



TIME Is Ticking for Cervical Cancer

Abstract
Simple Summary
Abstract
1. Introduction
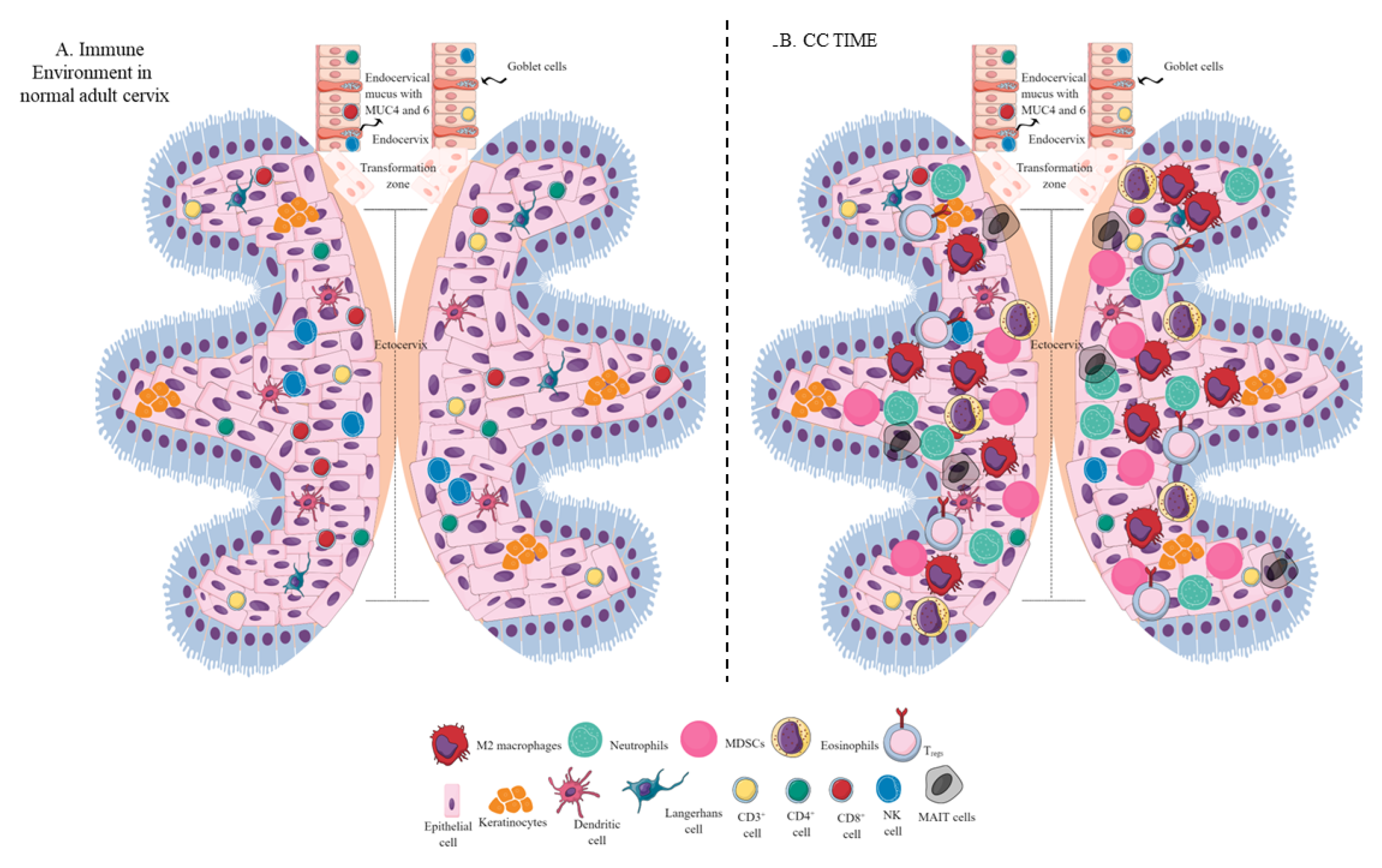
2. The Immune Environment of the Healthy Cervix
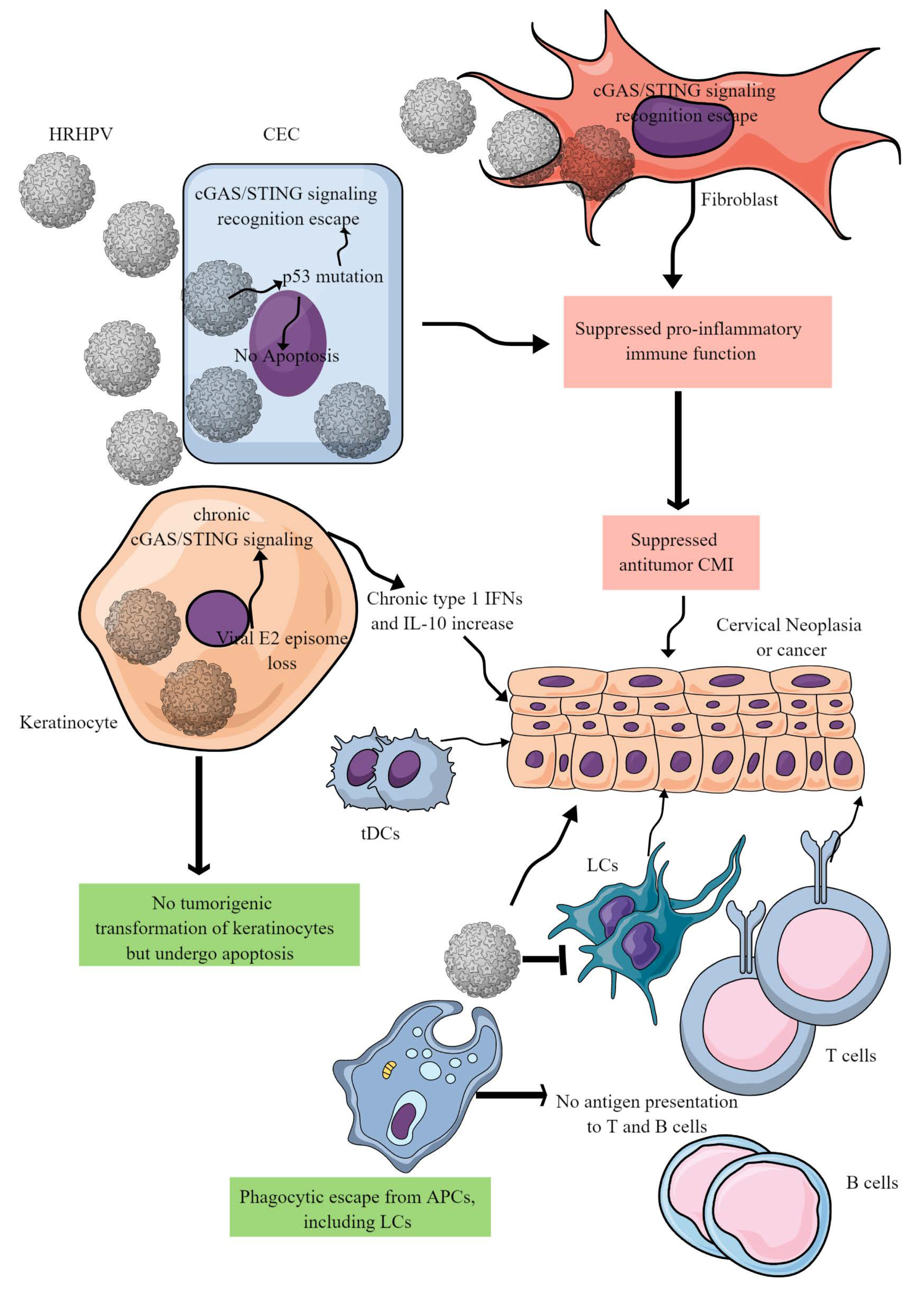
3. CC Immunopathogenesis
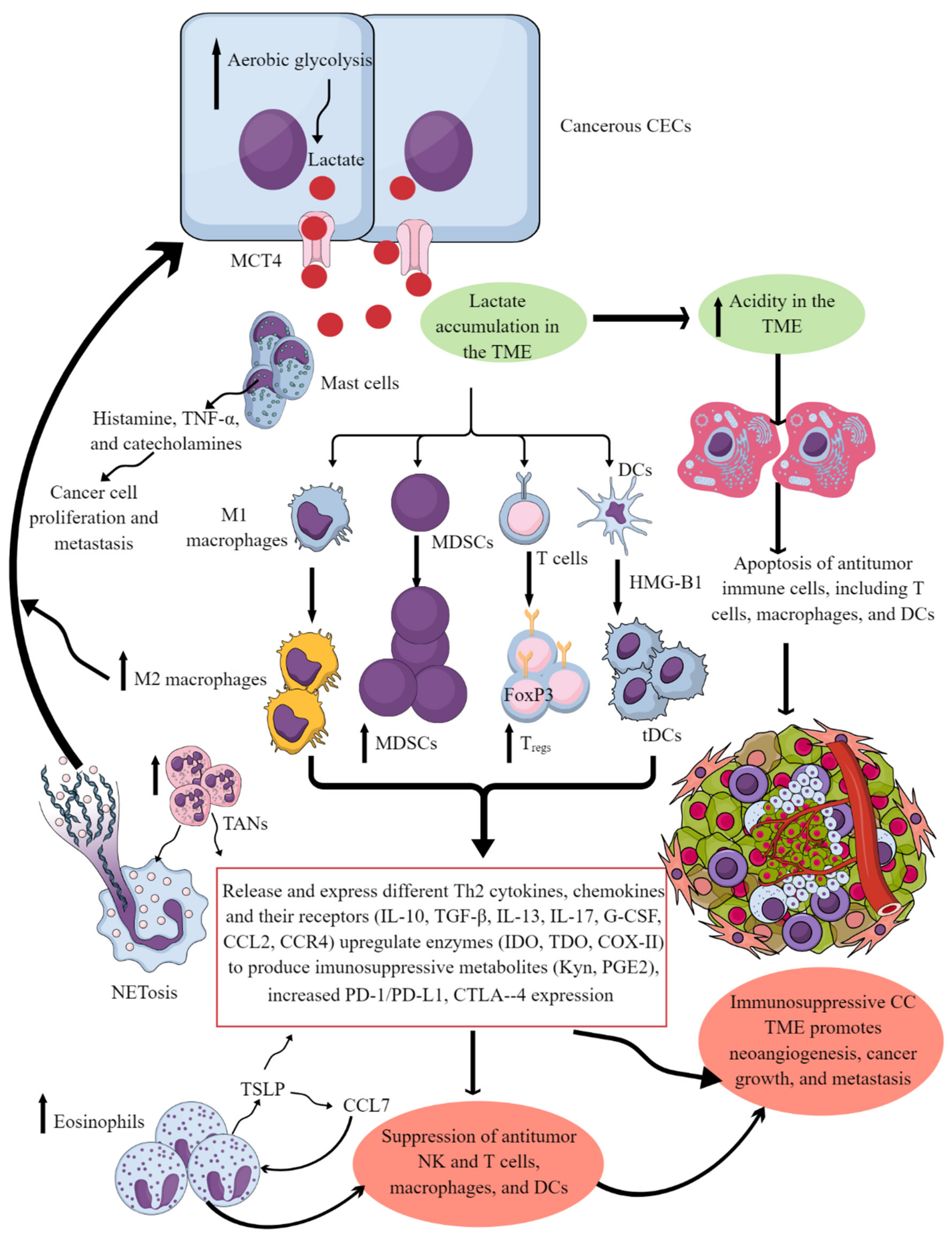
4. Factors Regulating CC Immunosuppressive TIME
5. Immune Cell Populations in the CC TIME
5.1. Tumor-Associated Macrophages (TAMs)
5.2. Neutrophils in the CC TIME
5.3. MDSCs in the CC TIME
5.4. MAIT Cells in the CC TIME
5.5. Mast Cells in the CC TIME
5.6. Eosinophils in the CC TIME
5.7. DCs in the CC TIME
5.8. NK Cells in the CC TIME
5.9. T Cells in the CC TIME
5.10. B Cells in the CC TIME
6. Targeting the CC TIME
7. Future Perspective and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AC | Adenocarcinoma |
| ACTS | Adoptive T-cell therapies |
| AHR | Aryl hydrocarbon receptor |
| AIF | Apoptosis inducible factor |
| AIM-2 | Absent in melanoma-2 |
| AMP | Antimicrobial peptides |
| APC | Antigen-presenting cell |
| BECs | Basal epithelial cells |
| CAC | Cervical adenocarcinoma |
| CAR-T cell | Chimeric antigen receptor T cell |
| CC | Cervical cancer |
| CECs | Cervical epithelial cells |
| cGAMP | Cyclic GMP-AMP |
| cGAS | cGMP-AMP synthase |
| CIN | Cervical intraepithelial neoplasia |
| CLR | C-type lectin receptors |
| CMI | Cell-mediated immunity |
| CTL | Cytotoxic T lymphocyte |
| DNAM-1 | DNAX accessory molecule-1 |
| DGE | Differential gene expression |
| FoxP3 | Forkhead box protein P3 |
| FRT | Female reproductive tract |
| HIF | Hypoxia-inducible factor |
| HR-HPV | High-risk HPV |
| hTERT | Human telomeres reverse transcriptase |
| ICIs | Immune checkpoint inhibitors |
| IDO | Indolamine dioxygenase |
| LCs | Langerhans cells |
| MAIT | Mucosal-associated invariant T |
| MIP | Macrophage inflammatory protein |
| MUC | Mucin |
| NET | Neutrophil extracellular trap |
| NKCC | NK-cell-mediated toxicity |
| NKG2D | Natural killer group 2D |
| NLR | Neutrophil lymphocyte ratio |
| NOD | Nucleotide-binding oligomerization domain |
| PRR | Pattern-recognition receptor |
| RGS | Regulator of G protein signaling |
| RICK | RIP-like interacting CLARP kinase |
| SCC | Squamous cell carcinoma |
| SIL | Squamous intraepithelial lesion |
| STING | Stimulating interferon genes |
| TCR | T-cell receptor |
| TCR-T cells | TCR-engineered/modified effector T cells |
| TDO | Tryptophan dioxygenase |
| TIME | Tumor immune microenvironment |
| TME | Tumor microenvironment |
| TGF-β | Transforming growth factor beta |
| TSLP | Thymic stromal lymphopoietin |
| TZ | Transformational Zone |
| VISTA | V-domain immunoglobulin suppressor of T cell activation |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Brey, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tantengco, O.A.G.; Menon, R. Contractile function of the cervix plays a role in normal and pathological pregnancy and parturition. Med. Hypotheses 2020, 145, 110336. [Google Scholar] [CrossRef] [PubMed]
- Francoeur, A.A.; Liao, C.-I.; Casear, M.A.; Chan, A.; Kapp, D.S.; Cohen, J.G.; Salani, R.; Chan, J.K. The increasing incidence of stage IV cervical cancer in the USA: What factors are related? Int. J. Gynecol. Cancer 2022, 33, 136. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.K.; Johansson, M.E.V. The role of goblet cells and mucus in intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 785–803. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Gallagher, A.L.; Grencis, R.K.; Thornton, D.J. A new role for mucins in immunity: Insights from gastrointestinal nematode infection. Int. J. Biochem. Cell Biol. 2013, 45, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, C.J.; Kim, D.-J.; Kang, J.-H. Immune Cells in the Female Reproductive Tract. Immune Netw. 2015, 15, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Taub, R.; Jensen, J.T. Cervical mucus and contraception: What we know and what we don’t. Contraception 2017, 96, 310–321. [Google Scholar] [CrossRef]
- Gipson, I.K. Mucins of the human endocervix. Front. Biosci. 2001, 6, D1245–D1255. [Google Scholar] [CrossRef]
- Gipson, I.K.; Spurr-Michaud, S.; Moccia, R.; Zhan, Q.; Toribara, N.; Ho, S.B.; Gargiulo, A.R.; Hill, J.A., 3rd. MUC4 and MUC5B transcripts are the prevalent mucin messenger ribonucleic acids of the human endocervix. Biol. Reprod. 1999, 60, 58–64. [Google Scholar] [CrossRef]
- Linden, S.K.; Sutton, P.; Karlsson, N.G.; Korolik, V.; McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008, 1, 183–197. [Google Scholar] [CrossRef]
- Sheng, Y.H.; Hasnain, S.Z. Mucus and Mucins: The Underappreciated Host Defence System. Front. Cell. Infect. Microbiol. 2022, 12, 856962. [Google Scholar] [CrossRef]
- Nasu, K.; Narahara, H. Pattern Recognition via the Toll-Like Receptor System in the Human Female Genital Tract. Mediat. Inflamm. 2010, 2010, 976024. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M.; Murphy, A.J.; Barrett, K.T.; Wira, C.R.; Guyre, P.M.; Pioli, P.A. Functional expression of pattern recognition receptors in tissues of the human female reproductive tract. J. Reprod. Immunol. 2009, 80, 33–40. [Google Scholar] [CrossRef] [PubMed]
- De Tomasi, J.B.; Opata, M.M.; Mowa, C.N. Immunity in the Cervix: Interphase between Immune and Cervical Epithelial Cells. J. Immunol. Res. 2019, 2019, 7693183. [Google Scholar] [CrossRef]
