

Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19



{kind=link}
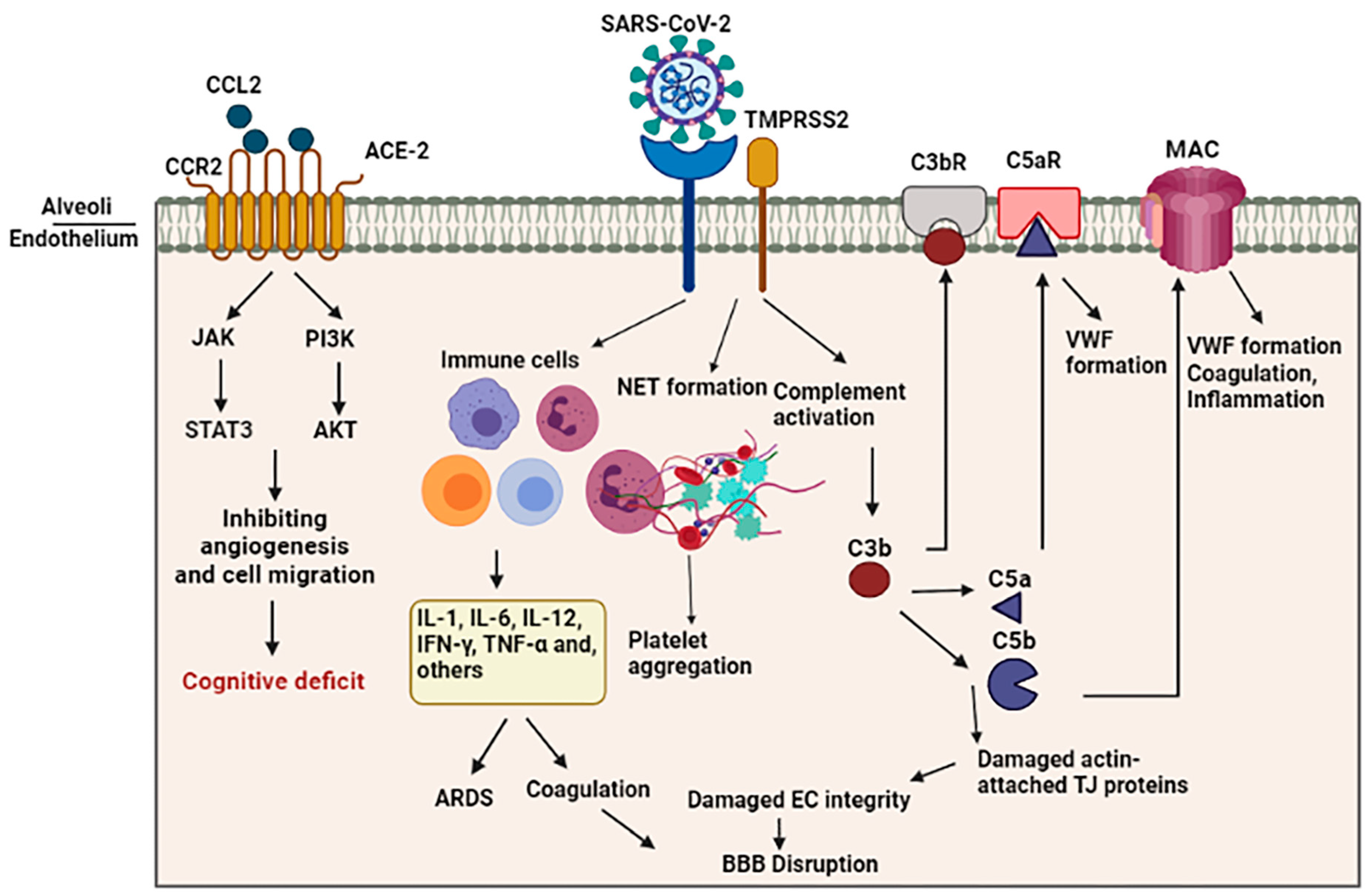{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Long-Term Neurological and Cognitive Dysfunction Following COVID-19
3. Molecules Contribute to COVID-19 Penetration into the CNS
4. Endothelial Cell Infection and Vascular Dysfunction in COVID-19
5. Degradation of Endothelial Glycocalyx Makes Them Vulnerable to SARS-CoV-2 Entry
6. Vascular Inflammation and Blood-Brain Barrier Disruption in COVID-19
7. Disseminating Intravascular Coagulation and BBB Disruption in COVID-19
8. Pneumonia and BBB Disruption in COVID-19
9. Vascular Dysfunction, Brain Inflammation, and Cognitive Impairment
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garg, A.; Marji, A.; Goyal, S.; Ismail, R. A case of COVID-19 with memory impairment and delayed presentation as stroke. Cureus 2020, 12, e10025. [Google Scholar]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar]
- Pilotto, A.; Cristillo, V.; Cotti Piccinelli, S.; Zoppi, N.; Bonzi, G.; Sattin, D.; Schiavolin, S.; Raggi, A.; Canale, A.; Gipponi, S. Long-term neurological manifestations of COVID-19: Prevalence and predictive factors. Neurol. Sci. 2021, 42, 4903–4907. [Google Scholar] [PubMed]
- Grant, M.C.; Geoghegan, L.; Arbyn, M.; Mohammed, Z.; McGuinness, L.; Clarke, E.L.; Wade, R.G. The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19): A systematic review and meta-analysis of 148 studies from 9 countries. PLoS ONE 2020, 15, e0234765. [Google Scholar]
- Dubé, M.; Le Coupanec, A.; Wong, A.H.; Rini, J.M.; Desforges, M.; Talbot, P.J. Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J. Virol. 2018, 92, e00404–e00418. [Google Scholar] [PubMed]
- Goërtz, Y.M.; Van Herck, M.; Delbressine, J.M.; Vaes, A.W.; Meys, R.; Machado, F.V.; Houben-Wilke, S.; Burtin, C.; Posthuma, R.; Franssen, F.M. Persistent symptoms 3 months after a SARS-CoV-2 infection: The post-COVID-19 syndrome? ERJ Open Res. 2020, 6, 542–2020. [Google Scholar]
- Raja, C.P.; Bharathi, B.; Chandrasekaran, C.V.; Muruganantham, N.; Deepak, M.; Amit, A. Science behind Usefulness of Bacopa monnieri for Memory and Cognition. In Phytopharmaceuticals for Brain Health; CRC Press: Boca Raton, FL, USA, 2017; pp. 225–250. [Google Scholar]
- Stefano, G.B.; Ptacek, R.; Ptackova, H.; Martin, A.; Kream, R.M. Selective neuronal mitochondrial targeting in SARS-CoV-2 infection affects cognitive processes to induce ‘brain fog’and results in behavioral changes that favor viral survival. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2021, 27, e930886-1. [Google Scholar]
- Bliddal, S.; Banasik, K.; Pedersen, O.B.; Nissen, J.; Cantwell, L.; Schwinn, M.; Tulstrup, M.; Westergaard, D.; Ullum, H.; Brunak, S. Acute and persistent symptoms in non-hospitalized PCR-confirmed COVID-19 patients. Sci. Rep. 2021, 11, 13153. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef]
- Semmler, A.; Widmann, C.N.; Okulla, T.; Urbach, H.; Kaiser, M.; Widman, G.; Mormann, F.; Weide, J.; Fliessbach, K.; Hoeft, A. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J. Neurol. Neurosurg. Psychiatry 2013, 84, 62–69. [Google Scholar] [CrossRef]
- Penninx, B.W.J.H. Psychiatric symptoms and cognitive impairment in “Long COVID”: The relevance of immunopsychiatry. World Psychiatry 2021, 20, 357. [Google Scholar] [PubMed]
