


Targeting Endothelial HIF2α/ARNT Expression for Ischemic Heart Disease Therapy

Abstract
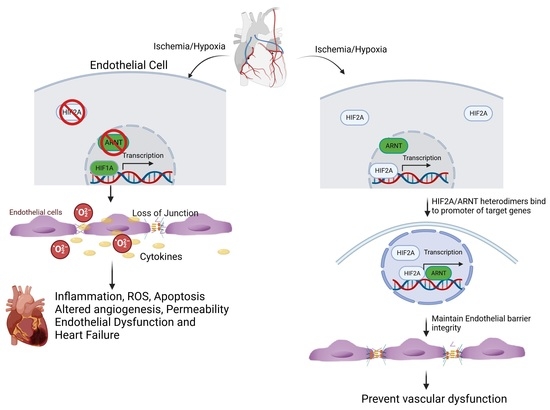
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Activation of HIF during Hypoxia
3. Role of HIF2α and ARNT in Vascular Endothelial Cells and Inflammation
3.1. HIF2a Expression and Inflammation
3.2. The Role of ARNT in Heterodimeric Transcription Factors and Endothelial Cell Function
3.3. The Anti-Inflammatory and Antioxidative Role of ARNT
4. Potential Role of HIF2α and ARNT in Ischemic Heart Disease
4.1. HIF2α and ARNT in Mouse Heart Development and Cardiovascular Function
4.2. Cardioprotective Potential of ARNT and HIF2α
5. Future Perspectives and Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopez-Barneo, J.; Pardal, R.; Ortega-Saenz, P. Cellular mechanism of oxygen sensing. Annu. Rev. Physiol. 2001, 63, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Ferdinand, P.; Roffe, C. Hypoxia after stroke: A review of experimental and clinical evidence. Exp. Transl. Stroke Med. 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Cornet, A.D.; Kooter, A.J.; Peters, M.J.; Smulders, Y.M. The potential harm of oxygen therapy in medical emergencies. Crit. Care 2013, 17, 313. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C.; Arnould, T.; Remacle, J. Endothelial cell responses to hypoxia: Initiation of a cascade of cellular interactions. Biochim. Biophys. Acta 2000, 1497, 1–10. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Wu, D.; Rastinejad, F. Structural characterization of mammalian bHLH-PAS transcription factors. Curr. Opin. Struct. Biol. 2017, 43, 1–9. [Google Scholar] [CrossRef]
- Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouyssegur, J. A dialogue between the hypoxia-inducible factor and the tumor microenvironment. Cancer Microenviron. 2008, 1, 53–68. [Google Scholar] [CrossRef]
- Downes, N.L.; Laham-Karam, N.; Kaikkonen, M.U.; Yla-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and characterization of hypoxia-inducible factor (HIF)-3α in human kidney: Suppression of HIF-mediated gene expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef]
- Imamura, T.; Kikuchi, H.; Herraiz, M.T.; Park, D.Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1α and HIF-2α have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Kruger, J.; Maierhofer, A.; Bottcher, Y.; Kloting, N.; El Hajj, N.; Schleinitz, D.; Schon, M.R.; Dietrich, A.; Fasshauer, M.; et al. Hypoxia-inducible factor 3A gene expression and methylation in adipose tissue is related to adipose tissue dysfunction. Sci. Rep. 2016, 6, 27969. [Google Scholar] [CrossRef]
- Huang, Y.; Hickey, R.P.; Yeh, J.L.; Liu, D.; Dadak, A.; Young, L.H.; Johnson, R.S.; Giordano, F.J. Cardiac myocyte-specific HIF-1α deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 2004, 18, 1138–1140. [Google Scholar] [CrossRef]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. bHLH-PAS proteins in cancer. Nat. Rev. Cancer 2013, 13, 827–841. [Google Scholar] [CrossRef]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar] [PubMed]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsila, M.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1α and HIF-2α expression in lung epithelial cells: Implication of natural antisense HIF-1α. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef]
- Scortegagna, M.; Morris, M.A.; Oktay, Y.; Bennett, M.; Garcia, J.A. The HIF family member EPAS1/HIF-2α is required for normal hematopoiesis in mice. Blood 2003, 102, 1634–1640. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Bartels, K.; Grenz, A.; Eltzschig, H.K. Hypoxia and inflammation are two sides of the same coin. Proc. Natl. Acad. Sci. USA 2013, 110, 18351–18352. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Invest. 2014, 124, 2396–2409. [Google Scholar] [CrossRef]