- Farage, M.A.; Miller, K.W.; Gerberick, G.F.; Saito, F.; Ledger, W.; Witkin, S. Innate immunity in the lower female mucosal tract. J. Steroids Horm. Sci. 2011, 2, 106. [Google Scholar] [CrossRef]
- Quayle, A.J. The innate and early immune response to pathogen challenge in the female genital tract and the pivotal role of epithelial cells. J. Reprod. Immunol. 2002, 57, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Fichorova, R.N.; Anderson, D.J. Differential expression of immunobiological mediators by immortalized human cervical and vaginal epithelial cells. Biol. Reprod. 1999, 60, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Wira, C.R.; Grant-Tschudy, K.S.; Crane-Godreau, M.A. Epithelial cells in the female reproductive tract: A central role as sentinels of immune protection. Am. J. Reprod. Immunol. 2005, 53, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Inki, P. Expression of syndecan-1 in female reproductive tract tissues and cultured keratinocytes. Mol. Hum. Reprod. 1997, 3, 299–305. [Google Scholar] [CrossRef]
- Gallo, R.L.; Nakatsuji, T. Microbial Symbiosis with the Innate Immune Defense System of the Skin. J. Investig. Dermatol. 2011, 131, 1974–1980. [Google Scholar] [CrossRef]
- Nelson, A.M.; Reddy, S.K.; Ratliff, T.S.; Hossain, M.Z.; Katseff, A.S.; Zhu, A.S.; Chang, E.; Resnik, S.R.; Page, C.; Kim, D.; et al. dsRNA Released by Tissue Damage Activates TLR3 to Drive Skin Regeneration. Cell Stem Cell 2015, 17, 139–151. [Google Scholar] [CrossRef]
- Wang, J.-N.; Li, M. The Immune Function of Keratinocytes in Anti-Pathogen Infection in the Skin. Int. J. Dermatol. Venereol. 2020, 3, 231–238. [Google Scholar] [CrossRef]
- Chieosilapatham, P.; Kiatsurayanon, C.; Umehara, Y.; Trujillo-Paez, J.V.; Peng, G.; Yue, H.; Nguyen, L.T.H.; Niyonsaba, F. Keratinocytes: Innate immune cells in atopic dermatitis. Clin. Exp. Immunol. 2021, 204, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The diverse contribution of keratinocytes to immune responses in skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Szatmári, T.; Mundt, F.; Kumar-Singh, A.; Möbus, L.; Ötvös, R.; Hjerpe, A.; Dobra, K. Molecular targets and signaling pathways regulated by nuclear translocation of syndecan-1. BMC Cell Biol. 2017, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Garcia, M.; Patel, M.V.; Shen, Z.; Fahey, J.V.; Biswas, N.; Mestecky, J.; Wira, C.R. Chapter 108—Mucosal Immunity in the Human Female Reproductive Tract. In Mucosal Immunology, 4th ed.; Mestecky, J., Strober, W., Russell, M.W., Kelsall, B.L., Cheroutre, H., Lambrecht, B.N., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 2097–2124. [Google Scholar]
- White, H.D.; Crassi, K.; Wira, C. Cytolytic functional activities of NK cells and cytotoxic T lymphocytes (CTL) are coordinately regulated in the human female reproductive tract. In Mucosal Solutions: Advances in Mucosal Immunology; The University of Sydney: Sydney, Australia, 1997; pp. 385–391. [Google Scholar]
- Trifonova, R.T.; Lieberman, J.; van Baarle, D. Distribution of Immune Cells in the Human Cervix and Implications for HIV Transmission. Am. J. Reprod. Immunol. 2014, 71, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Pudney, J.; Quayle, A.J.; Anderson, D.J. Immunological Microenvironments in the Human Vagina and Cervix: Mediators of Cellular Immunity Are Concentrated in the Cervical Transformation Zone1. Biol. Reprod. 2005, 73, 1253–1263. [Google Scholar] [CrossRef]
- Givan, A.L.; White, H.D.; Stern, J.E.; Colby, E.; Guyre, P.M.; Wira, C.R.; Gosselin, E.J. Flow Cytometric Analysis of Leukocytes in the Human Female Reproductive Tract: Comparison of Fallopian Tube, Uterus, Cervix, and Vagina. Am. J. Reprod. Immunol. 1997, 38, 350–359. [Google Scholar] [CrossRef]
- Kutteh, W.H.; Mestecky, J.; Wira, C. Mucosal Immunity in the Human Female Reproductive Tract; Academic Press: New York, NY, USA, 2005. [Google Scholar]
- Sakamoto, Y.; Moran, P.; Bulmer, J.N.; Searle, R.F.; Robson, S.C. Macrophages and not granulocytes are involved in cervical ripening. J. Reprod. Immunol. 2005, 66, 161–173. [Google Scholar] [CrossRef]
- Monin, L.; Whettlock, E.M.; Male, V. Immune responses in the human female reproductive tract. Immunology 2020, 160, 106–115. [Google Scholar] [CrossRef]
- Wira, C.R.; Fahey, J.V.; Rodriguez-Garcia, M.; Shen, Z.; Patel, M.V. Regulation of Mucosal Immunity in the Female Reproductive Tract: The Role of Sex Hormones in Immune Protection against Sexually Transmitted Pathogens. Am. J. Reprod. Immunol. 2014, 72, 236–258. [Google Scholar] [CrossRef] [PubMed]
- Maucort-Boulch, D.; Franceschi, S.; Plummer, M.; Group IHPSS. International correlation between human papillomavirus prevalence and cervical cancer incidence. Cancer Epidemiol. Biomark. Prev. 2008, 17, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Demarco, M.; Hyun, N.; Carter-Pokras, O.; Raine-Bennett, T.R.; Cheung, L.; Chen, X.; Hammer, A.; Campos, N.; Kinney, W.; Gage, J.C.; et al. A study of type-specific HPV natural history and implications for contemporary cervical cancer screening programs. eClinicalMedicine 2020, 22, 100293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jin, S.; Li, X.; Liu, L.; Xi, L.; Wang, F.; Zhang, S. Human Papillomavirus Type 16 Disables the Increased Natural Killer Cells in Early Lesions of the Cervix. J. Immunol. Res. 2019, 2019, 9182979. [Google Scholar] [CrossRef]