- Kozik, V.; Reuken, P.; Utech, I.; Gramlich, J.; Stallmach, Z.; Demeyere, N.; Rakers, F.; Schwab, M.; Stallmach, A.; Finke, K. Subtle cognitive impairments in memory, attention, and executive functioning in patients with post-COVID syndrome and their relationships with clinical variables and subjective complaints. medRxiv 2022. [Google Scholar] [CrossRef]
- Li, Y.; Ji, M.; Yang, J. Current understanding of long-term cognitive impairment after sepsis. Front. Immunol. 2022, 13, 855006. [Google Scholar] [CrossRef] [PubMed]
- Rass, V.; Beer, R.; Schiefecker, A.J.; Kofler, M.; Lindner, A.; Mahlknecht, P.; Heim, B.; Limmert, V.; Sahanic, S.; Pizzini, A. Neurological outcome and quality of life 3 months after COVID-19: A prospective observational cohort study. Eur. J. Neurol. 2021, 28, 3348–3359. [Google Scholar]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent symptoms in patients after acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M. Neurologic features in severe SARS-CoV-2 infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar]
- Mazza, M.G.; Palladini, M.; De Lorenzo, R.; Magnaghi, C.; Poletti, S.; Furlan, R.; Ciceri, F.; Rovere-Querini, P.; Benedetti, F.; The COVID-19 BioB Outpatient Clinic Study Group. Persistent psychopathology and neurocognitive impairment in COVID-19 survivors: Effect of inflammatory biomarkers at three-month follow-up. Brain Behav. Immun. 2021, 94, 138–147. [Google Scholar]
- Hellmuth, J.; Barnett, T.A.; Asken, B.M.; Kelly, J.D.; Torres, L.; Stephens, M.L.; Greenhouse, B.; Martin, J.N.; Chow, F.C.; Deeks, S.G. Persistent COVID-19-associated neurocognitive symptoms in non-hospitalized patients. J. Neurovirol. 2021, 27, 191–195. [Google Scholar] [CrossRef]
- Garrigues, E.; Janvier, P.; Kherabi, Y.; Le Bot, A.; Hamon, A.; Gouze, H.; Doucet, L.; Berkani, S.; Oliosi, E.; Mallart, E. Post-discharge persistent symptoms and health-related quality of life after hospitalization for COVID-19. J. Infect. 2020, 81, e4–e6. [Google Scholar] [CrossRef]
- Han, Q.; Zheng, B.; Daines, L.; Sheikh, A. Long-term sequelae of COVID-19: A systematic review and meta-analysis of one-year follow-up studies on post-COVID symptoms. Pathogens 2022, 11, 269. [Google Scholar]
- Ritchie, K.; Chan, D. The emergence of cognitive COVID. World Psychiatry 2021, 20, 52. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, S.; Chen, J.; Wei, N.; Wang, D.; Lyu, H.; Shi, C.; Hu, S. The landscape of cognitive function in recovered COVID-19 patients. J. Psychiatr. Res. 2020, 129, 98–102. [Google Scholar]
- Raman, B.; Cassar, M.P.; Tunnicliffe, E.M.; Filippini, N.; Griffanti, L.; Alfaro-Almagro, F.; Okell, T.; Sheerin, F.; Xie, C.; Mahmod, M. Medium-term effects of SARS-CoV-2 infection on multiple vital organs, exercise capacity, cognition, quality of life and mental health, post-hospital discharge. EClinicalMedicine 2021, 31, 100683. [Google Scholar] [PubMed]
- Woo, M.S.; Malsy, J.; Pöttgen, J.; Seddiq Zai, S.; Ufer, F.; Hadjilaou, A.; Schmiedel, S.; Addo, M.M.; Gerloff, C.; Heesen, C. Frequent neurocognitive deficits after recovery from mild COVID-19. Brain Commun. 2020, 2, fcaa205. [Google Scholar]
- Kumar, V.; Singla, S.; Gupta, N.; Bharati, S.J.; Garg, R.; Pandit, A.; Vig, S.; Mishra, S.; Bhatnagar, S. The incidence of anosmia in patients with laboratory-confirmed COVID 19 infection in India: An observational study. J. Anaesthesiol. Clin. Pharmacol. 2021, 37, 51. [Google Scholar] [PubMed]
