, S. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci. Rep. 2014, 4, 5396. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.J.; Choi, R.B.; Bullock, W.A.; Robling, A.G. Perspective: The current state of Cre driver mouse lines in skeletal research: Challenges and opportunities. Bone 2023, 170, 116719. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Orsulic, S. Mouse models of cancer. Annu. Rev. Pathol. 2011, 6, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, J.; Xu, S.; She, T.; Zhang, Y.; Sun, Y.; Wen, M.; Jiang, T.; Xiong, Y.; Lei, J. Experimental mouse models for translational human cancer research. Front. Immunol. 2023, 14, 1095388. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Kim, J. Autochthonous murine models for the study of smoker and never-smoker associated lung cancers. Transl. Lung Cancer Res. 2018, 7, 464–486. [Google Scholar] [CrossRef]
- Wong, E.; Yang, K.; Kuraguchi, M.; Werling, U.; Avdievich, E.; Fan, K.; Fazzari, M.; Jin, B.; Brown, A.M.; Lipkin, M.; et al. Mbd4 inactivation increases C→T transition mutations and promotes gastrointestinal tumor formation. Proc. Natl. Acad. Sci. USA 2002, 99, 14937–14942. [Google Scholar] [CrossRef]
- Millar, C.B.; Guy, J.; Sansom, O.J.; Selfridge, J.; MacDougall, E.; Hendrich, B.; Keightley, P.D.; Bishop, S.M.; Clarke, A.R.; Bird, A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 2002, 297, 403–405. [Google Scholar] [CrossRef]
- Evers, B.; Jonkers, J. Mouse models of BRCA1 and BRCA2 deficiency: Past lessons, current understanding and future prospects. Oncogene 2006, 25, 5885–5897. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Wang, Z. Cellular barcoding: From developmental tracing to anti-tumor drug discovery. Cancer Lett. 2023, 567, 216281. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, L.; Li, Y.; Pu, W.; Zhang, S.; Han, X.; Lui, K.O.; Zhou, B. Dual Cre and Dre recombinases mediate synchronized lineage tracing and cell subset ablation in vivo. J. Biol. Chem. 2022, 298, 101965. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, F.; Asselin-Labat, M.L.; Shackleton, M.; Forrest, N.C.; Lindeman, G.J.; Visvader, J.E. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008, 68, 7711–7717. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.W.; Wang, X.; Diehn, M.; Shedden, K.; Chen, G.Y.; Sherlock, G.; Gurney, A.; Lewicki, J.; Clarke, M.F. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells 2008, 26, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.; Wang, Z.; Liu, Y.; Yu, J.; Wang, W.; Chen, S.; Wu, W.; Wang, J.; Qian, G.; et al. Standardization of organoid culture in cancer research. Cancer Med. 2023, 12, 14375–14386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cong, L.; Cong, X. Patient-Derived Organoids in Precision Medicine: Drug Screening, Organoid-on-a-Chip and Living Organoid Biobank. Front. Oncol. 2021, 11, 762184. [Google Scholar] [CrossRef]
- Nam, C.; Ziman, B.; Sheth, M.; Zhao, H.; Lin, D.C. Genomic and Epigenomic Characterization of Tumor Organoid Models. Cancers 2022, 14, 4090. [Google Scholar] [CrossRef]
- Chen, C.; Xing, D.; Tan, L.; Li, H.; Zhou, G.; Huang, L.; Xie, X.S. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356, 189–194. [Google Scholar] [CrossRef]
- Chu, W.K.; Edge, P.; Lee, H.S.; Bansal, V.; Bafna, V.; Huang, X.; Zhang, K. Ultraaccurate genome sequencing and haplotyping of single human cells. Proc. Natl. Acad. Sci. USA 2017, 114, 12512–12517. [Google Scholar] [CrossRef]
- Fu, Y.; Li, C.; Lu, S.; Zhou, W.; Tang, F.; Xie, X.S.; Huang, Y. Uniform and accurate single-cell sequencing based on emulsion whole-genome amplification. Proc. Natl. Acad. Sci. USA 2015, 112, 11923–11928. [Google Scholar] [CrossRef]
- Xing, D.; Tan, L.; Chang, C.H.; Li, H.; Xie, X.S. Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc. Natl. Acad. Sci. USA 2021, 118, e2013106118. [Google Scholar] [CrossRef]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Eastel, J.M.; Lam, K.W.; Lee, N.L.; Lok, W.Y.; Tsang, A.H.F.; Pei, X.M.; Chan, A.K.C.; Cho, W.C.S.; Wong, S.C.C. Application of NanoString technologies in companion diagnostic development. Expert Rev. Mol. Diagn. 2019, 19, 591–598. [Google Scholar] [CrossRef]