- Bosch, F.X.; Burchell, A.N.; Schiffman, M.; Giuliano, A.R.; de Sanjose, S.; Bruni, L.; Tortolero-Luna, G.; Kjaer, S.K.; Muñoz, N. Epidemiology and Natural History of Human Papillomavirus Infections and Type-Specific Implications in Cervical Neoplasia. Vaccine 2008, 26, K1–K16. [Google Scholar] [CrossRef]
- Bruno, M.T.; Scalia, G.; Cassaro, N.; Boemi, S. Multiple HPV 16 infection with two strains: A possible marker of neoplastic progression. BMC Cancer 2020, 20, 444. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, X.; Molijn, A.; Jenkins, D.; Shi, J.-F.; Quint, W.; Schmidt, J.E.; Wang, P.; Liu, Y.-L.; Li, L.-K.; et al. Human papillomavirus type-distribution in cervical cancer in China: The importance of HPV 16 and 18. Cancer Causes Control 2009, 20, 1705–1713. [Google Scholar] [CrossRef]
- Haghshenas, M.; Golini-Moghaddam, T.; Rafiei, A.; Emadeian, O.; Shykhpour, A.; Ashrafi, G.H. Prevalence and type distribution of high-risk human papillomavirus in patients with cervical cancer: A population-based study. Infect. Agents Cancer 2013, 8, 20. [Google Scholar] [CrossRef]
- de Freitas, A.C.; de Oliveira, T.H.A.; Barros, M.R.; Venuti, A. hrHPV E5 oncoprotein: Immune evasion and related immunotherapies. J. Exp. Clin. Cancer Res. 2017, 36, 71. [Google Scholar] [CrossRef]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef]
- Basukala, O.; Banks, L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021, 13, 1892. [Google Scholar] [CrossRef]
- Kumar, V.; Stewart, J.H. Immunometabolic reprogramming, another cancer hallmark. Front. Immunol. 2023, 14, 1125874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shen, C.; Zhao, L.; Wang, J.; McCrae, M.; Chen, X.; Lu, F. Dysregulation of host cellular genes targeted by human papillomavirus (HPV) integration contributes to HPV-related cervical carcinogenesis. Int. J. Cancer 2016, 138, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Stanley Margaret, A. Epithelial Cell Responses to Infection with Human Papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef]
- Fritz, J.H.; Le Bourhis, L.; Magalhaes, J.G.; Philpott, D.J. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol. 2008, 29, 41–49. [Google Scholar] [CrossRef]
- Tindle, R.W. Immune evasion in human papillomavirus-associated cervical cancer. Nat. Rev. Cancer 2002, 2, 59–64. [Google Scholar] [CrossRef]
- Uhlorn, B.L.; Jackson, R.; Li, S.; Bratton, S.M.; Van Doorslaer, K.; Campos, S.K. Vesicular trafficking permits evasion of cGAS/STING surveillance during initial human papillomavirus infection. PLoS Pathog. 2020, 16, e1009028. [Google Scholar] [CrossRef]
- Berger, K.L.; Barriga, F.; Lace, M.J.; Turek, L.P.; Zamba, G.J.; Domann, F.E.; Lee, J.H.; Klingelhutz, A.J. Cervical keratinocytes containing stably replicating extrachromosomal HPV-16 are refractory to transformation by oncogenic H-Ras. Virology 2006, 356, 68–78. [Google Scholar] [CrossRef]
- Pett, M.R.; Herdman, M.T.; Palmer, R.D.; Yeo, G.S.H.; Shivji, M.K.; Stanley, M.A.; Coleman, N. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc. Natl. Acad. Sci. USA 2006, 103, 3822–3827. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Calderon, A.; Lieberman, P.M. Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol. 2020, 28, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef]
- Lou, M.; Huang, D.; Zhou, Z.; Shi, X.; Wu, M.; Rui, Y.; Su, J.; Zheng, W.; Yu, X.-F. DNA virus oncoprotein HPV18 E7 selectively antagonizes cGAS-STING-triggered innate immune activation. J. Med. Virol. 2023, 95, e28310. [Google Scholar] [CrossRef]
- Xiao, D.; Huang, W.; Ou, M.; Guo, C.; Ye, X.; Liu, Y.; Wang, M.; Zhang, B.; Zhang, N.; Huang, S.; et al. Interaction between susceptibility loci in cGAS-STING pathway, MHC gene and HPV infection on the risk of cervical precancerous lesions in Chinese population. Oncotarget 2016, 7, 84228–84238. [Google Scholar] [CrossRef]
- Berti, F.C.B.; Pereira, A.P.L.; Cebinelli, G.C.M.; Trugilo, K.P.; Brajão de Oliveira, K. The role of interleukin 10 in human papilloma virus infection and progression to cervical carcinoma. Cytokine Growth Factor. Rev. 2017, 34, 1–13. [Google Scholar] [CrossRef]
- Nees, M.; Geoghegan, J.M.; Munson, P.; Prabhu, V.; Liu, Y.; Androphy, E.; Woodworth, C.D. Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-beta2 in cervical keratinocytes. Cancer Res. 2000, 60, 4289–4298. [Google Scholar]
- Iglesias, M.; Yen, K.; Gaiotti, D.; Hildesheim, A.; Stoler, M.H.; Woodworth, C.D. Human papillomavirus type 16 E7 protein sensitizes cervical keratinocytes to apoptosis and release of interleukin-1alpha. Oncogene 1998, 17, 1195–1205. [Google Scholar] [CrossRef]
- Shimada, M.; Yamashita, A.; Saito, M.; Ichino, M.; Kinjo, T.; Mizuki, N.; Klinman, D.M.; Okuda, K. The human papillomavirus E6 protein targets apoptosis-inducing factor (AIF) for degradation. Sci. Rep. 2020, 10, 14195. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hua, K.A.-O. Cervical Cancer: Emerging Immune Landscape and Treatment. OncoTargets Ther. 2020, 13, 8037–8047. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Li, J.; Montrose, D.C.; Martinez, L.A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 2023, 83, 266–280.e266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, H.; Liu, J.; Tao, T.; Zeng, Z.; Wang, M. RGS1 and related genes as potential targets for immunotherapy in cervical cancer: Computational biology and experimental validation. J. Transl. Med. 2022, 20, 334. [Google Scholar] [CrossRef]