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-inducible factor 2-α-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat. Commun. 2018, 9, 816. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef]
- Pasupneti, S.; Tian, W.; Tu, A.B.; Dahms, P.; Granucci, E.; Gandjeva, A.; Xiang, M.; Butcher, E.C.; Semenza, G.L.; Tuder, R.M.; et al. Endothelial HIF-2α as a Key Endogenous Mediator Preventing Emphysema. Am. J. Respir. Crit. Care Med. 2020, 202, 983–995. [Google Scholar] [CrossRef]
- Zeng, H.; He, X.; Tuo, Q.H.; Liao, D.F.; Zhang, G.Q.; Chen, J.X. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci. Rep. 2016, 6, 20931. [Google Scholar] [CrossRef]
- Gong, H.; Rehman, J.; Tang, H.; Wary, K.; Mittal, M.; Chaturvedi, P.; Zhao, Y.Y.; Komarova, Y.A.; Vogel, S.M.; Malik, A.B. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Invest. 2015, 125, 652–664. [Google Scholar] [CrossRef]
- Wei, T.; Gao, J.; Huang, C.; Song, B.; Sun, M.; Shen, W. SIRT3 (Sirtuin-3) Prevents Ang II (Angiotensin II)-Induced Macrophage Metabolic Switch Improving Perivascular Adipose Tissue Function. Arter. Thromb. Vasc. Biol. 2021, 41, 714–730. [Google Scholar] [CrossRef]
- Zhang, C.; Li, N.; Suo, M.; Zhang, C.; Liu, J.; Liu, L.; Qi, Y.; Zheng, X.; Xie, L.; Hu, Y.; et al. Sirtuin 3 deficiency aggravates angiotensin II-induced hypertensive cardiac injury by the impairment of lymphangiogenesis. J. Cell. Mol. Med. 2021, 25, 7760–7771. [Google Scholar] [CrossRef]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Kohler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin α/β pathway. Biochim. Biophys. Acta 2008, 1783, 394–404. [Google Scholar] [CrossRef]
- Hoffman, E.C.; Reyes, H.; Chu, F.F.; Sander, F.; Conley, L.H.; Brooks, B.A.; Hankinson, O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 1991, 252, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Ullah, K.; Wu, R. Hypoxia-Inducible Factor Regulates Endothelial Metabolism in Cardiovascular Disease. Front. Physiol. 2021, 12, 670653. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Zheng, M.; Knapp, M.; Sladojevic, N.; Zhang, Q.; Ai, L.; Harrison, D.; Chen, A.; Sitikov, A.; Shen, L.; et al. Endothelial Aryl Hydrocarbon Receptor Nuclear Translocator Mediates the Angiogenic Response to Peripheral Ischemia in Mice With Type 2 Diabetes Mellitus. Front. Cell Dev. Biol. 2021, 9, 691801. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.M.; Gleadle, J.M.; Pugh, C.W.; Hankinson, O.; Ratcliffe, P.J. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J. Biol. Chem. 1996, 271, 15117–15123. [Google Scholar] [CrossRef] [PubMed]
- Maltepe, E.; Schmidt, J.V.; Baunoch, D.; Bradfield, C.A.; Simon, M.C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature 1997, 386, 403–407. [Google Scholar] [CrossRef]
- Han, Y.; Yang, K.; Proweller, A.; Zhou, G.; Jain, M.K.; Ramirez-Bergeron, D.L. Inhibition of ARNT severely compromises endothelial cell viability and function in response to moderate hypoxia. Angiogenesis 2012, 15, 409–420. [Google Scholar] [CrossRef]
- Libby, P. Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nutr. 2006, 83, 456S–460S. [Google Scholar] [CrossRef]
- Black, P.H.; Garbutt, L.D. Stress, inflammation and cardiovascular disease. J. Psychosom. Res. 2002, 52, 1–23. [Google Scholar] [CrossRef]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef]
- Øvrevik, J.; Låg, M.; Lecureur, V.; Gilot, D.; Lagadic-Gossmann, D.; Refsnes, M.; Schwarze, P.E.; Skuland, T.; Becher, R.; Holme, J.A. AhR and Arnt differentially regulate NF-κB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun. Signal. 2014, 12, 48. [Google Scholar] [CrossRef]