- Jaywant, A.; Vanderlind, W.M.; Alexopoulos, G.S.; Fridman, C.B.; Perlis, R.H.; Gunning, F.M. Frequency and profile of objective cognitive deficits in hospitalized patients recovering from COVID-19. Neuropsychopharmacology 2021, 46, 2235–2240. [Google Scholar] [PubMed]
- Villani, E.R.; Vetrano, D.L.; Damiano, C.; Paola, A.D.; Ulgiati, A.M.; Martin, L.; Hirdes, J.P.; Fratiglioni, L.; Bernabei, R.; Onder, G. Impact of COVID-19-related lockdown on psychosocial, cognitive, and functional well-being in adults with down syndrome. Front. Psychiatry 2020, 11, 578686. [Google Scholar] [PubMed]
- Zhou, J.; Liu, C.; Sun, Y.; Huang, W.; Ye, K. Cognitive disorders associated with hospitalization of COVID-19: Results from an observational cohort study. Brain Behav. Immun. 2021, 91, 383–392. [Google Scholar]
- Heesakkers, H.; van der Hoeven, J.G.; Corsten, S.; Janssen, I.; Ewalds, E.; Simons, K.S.; Westerhof, B.; Rettig, T.C.; Jacobs, C.; van Santen, S. Clinical outcomes among patients with 1-year survival following intensive care unit treatment for COVID-19. JAMA 2022, 327, 559–565. [Google Scholar]
- Del Brutto, O.H.; Wu, S.; Mera, R.M.; Costa, A.F.; Recalde, B.Y.; Issa, N.P. Cognitive decline among individuals with history of mild symptomatic SARS-CoV-2 infection: A longitudinal prospective study nested to a population cohort. Eur. J. Neurol. 2021, 28, 3245–3253. [Google Scholar] [CrossRef]
- Cristillo, V.; Pilotto, A.; Cotti Piccinelli, S.; Bonzi, G.; Canale, A.; Gipponi, S.; Bezzi, M.; Leonardi, M.; Padovani, A.; Neuro Covid Next Study Group. Premorbid vulnerability and disease severity impact on Long-COVID cognitive impairment. Aging Clin. Exp. Res. 2022, 34, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201. [Google Scholar]
- Shabani, Z. Demyelination as a result of an immune response in patients with COVID-19. Acta Neurol. Belg. 2021, 121, 859–866. [Google Scholar] [CrossRef]
- Doobay, M.F.; Talman, L.S.; Obr, T.D.; Tian, X.; Davisson, R.L.; Lazartigues, E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 292, R373–R381. [Google Scholar] [PubMed]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Seyran, M.; Takayama, K.; Uversky, V.N.; Lundstrom, K.; Palù, G.; Sherchan, S.P.; Attrish, D.; Rezaei, N.; Aljabali, A.A.; Ghosh, S. The structural basis of accelerated host cell entry by SARS-CoV-2. FEBS J. 2021, 288, 5010–5020. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J.; Kamel, H. Effects of COVID-19 on the nervous system. Cell 2020, 183, 16–27.e1. [Google Scholar] [CrossRef]
- Davies, J.; Randeva, H.S.; Chatha, K.; Hall, M.; Spandidos, D.A.; Karteris, E.; Kyrou, I. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol. Med. Rep. 2020, 22, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.; Pillat, M.M. CD147 as a target for COVID-19 treatment: Suggested effects of azithromycin and stem cell engagement. Stem Cell Rev. Rep. 2020, 16, 434–440. [Google Scholar] [PubMed]
- Istifli, E.S.; Tepe, A.Ş.; SarikÜrkcÜ, C.; Tepe, B. Interaction of certain monoterpenoid hydrocarbons with the receptor binding domain of 2019 novel coronavirus (2019-nCoV), transmembrane serine protease 2 (TMPRSS2), cathepsin B, and cathepsin L (CatB/L) and their pharmacokinetic properties. Turk. J. Biol. 2020, 44, 242–264. [Google Scholar]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Shabani, Z.; Schuerger, J.; Su, H. Cellular loci involved in the development of brain arteriovenous malformations. Front. Hum. Neurosci. 2022, 16, 968369. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Smith, K.; Yanek, L.; Amat-Alcaron, N.; Thiemann, D.R.; Cox, A.L.; Leucker, T.M.; Lowenstein, C.J. Markers of endothelial cell activation are associated with the severity of pulmonary disease in COVID-19. PLoS ONE 2022, 17, e0268296. [Google Scholar]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. Endothelial cell activation by sars-cov-2 spike s1 protein: A crosstalk between endothelium and innate immune cells. Biomedicines 2021, 9, 1220. [Google Scholar]