- Agenès, F.; Bosco, N.; Mascarell, L.; Fritah, S.; Ceredig, R. Differential expression of regulator of G-protein signalling transcripts and in vivo migration of CD4+ naïve and regulatory T cells. Immunology 2005, 115, 179–188. [Google Scholar] [CrossRef]
- Shi, G.X.; Harrison, K.; Han, S.B.; Moratz, C.; Kehrl, J.H. Toll-like receptor signaling alters the expression of regulator of G protein signaling proteins in dendritic cells: Implications for G protein-coupled receptor signaling. J. Immunol. 2004, 172, 5175–5184. [Google Scholar] [CrossRef]
- Bai, Y.; Hu, M.; Chen, Z.; Wei, J.; Du, H. Single-Cell Transcriptome Analysis Reveals RGS1 as a New Marker and Promoting Factor for T-Cell Exhaustion in Multiple Cancers. Front. Immunol. 2021, 12, 767070. [Google Scholar] [CrossRef]
- Kang, Y.; Huang, J.; Liu, Y.; Zhang, N.; Cheng, Q.; Zhang, Y. Integrated Analysis of Immune Infiltration Features for Cervical Carcinoma and Their Associated Immunotherapeutic Responses. Front. Cell Dev. Biol. 2021, 9, 573497. [Google Scholar] [CrossRef]
- Yamato, K.; Yamada, T.; Kizaki, M.; Ui-Tei, K.; Natori, Y.; Fujino, M.; Nishihara, T.; Ikeda, Y.; Nasu, Y.; Saigo, K.; et al. New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer. Cancer Gene Ther. 2008, 15, 140–153. [Google Scholar] [CrossRef]
- Bergot, A.S.; Ford, N.; Leggatt, G.R.; Wells, J.W.; Frazer, I.H.; Grimbaldeston, M.A. HPV16-E7 expression in squamous epithelium creates a local immune suppressive environment via CCL2- and CCL5- mediated recruitment of mast cells. PLoS Pathog. 2014, 10, e1004466. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef]
- Taghizadeh, E.; Jahangiri, S.; Rostami, D.; Taheri, F.; Renani, P.G.; Taghizadeh, H.; Gheibi Hayat, S.M. Roles of E6 and E7 Human Papillomavirus Proteins in Molecular Pathogenesis of Cervical Cancer. Curr. Protein Pept. Sci. 2019, 20, 926–934. [Google Scholar] [CrossRef]
- Romani, N.; Brunner, P.M.; Stingl, G. Changing Views of the Role of Langerhans Cells. J. Investig. Dermatol. 2012, 132, 872–881. [Google Scholar] [CrossRef]
- Doebel, T.; Voisin, B.; Nagao, K. Langerhans Cells—The Macrophage in Dendritic Cell Clothing. Trends Immunol. 2017, 38, 817–828. [Google Scholar] [CrossRef]
- Merad, M.; Ginhoux, F.; Collin, M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol. 2008, 8, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; Raff, A.B.; Raff, L.M.; Da Silva, D.M.; Yan, L.; Skeate, J.G.; Wong, M.K.; Lin, Y.G.; Kast, W.M. Inhibition of Langerhans cell maturation by human papillomavirus type 16: A novel role for the annexin A2 heterotetramer in immune suppression. J. Immunol. 2014, 192, 4748–4757. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, D.M.; Woodham, A.W.; Skeate, J.G.; Rijkee, L.K.; Taylor, J.R.; Brand, H.E.; Muderspach, L.I.; Roman, L.D.; Yessaian, A.A.; Pham, H.Q.; et al. Langerhans cells from women with cervical precancerous lesions become functionally responsive against human papillomavirus after activation with stabilized Poly-I:C. Clin. Immunol. 2015, 161, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Fahey, L.M.; Raff, A.B.; Da Silva, D.M.; Kast, W.M. Reversal of human papillomavirus-specific T cell immune suppression through TLR agonist treatment of Langerhans cells exposed to human papillomavirus type 16. J. Immunol. 2009, 182, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Fausch, S.C.; Da Silva, D.M.; Kast, W.M. Heterologous papillomavirus virus-like particles and human papillomavirus virus-like particle immune complexes activate human Langerhans cells. Vaccine 2005, 23, 1720–1729. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Kuchroo, V.K. IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef]
- Gee, K.; Guzzo, C.; Che Mat, N.F.; Ma, W.; Kumar, A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm. Allergy Drug. Targets 2009, 8, 40–52. [Google Scholar] [CrossRef]
- García-Piñeres, A.J.; Hildesheim, A.; Trivett, M.; Williams, M.; Wu, L.; Kewalramani, V.N.; Pinto, L.A. Role of DC-SIGN in the activation of dendritic cells by HPV-16 L1 virus-like particle vaccine. Eur. J. Immunol. 2006, 36, 437–445. [Google Scholar] [CrossRef]
- Woodham, A.W.; Yan, L.; Skeate, J.G.; van der Veen, D.; Brand, H.H.; Wong, M.K.; Da Silva, D.M.; Kast, W.M. T cell ignorance is bliss: T cells are not tolerized by Langerhans cells presenting human papillomavirus antigens in the absence of costimulation. Papillomavirus Res. 2016, 2, 21–30. [Google Scholar] [CrossRef]
- Reyna-Hernández, M.A.; Alarcón-Romero, L.D.C.; Ortiz-Ortiz, J.; Illades-Aguiar, B.; Jiménez-López, M.A.; Ocampo-Bárcenas, A.; Morrugares-Ixtepan, M.O.; Torres-Rojas, F.I. GLUT1, LDHA, and MCT4 Expression Is Deregulated in Cervical Cancer and Precursor Lesions. J. Histochem. Cytochem. 2022, 70, 437–446. [Google Scholar] [CrossRef]