- Wright, C.W.; Duckett, C.S. The Aryl Hydrocarbon Nuclear Translocator Alters CD30-Mediated NF-κB—Dependent Transcription. Science 2009, 323, 251–255. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Scott, C.; Stokes, R.; Cha, K.M.; Clouston, A.; Eslam, M.; Metwally, M.; Swarbrick, M.M.; George, J.; Gunton, J.E. Myeloid cell deletion of Aryl hydrocarbon Receptor Nuclear Translocator (ARNT) induces non-alcoholic steatohepatitis. PLoS ONE 2019, 14, e0225332. [Google Scholar] [CrossRef]
- Glover, L.; Irizarry, K.; Scully, M.; Campbell, E.; Bowers, B.; Aherne, C.; Kominsky, D.; Macmanus, C.; Colgan, S. IFN- Attenuates Hypoxia-Inducible Factor (HIF) Activity in Intestinal Epithelial Cells through Transcriptional Repression of HIF-1β. J. Immunol. 2011, 186, 1790–1798. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Ishihara, Y.; Campbell, C.E.; Kado, S.Y.; Nguyen-Chi, A.; Sweeney, C.; Pollet, M.; Haarmann-Stemmann, T.; Tuscano, J.M. A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth. Cancers 2019, 11, 589. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor repressor—More than a simple feedback inhibitor of AhR signaling: Clues for its role in inflammation and cancer. Curr. Opin. Toxicol. 2017, 2, 109–119. [Google Scholar] [CrossRef]
- Shieh, J.-M.; Shen, C.-J.; Chang, W.-C.; Cheng, H.-C.; Chan, Y.-Y.; Huang, W.-C.; Chang, W.-C.; Chen, B.-K. An Increase in Reactive Oxygen Species by Deregulation of ARNT Enhances Chemotherapeutic Drug-Induced Cancer Cell Death. PLoS ONE 2014, 9, e99242. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Belmadani, S.; Wu, J.; Xu, X.; Elford, H.; Potter, B.J.; Zhang, C.; Lee, S.; Chen, J.; et al. Role of TNF-α-induced reactive oxygen species in endothelial dysfunction during reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2242–H2249. [Google Scholar] [CrossRef]
- Benincasa, G.; Coscioni, E.; Napoli, C. Cardiovascular risk factors and molecular routes underlying endothelial dysfunction: Novel opportunities for primary prevention. Biochem. Pharmacol. 2022, 202, 115108. [Google Scholar] [CrossRef]
- He, F.; Zuo, L. Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases. Int. J. Mol. Sci. 2015, 16, 27770–27780. [Google Scholar] [CrossRef] [PubMed]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef] [PubMed]
- Sugamura, K.; Keaney, J.F. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-R.; Chang, T.-W.; Lee, C.-T.; Shen, C.-J.; Chang, W.-C.; Chen, B.-K. ARNT deficiency represses pyruvate dehydrogenase kinase 1 to trigger ROS production and melanoma metastasis. Oncogenesis 2021, 10, 11. [Google Scholar] [CrossRef]
- Gu, C.; Gonzalez, J.; Zhang, T.; Kamel-Reid, S.; Wells, R.A. The aryl hydrocarbon receptor nuclear translocator (ARNT) modulates the antioxidant response in AML cells. Leuk. Res. 2013, 37, 1750–1756. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Dunwoodie, S.L. The role of hypoxia in development of the Mammalian embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef]
- Duan, L.J.; Zhang-Benoit, Y.; Fong, G.H. Endothelium-intrinsic requirement for Hif-2α during vascular development. Circulation 2005, 111, 2227–2232. [Google Scholar] [CrossRef]
- Patterson, A.J.; Zhang, L. Hypoxia and fetal heart development. Curr. Mol. Med. 2010, 10, 653–666. [Google Scholar] [CrossRef]
- Aitola, M.H.; Pelto-Huikko, M.T. Expression of Arnt and Arnt2 mRNA in developing murine tissues. J. Histochem. Cytochem. 2003, 51, 41–54. [Google Scholar] [CrossRef]
- Xu, B.; Doughman, Y.; Turakhia, M.; Jiang, W.; Landsettle, C.E.; Agani, F.H.; Semenza, G.L.; Watanabe, M.; Yang, Y.C. Partial rescue of defects in Cited2-deficient embryos by HIF-1α heterozygosity. Dev. Biol. 2007, 301, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, E.J.; Bischoff, J. Heart valve development: Endothelial cell signaling and differentiation. Circ. Res. 2004, 95, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Tomanek, R.J. Formation of the coronary vasculature during development. Angiogenesis 2005, 8, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Licht, A.H.; Muller-Holtkamp, F.; Flamme, I.; Breier, G. Inhibition of hypoxia-inducible factor activity in endothelial cells disrupts embryonic cardiovascular development. Blood 2006, 107, 584–590. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell Biol. 2016, 36, 1584–1594. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, L.; Drysdale, L.; Fong, G.H. The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 8386–8391. [Google Scholar] [CrossRef]