- Szekely, L.; Bozoky, B.; Bendek, M.; Ostad, M.; Lavignasse, P.; Haag, L.; Wu, J.; Jing, X.; Gupta, S.; Saccon, E. Pulmonary stromal expansion and intra-alveolar coagulation are primary causes of COVID-19 death. Heliyon 2021, 7, e07134. [Google Scholar] [PubMed]
- Bhatnagar, J.; Gary, J.; Reagan-Steiner, S.; Estetter, L.B.; Tong, S.; Tao, Y.; Denison, A.M.; Lee, E.; DeLeon-Carnes, M.; Li, Y. Evidence of severe acute respiratory syndrome coronavirus 2 replication and tropism in the lungs, airways, and vascular endothelium of patients with fatal coronavirus disease 2019: An autopsy case series. J. Infect. Dis. 2021, 223, 752–764. [Google Scholar]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.v.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2004, 203, 631–637. [Google Scholar]
- Chen, L.; Hao, G. The role of angiotensin-converting enzyme 2 in coronaviruses/influenza viruses and cardiovascular disease. Cardiovasc. Res. 2020, 116, 1932–1936. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Maccio, U.; Zinkernagel, A.S.; Shambat, S.M.; Zeng, X.; Cathomas, G.; Ruschitzka, F.; Schuepbach, R.A.; Moch, H.; Varga, Z. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine 2021, 63, 103182. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kanevsky, I.; Yildiz, S.; Sellers, R.S.; Swanson, K.A.; Franks, T.; Rathnasinghe, R.; Munoz-Moreno, R.; Jangra, S.; Gonzalez, O. Modeling SARS-CoV-2: Comparative pathology in rhesus macaque and Golden Syrian hamster models. Toxicol. Pathol. 2022, 50, 280–293. [Google Scholar] [PubMed]
- Adesse, D.; Gladulich, L.; Alvarez-Rosa, L.; Siqueira, M.; Marcos, A.C.; Heider, M.; Motta, C.S.; Torices, S.; Toborek, M.; Stipursky, J. Role of aging in Blood–Brain Barrier dysfunction and susceptibility to SARS-CoV-2 infection: Impacts on neurological symptoms of COVID-19. Fluids Barriers CNS 2022, 19, 63. [Google Scholar]
- Motta, C.S.; Torices, S.; da Rosa, B.G.; Marcos, A.C.; Alvarez-Rosa, L.; Siqueira, M.; Moreno-Rodriguez, T.; Matos, A.d.R.; Caetano, B.C.; Martins, J.S.C.d.C. Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling. Viruses 2023, 15, 745. [Google Scholar] [CrossRef]
- Qin, Z.; Liu, F.; Blair, R.; Wang, C.; Yang, H.; Mudd, J.; Currey, J.M.; Iwanaga, N.; He, J.; Mi, R. Endothelial cell infection and dysfunction, immune activation in severe COVID-19. Theranostics 2021, 11, 8076. [Google Scholar] [CrossRef]
- Werlein, C.; Ackermann, M.; Stark, H.; Shah, H.R.; Tzankov, A.; Haslbauer, J.D.; von Stillfried, S.; Bülow, R.D.; El-Armouche, A.; Kuenzel, S. Inflammation and vascular remodeling in COVID-19 hearts. Angiogenesis 2022, 26, 233–248. [Google Scholar]
- Potje, S.R.; Costa, T.J.; Fraga-Silva, T.F.; Martins, R.B.; Benatti, M.N.; Almado, C.E.; de Sa, K.S.; Bonato, V.L.; Arruda, E.; Louzada-Junior, P. Heparin prevents in vitro glycocalyx shedding induced by plasma from COVID-19 patients. Life Sci. 2021, 276, 119376. [Google Scholar]