- Bedoya, A.M.; Tate, D.J.; Baena, A.; Córdoba, C.M.; Borrero, M.; Pareja, R.; Rojas, F.; Patterson, J.R.; Herrero, R.; Zea, A.H.; et al. Immunosuppression in cervical cancer with special reference to arginase activity. Gynecol. Oncol. 2014, 135, 74–80. [Google Scholar] [CrossRef]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Gerner, E.W.; Meyskens, F.L., Jr. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Souid, M.; Ghedira, R.; Souissi, S.; Bouzgarrou, N.; Gabbouj, S.; Shini-Hadhri, S.; Rhim, M.-S.; Boukadida, A.; Toumi, D.; Faleh, R.; et al. Arginase is involved in cervical lesions progression and severity. Immunobiology 2022, 227, 152189. [Google Scholar] [CrossRef] [PubMed]
- Venancio, P.A.; Consolaro, M.E.L.; Derchain, S.F.; Boccardo, E.; Villa, L.L.; Maria-Engler, S.S.; Campa, A.; Discacciati, M.G. Indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase expression in HPV infection, SILs, and cervical cancer. Cancer Cytopathol. 2019, 127, 586–597. [Google Scholar] [CrossRef]
- Hascitha, J.; Priya, R.; Jayavelu, S.; Dhandapani, H.; Selvaluxmy, G.; Sunder Singh, S.; Rajkumar, T. Analysis of Kynurenine/Tryptophan ratio and expression of IDO1 and 2 mRNA in tumour tissue of cervical cancer patients. Clin. Biochem. 2016, 49, 919–924. [Google Scholar] [CrossRef]
- Ferns, D.M.; Kema, I.P.; Buist, M.R.; Nijman, H.W.; Kenter, G.G.; Jordanova, E.S. Indoleamine-2,3-dioxygenase (IDO) metabolic activity is detrimental for cervical cancer patient survival. OncoImmunology 2015, 4, e981457. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Tan, H.X.; Niu, T.T.; Liu, Y.K.; Gu, C.J.; Li, D.J.; Li, M.Q.; Wang, H.Y. The IFN-γ-IDO1-kynureine pathway-induced autophagy in cervical cancer cell promotes phagocytosis of macrophage. Int. J. Biol. Sci. 2021, 17, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, Z.; Tang, H. Tryptophan 2,3-dioxygenase may be a potential prognostic biomarker and immunotherapy target in cancer: A meta-analysis and bioinformatics analysis. Front. Oncol. 2022, 12, 977640. [Google Scholar] [CrossRef]
- Wang, J.; Mijiti, Y.; Chen, Y.; Liu, Z. Aryl hydrocarbon receptor is a prognostic biomarker and is correlated with immune responses in cervical cancer. Bioengineered 2021, 12, 11922–11935. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Feng, S.; Cao, Z.; Wang, X. Role of aryl hydrocarbon receptor in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2013, 1836, 197–210. [Google Scholar] [CrossRef]
- Gasiewicz, T.A.; Henry, E.C.; Collins, L.L. Expression and Activity of Aryl Hydrocarbon Receptors in Development and Cancer. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 279–321. [Google Scholar] [CrossRef]
- Marvin, D.L.; Spaans, V.M.; de Kroon, C.D.; Slieker, R.C.; Khelil, M.; Ten Dijke, P.; Ritsma, L.; Jordanova, E.S. Low Transforming Growth Factor-β Pathway Activity in Cervical Adenocarcinomas. Front. Oncol. 2022, 12, 797453. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef]
- Falcomatà, C.; Bärthel, S.; Schneider, G.; Rad, R.; Schmidt-Supprian, M.; Saur, D. Context-Specific Determinants of the Immunosuppressive Tumor Microenvironment in Pancreatic Cancer. Cancer Discov. 2023, 13, 278–297. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef] [PubMed]
- Piersma, S.J. Immunosuppressive tumor microenvironment in cervical cancer patients. Cancer Microenviron. 2011, 4, 361–375. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, W.; Zhu, W. Profiling of immune responses by lactate modulation in cervical cancer reveals key features driving clinical outcome. Heliyon 2023, 9, e14896. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, Y.; Wei, X.; Zhang, Q.; Jia, H.; Quan, S.; Cao, D.; Wang, L.; Yang, T.; Zhao, J.; et al. Negative immune factors might predominate local tumor immune status and promote carcinogenesis in cervical carcinoma. Virol. J. 2017, 14, 5. [Google Scholar] [CrossRef]
- Xu, J.; Huang, Z.; Wang, Y.; Xiang, Z.; Xiong, B. Identification of Novel Tumor Microenvironment Regulating Factor That Facilitates Tumor Immune Infiltration in Cervical Cancer. Front. Oncol. 2022, 12, 846786. [Google Scholar] [CrossRef]
- Miyata, Y.; Ogo, E.; Abe, T.; Hirata, H.; Tsuda, N.; Ushijima, K.; Kawahara, A.; Akiba, J.; Obara, H.; Kakuma, T. Dynamics in the expression of programmed death ligand 1 and cluster of differentiation 163 in the tumor microenvironment of uterine cervical cancer: A single-center retrospective study. Radiat. Oncol. 2023, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, J.; Xu, K.; Chen, H.; Bian, C. PD-1/PD-L1 inhibitors for advanced or metastatic cervical cancer: From bench to bed. Front. Oncol. 2022, 12, 849352. [Google Scholar] [CrossRef]
- Mezache, L.; Paniccia, B.; Nyinawabera, A.; Nuovo, G.J. Enhanced expression of PD L1 in cervical intraepithelial neoplasia and cervical cancers. Mod. Pathol. 2015, 28, 1594–1602. [Google Scholar] [CrossRef]
- Liu, C.; Lu, J.; Tian, H.; Du, W.; Zhao, L.; Feng, J.; Yuan, D.; Li, Z. Increased expression of PD-L1 by the human papillomavirus 16 E7 oncoprotein inhibits anticancer immunity. Mol. Med. Rep. 2017, 15, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Y.P.; Yang, Y.Z.; Kang, J.R.; Jin, Y.D.; Wang, H.W. Expressions of programmed death (PD)-1 and PD-1 ligand (PD-L1) in cervical intraepithelial neoplasia and cervical squamous cell carcinomas are of prognostic value and associated with human papillomavirus status. J. Obs. Gynaecol. Res. 2017, 43, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Liang, H.; Hu, J.; Liu, S.; Hao, X.; Wong, M.S.K.; Li, X.; Hu, L. PD-L1 Expression Correlates with Tumor Infiltrating Lymphocytes and Response to Neoadjuvant Chemotherapy in Cervical Cancer. J. Cancer 2018, 9, 2938–2945. [Google Scholar] [CrossRef]