- Bishop, T.; Ratcliffe, P.J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 2015, 117, 65–79. [Google Scholar] [CrossRef]
- Jiang, X.; Tian, W.; Tu, A.B.; Pasupneti, S.; Shuffle, E.; Dahms, P.; Zhang, P.; Cai, H.; Dinh, T.T.; Liu, B.; et al. Endothelial Hypoxia-Inducible Factor-2α Is Required for the Maintenance of Airway Microvasculature. Circulation 2019, 139, 502–517. [Google Scholar] [CrossRef]
- Abe, H.; Semba, H.; Takeda, N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J. Atheroscler. Thromb. 2017, 24, 884–894. [Google Scholar] [CrossRef]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Invest. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef]
- Chen, L.; Endler, A.; Uchida, K.; Horiguchi, S.; Morizane, Y.; Iijima, O.; Toi, M.; Shibasaki, F.; Aarup, A.; Pedersen, T.X.; et al. Int6/eIF3e silencing promotes functional blood vessel outgrowth and enhances wound healing by upregulating hypoxia-induced factor 2α expression. Circulation 2010, 122, 910–919. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, X.; Cui, H.; Shi, J.; Yuan, G.; Shi, S.; Hu, Y. The Role of the VEGF Family in Coronary Heart Disease. Front. Cardiovasc. Med. 2021, 8, 738325. [Google Scholar] [CrossRef]
- Ullah, K.; Ai, L.; Li, Y.; Liu, L.; Zhang, Q.; Pan, K.; Humayun, Z.; Sitikov, A.; Su, Q.; Zhao, Q.; et al. A Novel HIF-2α/ARNT Signaling Pathway Protects Against Microvascular Dysfunction and heart failure After Myocardial Infarction. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lähteenvuo, J.; Rosenzweig, A. Effects of aging on angiogenesis. Circ. Res. 2012, 110, 1252–1264. [Google Scholar] [CrossRef]
- Moriya, J.; Minamino, T. Angiogenesis, Cancer, and Vascular Aging. Front. Cardiovasc. Med. 2017, 4, 65. [Google Scholar] [CrossRef]
- Khurana, R.; Simons, M.; Martin, J.F.; Zachary, I.C. Role of angiogenesis in cardiovascular disease: A critical appraisal. Circulation 2005, 112, 1813–1824. [Google Scholar] [CrossRef]
- Liao, Y.-Y.; Chen, Z.-Y.; Wang, Y.-X.; Lin, Y.; Yang, F.; Zhou, Q.-L. New Progress in Angiogenesis Therapy of Cardiovascular Disease by Ultrasound Targeted Microbubble Destruction. BioMed Res. Int. 2014, 2014, 872984. [Google Scholar] [CrossRef]
- Wondimu, A.; Weir, L.; Robertson, D.; Mezentsev, A.; Kalachikov, S.; Panteleyev, A.A. Loss of Arnt (Hif1β) in mouse epidermis triggers dermal angiogenesis, blood vessel dilation and clotting defects. Lab. Investig. 2012, 92, 110–124. [Google Scholar] [CrossRef]
- Han, Y.; Tao, J.; Gomer, A.; Ramirez-Bergeron, D.L. Loss of endothelial-ARNT in adult mice contributes to dampened circulating proangiogenic cells and delayed wound healing. Vasc. Med. 2014, 19, 429–441. [Google Scholar] [CrossRef]
- Carreira, V.S.; Fan, Y.; Kurita, H.; Wang, Q.; Ko, C.-I.; Naticchioni, M.; Jiang, M.; Koch, S.; Zhang, X.; Biesiada, J.; et al. Disruption of Ah Receptor Signaling during Mouse Development Leads to Abnormal Cardiac Structure and Function in the Adult. PLoS ONE 2015, 10, e0142440. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.H.; Shah, Y.; Tomita, S.; Morris, H.D.; Gavrilova, O.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Disruption of the Arnt gene in endothelial cells causes hepatic vascular defects and partial embryonic lethality in mice. Hepatology 2006, 44, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.E.; Souder, J.P.; Gorelick, D.A. Hemato-vascular specification requires arnt1 and arnt2 genes in zebrafish embryos. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Kozak, K.R.; Abbott, B.; Hankinson, O. ARNT-Deficient Mice and Placental Differentiation. Dev. Biol. 1997, 191, 297–305. [Google Scholar] [CrossRef]