- du Preez, H.N.; Aldous, C.; Hayden, M.R.; Kruger, H.G.; Lin, J. Pathogenesis of COVID-19 described through the lens of an undersulfated and degraded epithelial and endothelial glycocalyx. FASEB J. 2022, 36, e22052. [Google Scholar] [CrossRef]
- Targosz-Korecka, M.; Kubisiak, A.; Kloska, D.; Kopacz, A.; Grochot-Przeczek, A.; Szymonski, M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci. Rep. 2021, 11, 12157. [Google Scholar] [CrossRef]
- Vollenberg, R.; Tepasse, P.-R.; Ochs, K.; Floer, M.; Strauss, M.; Rennebaum, F.; Kabar, I.; Rovas, A.; Nowacki, T. Indications of persistent glycocalyx damage in convalescent COVID-19 patients: A prospective multicenter study and hypothesis. Viruses 2021, 13, 2324. [Google Scholar] [CrossRef] [PubMed]
- Langen, U.H.; Ayloo, S.; Gu, C. Development and cell biology of the blood-brain barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef] [PubMed]
- Bleau, C.; Filliol, A.; Samson, M.; Lamontagne, L. Brain invasion by mouse hepatitis virus depends on impairment of tight junctions and beta interferon production in brain microvascular endothelial cells. J. Virol. 2015, 89, 9896–9908. [Google Scholar]
- Alquisiras-Burgos, I.; Peralta-Arrieta, I.; Alonso-Palomares, L.A.; Zacapala-Gomez, A.E.; Salmeron-Barcenas, E.G.; Aguilera, P. Neurological complications associated with the blood-brain barrier damage induced by the inflammatory response during SARS-CoV-2 infection. Mol. Neurobiol. 2021, 58, 520–535. [Google Scholar]
- Yang, R.-C.; Huang, K.; Zhang, H.-P.; Li, L.; Zhang, Y.-F.; Tan, C.; Chen, H.-C.; Jin, M.-L.; Wang, X.-R. SARS-CoV-2 productively infects human brain microvascular endothelial cells. J. Neuroinflamm. 2022, 19, 149. [Google Scholar] [CrossRef]
- Almutairi, M.M.; Gong, C.; Xu, Y.G.; Chang, Y.; Shi, H. Factors controlling permeability of the blood–brain barrier. Cell. Mol. Life Sci. 2016, 73, 57–77. [Google Scholar] [CrossRef]
- Ranaivo, H.R.; Zunich, S.M.; Choi, N.; Hodge, J.N.; Wainwright, M.S. Mild stretch-induced injury increases susceptibility to interleukin-1β-induced release of matrix metalloproteinase-9 from astrocytes. J. Neurotrauma 2011, 28, 1757–1766. [Google Scholar] [CrossRef]
- Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the blood–brain barrier. Int. J. Mol. Sci. 2021, 22, 2681. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A. Effect of SARS-CoV-2 proteins on vascular permeability. Elife 2021, 10, e69314. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Neuroimmune axes of the blood–brain barriers and blood–brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Wang, Y.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J. 2006, 20, 1185. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.J.; Feigen, C.M.; Vazquez, J.P.; Kobets, A.J.; Altschul, D.J. Neurological sequelae of COVID-19. J. Integr. Neurosci. 2022, 21, 77. [Google Scholar] [CrossRef]
- Staekenborg, S.S.; Van der Flier, W.M.; Van Straaten, E.C.; Lane, R.; Barkhof, F.; Scheltens, P. Neurological signs in relation to type of cerebrovascular disease in vascular dementia. Stroke 2008, 39, 317–322. [Google Scholar] [CrossRef]
- Miners, S.; Kehoe, P.G.; Love, S. Cognitive impact of COVID-19: Looking beyond the short term. Alzheimer’s Res. Ther. 2020, 12, 1–16. [Google Scholar]
- Loo, J.; Spittle, D.A.; Newnham, M. COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax 2021, 76, 412–420. [Google Scholar]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 170. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar]