- Yang-Chun, F.; Zhen-Zhen, C.; Yan-Chun, H.; Xiu-Min, M. Association between PD-L1 and HPV status and the prognostic value for HPV treatment in premalignant cervical lesion patients. Medicine 2017, 96, e7270. [Google Scholar] [CrossRef]
- Yao, S.; Zhao, L.; Chen, S.; Wang, H.; Gao, Y.; Shao, N.-Y.; Dai, M.; Cai, H. Cervical cancer immune infiltration microenvironment identification, construction of immune scores, assisting patient prognosis and immunotherapy. Front. Immunol. 2023, 14, 1135657. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.K. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef]
- Imazeki, H.; Ogiwara, Y.; Kawamura, M.; Boku, N.; Kudo-Saito, C. CD11b(+)CTLA4(+) myeloid cells are a key driver of tumor evasion in colorectal cancer. J. Immunother. Cancer 2021, 9, e002841. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e718. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Li, M.; Zhu, W. ERBB3 methylation and immune infiltration in tumor microenvironment of cervical cancer. Sci. Rep. 2022, 12, 8112. [Google Scholar] [CrossRef]
- Audirac-Chalifour, A.; Torres-Poveda, K.; Bahena-Román, M.; Téllez-Sosa, J.; Martínez-Barnetche, J.; Cortina-Ceballos, B.; López-Estrada, G.; Delgado-Romero, K.; Burguete-García, A.I.; Cantú, D.; et al. Cervical Microbiome and Cytokine Profile at Various Stages of Cervical Cancer: A Pilot Study. PLoS ONE 2016, 11, e0153274. [Google Scholar] [CrossRef]
- Kyrgiou, M.; Moscicki, A.-B. Vaginal microbiome and cervical cancer. Semin. Cancer Biol. 2022, 86, 189–198. [Google Scholar] [CrossRef]
- Vaccarella, S.; Lortet-Tieulent, J.; Plummer, M.; Franceschi, S.; Bray, F. Worldwide trends in cervical cancer incidence: Impact of screening against changes in disease risk factors. Eur. J. Cancer 2013, 49, 3262–3273. [Google Scholar] [CrossRef]
- Castanheira, C.P.; Sallas, M.L.; Nunes, R.A.L.; Lorenzi, N.P.C.; Termini, L. Microbiome and Cervical Cancer. Pathobiology 2021, 88, 187–197. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, A.X.J.; Chen, G.; Wu, Y.; Gu, W. Prognostic and therapeutic TILs of cervical cancer—Current advances and future perspectives. Mol. Ther.-Oncolytics 2021, 22, 410–430. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Frankel, T.; Lanfranca, M.P.; Zou, W. The Role of Tumor Microenvironment in Cancer Immunotherapy. Adv. Exp. Med. Biol. 2017, 1036, 51–64. [Google Scholar]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Ma, R.-Y.; Black, A.; Qian, B.-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022, 43, 546–563. [Google Scholar] [CrossRef]
- Kumar, V. Macrophages: The Potent Immunoregulatory Innate Immune Cells. In Macrophage Activation; Khalid Hussain, B., Ed.; IntechOpen: Rijeka, Croatia, 2019; Chapter 1. [Google Scholar]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hua, K. Dissecting the Single-Cell Transcriptome Network of Immune Environment Underlying Cervical Premalignant Lesion, Cervical Cancer and Metastatic Lymph Nodes. Front. Immunol. 2022, 13, 897366. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug. Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, S.Y.; Ziblat, A.; Secchiari, F.; Torres, N.I.; Sierra, J.M.; Raffo Iraolagoitia, X.L.; Araya, R.E.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Human M2 Macrophages Limit NK Cell Effector Functions through Secretion of TGF-β and Engagement of CD85j. J. Immunol. 2018, 200, 1008–1015. [Google Scholar] [CrossRef]
- Guo, F.; Feng, Y.C.; Zhao, G.; Zhang, R.; Cheng, Z.Z.; Kong, W.N.; Wu, H.L.; Xu, B.; Lv, X.; Ma, X.M. Tumor-Associated CD163(+) M2 Macrophage Infiltration is Highly Associated with PD-L1 Expression in Cervical Cancer. Cancer Manag. Res. 2020, 12, 5831–5843. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-J.; Wu, S.; Yan, R.-M.; Fan, L.-S.; Yu, L.; Zhang, Y.-M.; Wei, W.-F.; Zhou, C.-F.; Wu, X.-G.; Zhong, M.; et al. The role of the hypoxia-Nrp-1 axis in the activation of M2-like tumor-associated macrophages in the tumor microenvironment of cervical cancer. Mol. Carcinog. 2019, 58, 388–397. [Google Scholar] [CrossRef]
- Ren, J.; Li, L.; Yu, B.; Xu, E.; Sun, N.; Li, X.; Xing, Z.; Han, X.; Cui, Y.; Wang, X.; et al. Extracellular vesicles mediated proinflammatory macrophage phenotype induced by radiotherapy in cervical cancer. BMC Cancer 2022, 22, 88. [Google Scholar] [CrossRef] [PubMed]
- Krneta, T.; Gillgrass, A.; Poznanski, S.; Chew, M.; Lee, A.J.; Kolb, M.; Ashkar, A.A. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J. Leukoc. Biol. 2017, 101, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, Y.; Ma, P.; Zha, Y.; Zhang, J.; Lei, A.; Li, N. CAR-macrophage: An extensive immune enhancer to fight cancer. eBioMedicine 2022, 76, 103873. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat. Rev. Immunol. 2022, 22, 173–187. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef]