- Su, E.J.; Xin, H.; Yin, P.; Dyson, M.; Coon, J.; Farrow, K.N.; Mestan, K.K.; Ernst, L.M. Impaired Fetoplacental Angiogenesis in Growth-Restricted Fetuses With Abnormal Umbilical Artery Doppler Velocimetry Is Mediated by Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT). J. Clin. Endocrinol. Metab. 2015, 100, E30–E40. [Google Scholar] [CrossRef]
- Wu, R.; Chang, H.C.; Khechaduri, A.; Chawla, K.; Tran, M.; Chai, X.; Wagg, C.; Ghanefar, M.; Jiang, X.; Bayeva, M.; et al. Cardiac-specific ablation of ARNT leads to lipotoxicity and cardiomyopathy. J. Clin. Invest. 2014, 124, 4795–4806. [Google Scholar] [CrossRef]
- Knapp, M.; Zheng, M.; Sladojevic, N.; Zhao, Q.; Liao, J.K.; Wu, R. Abstract 20699: Reduction of Endothelial Arnt Mediates Vascular Dysfunction in Diabetes. Circulation 2016, 134, A20699. [Google Scholar]
- Nyamsuren, G.; Rapp, G.; Dihazi, H.; Zeisberg, E.M.; Tampe, D.; Tampe, B.; Zeisberg, M. PP2A phosphatase inhibition is anti-fibrotic through Ser77 phosphorylation-mediated ARNT/ARNT homodimer formation. Sci. Rep. 2021, 11, 24075. [Google Scholar] [CrossRef]
- Allison, S.J. Targeting ARNT to attenuate renal fibrosis. Nat. Rev. Nephrol. 2018, 14, 535. [Google Scholar] [CrossRef]
- Tampe, B.; Tampe, D.; Nyamsuren, G.; Klöpper, F.; Rapp, G.; Kauffels, A.; Lorf, T.; Zeisberg, E.M.; Müller, G.A.; Kalluri, R.; et al. Pharmacological induction of hypoxia-inducible transcription factor ARNT attenuates chronic kidney failure. J. Clin. Investig. 2018, 128, 3053–3070. [Google Scholar] [CrossRef]
- Bernhardt, W.M.; Campean, V.; Kany, S.; Jurgensen, J.S.; Weidemann, A.; Warnecke, C.; Arend, M.; Klaus, S.; Gunzler, V.; Amann, K.; et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol. 2006, 17, 1970–1978. [Google Scholar] [CrossRef]
- Hill, P.; Shukla, D.; Tran, M.G.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 39–46. [Google Scholar] [CrossRef]
- Eckle, T.; Kohler, D.; Lehmann, R.; El Kasmi, K.; Eltzschig, H.K. Hypoxia-inducible factor-1 is central to cardioprotection: A new paradigm for ischemic preconditioning. Circulation 2008, 118, 166–175. [Google Scholar] [CrossRef]
- Bautista, L.; Castro, M.J.; Lopez-Barneo, J.; Castellano, A. Hypoxia inducible factor-2α stabilization and maxi-K+ channel beta1-subunit gene repression by hypoxia in cardiac myocytes: Role in preconditioning. Circ. Res. 2009, 104, 1364–1372. [Google Scholar] [CrossRef]
- Hyvarinen, J.; Hassinen, I.E.; Sormunen, R.; Maki, J.M.; Kivirikko, K.I.; Koivunen, P.; Myllyharju, J. Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J. Biol. Chem. 2010, 285, 13646–13657. [Google Scholar] [CrossRef]
- Motta, S.; Minici, C.; Corrada, D.; Bonati, L.; Pandini, A. Ligand-induced perturbation of the HIF-2α:ARNT dimer dynamics. PLoS Comput. Biol. 2018, 14, e1006021. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, K.; Ai, L.; Humayun, Z.; Wu, R. Targeting Endothelial HIF2α/ARNT Expression for Ischemic Heart Disease Therapy. Biology 2023, 12, 995. https://doi.org/10.3390/biology12070995
Ullah K, Ai L, Humayun Z, Wu R. Targeting Endothelial HIF2α/ARNT Expression for Ischemic Heart Disease Therapy. Biology. 2023; 12(7):995. https://doi.org/10.3390/biology12070995
Chicago/Turabian StyleUllah, Karim, Lizhuo Ai, Zainab Humayun, and Rongxue Wu. 2023. "Targeting Endothelial HIF2α/ARNT Expression for Ischemic Heart Disease Therapy" Biology 12, no. 7: 995. https://doi.org/10.3390/biology12070995
APA StyleUllah, K., Ai, L., Humayun, Z., & Wu, R. (2023). Targeting Endothelial HIF2α/ARNT Expression for Ischemic Heart Disease Therapy. Biology, 12(7), 995. https://doi.org/10.3390/biology12070995