- Harris, A.G.; Skalak, T.C. Leukocyte cytoskeletal structure determines capillary plugging and network resistance. Am. J. Physiol.-Heart Circ. Physiol. 1993, 265, H1670–H1675. [Google Scholar] [CrossRef]
- Cruz Hernández, J.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, M.; Berg, M.; Park, L.; Vinarcsik, L.K.; Ivasyk, I.; Rivera, D.A. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, C.; Chen, Z.; Bankaitis, V.; Tzima, E.; Sheibani, N.; Burridge, K. Pericytes regulate vascular basement membrane remodeling and govern neutrophil extravasation during inflammation. PLoS ONE 2012, 7, e45499. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Zhang, Z.G.; Eliceiri, B.P.; Jiang, Q.; Boccia, A.D.; Zhang, R.L.; Chopp, M.; Cheresh, D.A. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 2001, 7, 222–227. [Google Scholar] [CrossRef]
- Liu, D.Z.; Ander, B.P.; Xu, H.; Shen, Y.; Kaur, P.; Deng, W.; Sharp, F.R. Blood–brain barrier breakdown and repair by Src after thrombin-induced injury. Ann. Neurol. 2010, 67, 526–533. [Google Scholar] [CrossRef]
- Tyagi, N.; Roberts, A.M.; Dean, W.L.; Tyagi, S.C.; Lominadze, D. Fibrinogen induces endothelial cell permeability. Mol. Cell. Biochem. 2008, 307, 13–22. [Google Scholar] [CrossRef]
- Trepat, X.; Grabulosa, M.; Buscemi, L.; Rico, F.; Farré, R.; Navajas, D. Thrombin and histamine induce stiffening of alveolar epithelial cells. J. Appl. Physiol. 2005, 98, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Yepes, M.; Sandkvist, M.; Moore, E.G.; Bugge, T.H.; Strickland, D.K.; Lawrence, D.A. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor–related protein. J. Clin. Investig. 2003, 112, 1533–1540. [Google Scholar] [CrossRef]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimer’s Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef]
- Shah, F.A.; Pike, F.; Alvarez, K.; Angus, D.; Newman, A.B.; Lopez, O.; Tate, J.; Kapur, V.; Wilsdon, A.; Krishnan, J.A. Bidirectional relationship between cognitive function and pneumonia. Am. J. Respir. Crit. Care Med. 2013, 188, 586–592. [Google Scholar] [CrossRef]
- Salive, M.E.; Satterfield, S.; Ostfeld, A.M.; Wallace, R.B.; Havlik, R.J. Disability and cognitive impairment are risk factors for pneumonia-related mortality in older adults. Public Health Rep. 1993, 108, 314. [Google Scholar]
- Herridge, M.S.; Moss, M.; Hough, C.L.; Hopkins, R.O.; Rice, T.W.; Bienvenu, O.J.; Azoulay, E. Recovery and outcomes after the acute respiratory distress syndrome (ARDS) in patients and their family caregivers. Intensive Care Med. 2016, 42, 725–738. [Google Scholar] [CrossRef]
- Hopkins, R.O.; Jackson, J.C. Long-term neurocognitive function after critical illness. Chest 2006, 130, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.A.; Snitz, B.E.; Alvarez, K.A.; Nahin, R.L.; Weissfeld, L.A.; Lopez, O.; Angus, D.C.; Shah, F.; Ives, D.G.; Fitzpatrick, A.L. Infection hospitalization increases risk of dementia in the elderly. Crit. Care Med. 2014, 42, 1037. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.E.; Brummel, N.E.; Archer, K.; Ely, E.W.; Jackson, J.C.; Hopkins, R.O. Cognitive dysfunction in ICU patients: Risk factors, predictors, and rehabilitation interventions. Crit. Care Med. 2013, 41, S81–S98. [Google Scholar] [CrossRef] [PubMed]
- Cervos-Navarro, J.; Diemer, N. Selective vulnerability in brain hypoxia. Crit. Rev. Neurobiol. 1991, 6, 149–182. [Google Scholar] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef]