- Veldman-Jones, M.H.; Brant, R.; Rooney, C.; Geh, C.; Emery, H.; Harbron, C.G.; Wappett, M.; Sharpe, A.; Dymond, M.; Barrett, J.C.; et al. Evaluating Robustness and Sensitivity of the NanoString Technologies nCounter Platform to Enable Multiplexed Gene Expression Analysis of Clinical Samples. Cancer Res. 2015, 75, 2587–2593. [Google Scholar] [CrossRef]
- Rajurkar, M.; Parikh, A.R.; Solovyov, A.; You, E.; Kulkarni, A.S.; Chu, C.; Xu, K.H.; Jaicks, C.; Taylor, M.S.; Wu, C.; et al. Reverse Transcriptase Inhibition Disrupts Repeat Element Life Cycle in Colorectal Cancer. Cancer Discov. 2022, 12, 1462–1481. [Google Scholar] [CrossRef]
- Bergholtz, H.; Carter, J.M.; Cesano, A.; Cheang, M.C.U.; Church, S.E.; Divakar, P.; Fuhrman, C.A.; Goel, S.; Gong, J.; Guerriero, J.L.; et al. Best Practices for Spatial Profiling for Breast Cancer Research with the GeoMx® Digital Spatial Profiler. Cancers 2021, 13, 4456. [Google Scholar] [CrossRef]
- Hernandez, S.; Lazcano, R.; Serrano, A.; Powell, S.; Kostousov, L.; Mehta, J.; Khan, K.; Lu, W.; Solis, L.M. Challenges and Opportunities for Immunoprofiling Using a Spatial High-Plex Technology: The NanoString GeoMx® Digital Spatial Profiler. Front. Oncol. 2022, 12, 890410. [Google Scholar] [CrossRef]
- Kruse, H.; Šponer, J. Towards biochemically relevant QM computations on nucleic acids: Controlled electronic structure geometry optimization of nucleic acid structural motifs using penalty restraint functions. Phys. Chem. Chem. Phys. 2015, 17, 1399–1410. [Google Scholar] [CrossRef]
- Zou, H.; Wen, S.; Wu, X.; Wong, K.W.; Yam, C. DNA sequencing based on electronic tunneling in a gold nanogap: A first-principles study. Phys. Chem. Chem. Phys. 2022, 24, 5748–5754. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heterogeneity Category | Assessment Technique | Output |
|---|---|---|
| Genetic | Next-generation sequencing | Genetic variants, mutations, copy number alterations [63,64,65,66,67,68,69] |
| Epigenetic | Next-generation sequencing | DNA methylation patterns, histone modifications [3,4,5,6,45,46,47,48] |
| Angiogenic | Ultrasound, magnetic resonance imaging (MRI) | Density and distribution of blood vessels [17,18,19,20,21] |
| Metabolic | Positron emission tomography | Uptake of glucose [49,50,51,52,53] |
| Morphological | Radiomics | Scores that combine multiple morphological features [54,55,56,57] |
| Morphological and genetic | Spatial transcriptomics thorugh in situ hybridization and single-cell RNA sequencing | Genetic characterization of single cell in its biological context [21,22,23,24,25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaiano, A.; Ianniello, M.; Santorsola, M.; Ruggiero, R.; Sirica, R.; Sabbatino, F.; Perri, F.; Cascella, M.; Di Marzo, M.; Berretta, M.; et al. From Chaos to Opportunity: Decoding Cancer Heterogeneity for Enhanced Treatment Strategies. Biology 2023, 12, 1183. https://doi.org/10.3390/biology12091183
Ottaiano A, Ianniello M, Santorsola M, Ruggiero R, Sirica R, Sabbatino F, Perri F, Cascella M, Di Marzo M, Berretta M, et al. From Chaos to Opportunity: Decoding Cancer Heterogeneity for Enhanced Treatment Strategies. Biology. 2023; 12(9):1183. https://doi.org/10.3390/biology12091183
Chicago/Turabian StyleOttaiano, Alessandro, Monica Ianniello, Mariachiara Santorsola, Raffaella Ruggiero, Roberto Sirica, Francesco Sabbatino, Francesco Perri, Marco Cascella, Massimiliano Di Marzo, Massimiliano Berretta, and et al. 2023. "From Chaos to Opportunity: Decoding Cancer Heterogeneity for Enhanced Treatment Strategies" Biology 12, no. 9: 1183. https://doi.org/10.3390/biology12091183
APA StyleOttaiano, A., Ianniello, M., Santorsola, M., Ruggiero, R., Sirica, R., Sabbatino, F., Perri, F., Cascella, M., Di Marzo, M., Berretta, M., Caraglia, M., Nasti, G., & Savarese, G. (2023). From Chaos to Opportunity: Decoding Cancer Heterogeneity for Enhanced Treatment Strategies. Biology, 12(9), 1183. https://doi.org/10.3390/biology12091183