- Nomelini, R.S.; Mota, S.D.S.; Murta, E.F.C. Absolute band neutrophils count is a predictor of overall survival in advanced uterine cervical cancer. Arch. Gynecol. Obstet. 2022, 306, 1697–1701. [Google Scholar] [CrossRef]
- Carus, A.; Ladekarl, M.; Hager, H.; Nedergaard, B.S.; Donskov, F. Tumour-associated CD66b+ neutrophil count is an independent prognostic factor for recurrence in localised cervical cancer. Br. J. Cancer 2013, 108, 2116–2122. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Dai, X.; Ma, Q.; Wu, X. Stromal Neutrophil Extracellular Trap Density Is an Independent Prognostic Factor for Cervical Cancer Recurrence. Front. Oncol. 2021, 11, 659445. [Google Scholar] [CrossRef] [PubMed]
- Punt, S.; Fleuren, G.J.; Kritikou, E.; Lubberts, E.; Trimbos, J.B.; Jordanova, E.S.; Gorter, A. Angels and demons: Th17 cells represent a beneficial response, while neutrophil IL-17 is associated with poor prognosis in squamous cervical cancer. Oncoimmunology 2015, 4, e984539. [Google Scholar] [CrossRef] [PubMed]
- Ittiamornlert, P.; Ruengkhachorn, I. Neutrophil-lymphocyte ratio as a predictor of oncologic outcomes in stage IVB, persistent, or recurrent cervical cancer patients treated by chemotherapy. BMC Cancer 2019, 19, 51. [Google Scholar] [CrossRef] [PubMed]
- Calo, C.A.; Barrington, D.A.; Brown, M.; Gonzalez, L.; Baek, J.; Huffman, A.; Benedict, J.; Backes, F.; Chambers, L.; Cohn, D.; et al. High pre-treatment neutrophil-to-lymphocyte ratio as a prognostic marker for worse survival in patients with recurrent/metastatic cervical cancer treated with immune checkpoint inhibitors. Gynecol. Oncol. Rep. 2022, 42, 101040. [Google Scholar] [CrossRef]
- Gruijs, M.; Sewnath, C.A.N.; van Egmond, M. Therapeutic exploitation of neutrophils to fight cancer. Semin. Immunol. 2021, 57, 101581. [Google Scholar] [CrossRef]
- Carnevale, S.; Ghasemi, S.; Rigatelli, A.; Jaillon, S. The complexity of neutrophils in health and disease: Focus on cancer. Semin. Immunol. 2020, 48, 101409. [Google Scholar] [CrossRef]
- Geh, D.; Leslie, J.; Rumney, R.; Reeves, H.L.; Bird, T.G.; Mann, D.A. Neutrophils as potential therapeutic targets in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Gaggar, S.; Gögenur, I. Neutrophils and polymorphonuclear myeloid-derived suppressor cells: An emerging battleground in cancer therapy. Oncogenesis 2022, 11, 22. [Google Scholar] [CrossRef]
- Wesolowski, R.; Markowitz, J.; Carson, W.E. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunotherapy Cancer 2013, 1, 10. [Google Scholar] [CrossRef]
- Kawano, M.; Mabuchi, S.; Matsumoto, Y.; Sasano, T.; Takahashi, R.; Kuroda, H.; Kozasa, K.; Hashimoto, K.; Isobe, A.; Sawada, K.; et al. The significance of G-CSF expression and myeloid-derived suppressor cells in the chemoresistance of uterine cervical cancer. Sci. Rep. 2015, 5, 18217. [Google Scholar] [CrossRef]
- Sasano, T.; Mabuchi, S.; Kozasa, K.; Kuroda, H.; Kawano, M.; Takahashi, R.; Komura, N.; Yokoi, E.; Matsumoto, Y.; Hashimoto, K.; et al. The Highly Metastatic Nature of Uterine Cervical/Endometrial Cancer Displaying Tumor-Related Leukocytosis: Clinical and Preclinical Investigations. Clin. Cancer Res. 2018, 24, 4018–4029. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, K.H.; Yoon, H.I.; Kim, G.E.; Kim, Y.B. Tumor-related leukocytosis is associated with poor radiation response and clinical outcome in uterine cervical cancer patients. Ann. Oncol. 2016, 27, 2067–2074. [Google Scholar] [CrossRef]
- Nagaraj, S.; Youn, J.I.; Gabrilovich, D.I. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J. Immunol. 2013, 191, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhu, M.; Marley, J.L.; Bi, K.; Wang, K.; Zhai, M.; Hu, H.; Guo, P.; Li, C.; Xu, Y.; et al. The combined action of monocytic myeloid-derived suppressor cells and mucosal-associated invariant T cells promotes the progression of cervical cancer. Int. J. Cancer 2021, 148, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, W.; Zhu, X.; Yu, M.; Zhou, C. Inhibition of myeloid-derived suppressive cell function with all-trans retinoic acid enhanced anti-PD-L1 efficacy in cervical cancer. Sci. Rep. 2022, 12, 9619. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ahmad, A. Role of MAIT cells in the immunopathogenesis of inflammatory diseases: New players in old game. Int. Rev. Immunol. 2018, 37, 90–110. [Google Scholar] [CrossRef]
- Ussher, J.E.; Willberg, C.B.; Klenerman, P. MAIT cells and viruses. Immunol. Cell Biol. 2018, 96, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Rouxel, O.; Lehuen, A. Mucosal-associated invariant T cells in autoimmune and immune-mediated diseases. Immunol. Cell Biol. 2018, 96, 618–629. [Google Scholar] [CrossRef]
- Hinks, T.S.C.; van Wilgenburg, B.; Wang, H.; Loh, L.; Koutsakos, M.; Kedzierska, K.; Corbett, A.J.; Chen, Z. Study of MAIT Cell Activation in Viral Infections In Vivo. Methods Mol. Biol. 2020, 2098, 261–281. [Google Scholar]
- Zumwalde, N.A.; Gumperz, J.E. Mucosal-Associated Invariant T Cells in Tumors of Epithelial Origin. Adv. Exp. Med. Biol. 2020, 1224, 63–77. [Google Scholar]
- Berzins, S.P.; Wallace, M.E.; Kannourakis, G.; Kelly, J. A Role for MAIT Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 949. [Google Scholar] [CrossRef]