- Yang, Y.; Rosenberg, G.A. Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef]
- Esch, T.; Stefano, G.B.; Ptacek, R.; Kream, R.M. Emerging roles of blood-borne intact and respiring mitochondria as bidirectional mediators of pro-and anti-inflammatory processes. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e924331–e924337. [Google Scholar] [CrossRef]
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol.-Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef] [PubMed]
- Ptacek, R.; Ptackova, H.; Martin, A.; Stefano, G.B. Psychiatric manifestations of COVID-19 and their social significance. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e930340–e930341. [Google Scholar] [CrossRef] [PubMed]
- Bostancıklıoğlu, M. SARS-CoV2 entry and spread in the lymphatic drainage system of the brain. Brain Behav. Immun. 2020, 87, 122. [Google Scholar] [CrossRef] [PubMed]
- Steardo, L.; Steardo Jr, L.; Zorec, R.; Verkhratsky, A. Neuroinfection may contribute to pathophysiology and clinical manifestations of COVID-19. Acta Physiol. 2020, 229, e13473. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Schweblin, C.; Lambeng, N.; Cherpillod, P.; Vazquez, J.; Lalive, P.H.; Schibler, M.; Deffert, C. Cerebrospinal fluid features in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reverse transcription polymerase chain reaction (RT-PCR) positive patients. Clin. Infect. Dis. 2021, 73, e3102–e3105. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Wang, L.; Zhang, L.; Qin, C.; Song, Y.; Ma, Y.; Chen, Y.; Chen, S.; Wang, Y.; Zhang, Z. Blood-brain barrier disruption induced cognitive impairment is associated with increase of inflammatory cytokine. Front. Aging Neurosci. 2018, 10, 129. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Insel, P.S.; Donohue, M.C.; Sperling, R.; Hansson, O.; Mattsson-Carlgren, N. The A4 study: β-amyloid and cognition in 4432 cognitively unimpaired adults. Ann. Clin. Transl. Neurol. 2020, 7, 776–785. [Google Scholar] [CrossRef]
- Ma, G.; Zhang, D.-F.; Zou, Q.-C.; Xie, X.; Xu, L.; Feng, X.-L.; Li, X.; Han, J.-B.; Yu, D.; Deng, Z.-H.; et al. SARS-CoV-2 Spike protein S2 subunit modulates γ-secretase and enhances amyloid-β production in COVID-19 neuropathy. Cell Discov. 2022, 8, 99. [Google Scholar] [CrossRef]
- Shen, W.-B.; Elahi, M.; Logue, J.; Yang, P.; Baracco, L.; Reece, E.A.; Wang, B.; Li, L.; Blanchard, T.G.; Han, Z.; et al. SARS-CoV-2 invades cognitive centers of the brain and induces Alzheimer’s-like neuropathology. BioRxiv 2022. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Abate, G.; Memo, M.; Uberti, D. Impact of COVID-19 on Alzheimer’s disease risk: Viewpoint for research action. MDPI 2020, 8, 286. [Google Scholar] [CrossRef]
- Patel, A.B.; Tsilioni, I.; Leeman, S.E.; Theoharides, T.C. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. USA 2016, 113, E7049–E7058. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Kempuraj, D. Role of SARS-CoV-2 Spike-Protein-Induced Activation of Microglia and Mast Cells in the Pathogenesis of Neuro-COVID. Cells 2023, 12, 688. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.M. Immunotherapies and COVID-19 related neurological manifestations: A comprehensive review article. J. Immunoass. Immunochem. 2020, 41, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Alnefeesi, Y.; Siegel, A.; Lui, L.M.; Teopiz, K.M.; Ho, R.; Lee, Y.; Nasri, F.; Gill, H.; Lin, K.; Cao, B. Impact of SARS-CoV-2 infection on cognitive function: A systematic review. Front. Psychiatry 2021, 11, 1629. [Google Scholar] [CrossRef]
- Gao, W.; Li, Y.; Zhang, T.; Lu, J.; Pan, J.; Qi, Q.; Dong, S.; Chen, X.; Su, Z.; Li, J. Systematic analysis of chemokines reveals CCL18 is a prognostic biomarker in glioblastoma. J. Inflamm. Res. 2022, 15, 2731–2743. [Google Scholar] [CrossRef]
- Kolodziej, A.; Schulz, S.; Guyon, A.; Wu, D.-F.; Pfeiffer, M.; Odemis, V.; Höllt, V.; Stumm, R. Tonic activation of CXC chemokine receptor 4 in immature granule cells supports neurogenesis in the adult dentate gyrus. J. Neurosci. 2008, 28, 4488–4500. [Google Scholar] [CrossRef] [PubMed]
- Senf, K.; Karius, J.; Stumm, R.; Neuhaus, E.M. Chemokine signaling is required for homeostatic and injury-induced neurogenesis in the olfactory epithelium. Stem Cells 2021, 39, 617–635. [Google Scholar] [CrossRef] [PubMed]
- Karimabad, M.N.; Hassanshahi, G.; Kounis, N.G.; Mplani, V.; Roditis, P.; Gogos, C.; Lagadinou, M.; Assimakopoulos, S.F.; Dousdampanis, P.; Koniari, I. The Chemokines CXC, CC and C in the Pathogenesis of COVID-19 Disease and as Surrogates of Vaccine-Induced Innate and Adaptive Protective Responses. Vaccines 2022, 10, 1299. [Google Scholar] [CrossRef]
- Yasui, H.; Kajiyama, H.; Tamauchi, S.; Suzuki, S.; Peng, Y.; Yoshikawa, N.; Sugiyama, M.; Nakamura, K.; Kikkawa, F. CCL2 secreted from cancer-associated mesothelial cells promotes peritoneal metastasis of ovarian cancer cells through the P38-MAPK pathway. Clin. Exp. Metastasis 2020, 37, 145–158. [Google Scholar] [CrossRef]
- Parajuli, B.; Horiuchi, H.; Mizuno, T.; Takeuchi, H.; Suzumura, A. CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia. Glia 2015, 63, 2274–2284. [Google Scholar] [CrossRef] [PubMed]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. Mechanistic insights into Long COVID in hamsters. Nat. Rev. Immunol. 2022, 22, 463. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lee, E.E.; Martin, A.S.; Soontornniyomkij, B.; Soontornniyomkij, V.; Achim, C.L.; Reuter, C.; Irwin, M.R.; Eyler, L.T.; Jeste, D.V. Abnormalities in chemokine levels in schizophrenia and their clinical correlates. Schizophr. Res. 2017, 181, 63–69. [Google Scholar] [CrossRef]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.-H.; Wood, J.; Yalçın, B.; Taylor, K.R.; Dutton, S.; Acosta-Alvarez, L. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. bioRxiv 2022. [Google Scholar] [CrossRef]
- Xu, J.; Dong, H.; Qian, Q.; Zhang, X.; Wang, Y.; Jin, W.; Qian, Y. Astrocyte-derived CCL2 participates in surgery-induced cognitive dysfunction and neuroinflammation via evoking microglia activation. Behav. Brain Res. 2017, 332, 145–153. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 111–118. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shabani, Z.; Liu, J.; Su, H. Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19. Biology 2023, 12, 1106. https://doi.org/10.3390/biology12081106
Shabani Z, Liu J, Su H. Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19. Biology. 2023; 12(8):1106. https://doi.org/10.3390/biology12081106
Chicago/Turabian StyleShabani, Zahra, Jialing Liu, and Hua Su. 2023. "Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19" Biology 12, no. 8: 1106. https://doi.org/10.3390/biology12081106
APA StyleShabani, Z., Liu, J., & Su, H. (2023). Vascular Dysfunctions Contribute to the Long-Term Cognitive Deficits Following COVID-19. Biology, 12(8), 1106. https://doi.org/10.3390/biology12081106