- Shaler, C.R.; Tun-Abraham, M.E.; Skaro, A.I.; Khazaie, K.; Corbett, A.J.; Mele, T.; Hernandez-Alejandro, R.; Haeryfar, S.M.M. Mucosa-associated invariant T cells infiltrate hepatic metastases in patients with colorectal carcinoma but are rendered dysfunctional within and adjacent to tumor microenvironment. Cancer Immunol. Immunother. 2017, 66, 1563–1575. [Google Scholar] [CrossRef]
- Haeryfar, S.M.M.; Shaler, C.R.; Rudak, P.T. Mucosa-associated invariant T cells in malignancies: A faithful friend or formidable foe? Cancer Immunol. Immunother. 2018, 67, 1885–1896. [Google Scholar] [CrossRef]
- Melo, A.M.; O’Brien, A.M.; Phelan, J.J.; Kennedy, S.A.; Wood, N.A.W.; Veerapen, N.; Besra, G.S.; Clarke, N.E.; Foley, E.K.; Ravi, A.; et al. Mucosal-Associated Invariant T Cells Display Diminished Effector Capacity in Oesophageal Adenocarcinoma. Front. Immunol. 2019, 10, 1580. [Google Scholar] [CrossRef]
- O’Neill, C.; Cassidy, F.C.; O’Shea, D.; Hogan, A.E. Mucosal Associated Invariant T Cells in Cancer-Friend or Foe? Cancers 2021, 13, 1582. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Zhou, K.; Wilson, M.; Kramer, A.; Zhu, Y.; Dawson, N.; Yang, L. Mucosal-associated invariant T cells for cancer immunotherapy. Mol. Ther. 2023, 31, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Hsiao, Y.-C.; Wu, C.-C.; Hsu, Y.-T.; Chang, C.-L. Less circulating mucosal-associated invariant T cells in patients with cervical cancer. Taiwan. J. Obstet. Gynecol. 2019, 58, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Allen, S.; McDonald, E.; Das, I.; Mak, J.Y.W.; Liu, L.; Fairlie, D.P.; Meehan, B.S.; Chen, Z.; Corbett, A.J.; et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov. 2020, 10, 124–141. [Google Scholar] [CrossRef]
- Ruf, B.; Catania, V.V.; Wabitsch, S.; Ma, C.; Diggs, L.P.; Zhang, Q.; Heinrich, B.; Subramanyam, V.; Cui, L.L.; Pouzolles, M.; et al. Activating Mucosal-Associated Invariant T Cells Induces a Broad Antitumor Response. Cancer Immunol. Res. 2021, 9, 1024–1034. [Google Scholar] [CrossRef]
- Kolkhir, P.; Elieh-Ali-Komi, D.; Metz, M.; Siebenhaar, F.; Maurer, M. Understanding human mast cells: Lesson from therapies for allergic and non-allergic diseases. Nat. Rev. Immunol. 2022, 22, 294–308. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, A. Mast cells: Emerging sentinel innate immune cells with diverse role in immunity. Mol. Immunol. 2010, 48, 14–25. [Google Scholar] [CrossRef]
- Abraham, S.N.; St John, A.L. Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 2010, 10, 440–452. [Google Scholar] [CrossRef]
- Galli, S.J.; Nakae, S.; Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005, 6, 135–142. [Google Scholar] [CrossRef]
- Aponte-López, A.; Muñoz-Cruz, S. Mast Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1273, 159–173. [Google Scholar]
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2020, 58, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas-Saez, A.; Schalper, J.A.; Nicovani, S.M.; Rudolph, M.I. Characterization of mast cells according to their content of tryptase and chymase in normal and neoplastic human uterine cervix. Int. J. Gynecol. Cancer 2002, 12, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.; Pai, M.R.; Poornima Baliga, B.; Nayak, K.S.; Shankarnarayana; Dighe, P. Mast cell profile in uterine cervix. Indian J. Pathol. Microbiol. 2004, 47, 178–180. [Google Scholar] [PubMed]
- Kalyani, R.; Rajeshwari, G. Significance of mast cells in non-neoplastic and neoplastic lesions of uterine cervix. Biomed. Res. Ther. 2016, 3, 3. [Google Scholar] [CrossRef]
- Guo, F.; Kong, W.N.; Li, D.W.; Zhao, G.; Wu, H.L.; Anwar, M.; Shang, X.Q.; Sun, Q.N.; Ma, C.L.; Ma, X.M. Low Tumor Infiltrating Mast Cell Density Reveals Prognostic Benefit in Cervical Carcinoma. Technol. Cancer Res. Treat. 2022, 21, 15330338221106530. [Google Scholar] [CrossRef]
- Benítez-Bribiesca, L.; Wong, A.; Utrera, D.; Castellanos, E. The role of mast cell tryptase in neoangiogenesis of premalignant and malignant lesions of the uterine cervix. J. Histochem. Cytochem. 2001, 49, 1061–1062. [Google Scholar] [CrossRef]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef]
- Li, J.; Peng, G.; Zhu, K.; Jie, X.; Xu, Y.; Rao, X.; Xu, Y.; Chen, Y.; Xing, B.; Wu, G.; et al. PD-1+ mast cell enhanced by PD-1 blocking therapy associated with resistance to immunotherapy. Cancer Immunol. Immunother. 2023, 72, 633–645. [Google Scholar] [CrossRef]
- Shamri, R.; Xenakis, J.J.; Spencer, L.A. Eosinophils in innate immunity: An evolving story. Cell Tissue Res. 2011, 343, 57–83. [Google Scholar] [CrossRef]
- Travers, J.; Rothenberg, M.E. Eosinophils in mucosal immune responses. Mucosal Immunol. 2015, 8, 464–475. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174. [Google Scholar] [CrossRef]
- Kita, H. Eosinophils: Multifaceted biological properties and roles in health and disease. Immunol. Rev. 2011, 242, 161–177. [Google Scholar] [CrossRef]
- Wen, T.; Rothenberg, M.E. The Regulatory Function of Eosinophils. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Ravin, K.A.; Loy, M. The Eosinophil in Infection. Clin. Rev. Allergy Immunol. 2016, 50, 214–227. [Google Scholar] [CrossRef]
- Blanchard, C.; Rothenberg, M.E. Chapter 3 Biology of the Eosinophil. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2009; pp. 81–121. [Google Scholar]
- Kurose, N.; Mizuguchi, S.; Ohkanemasa, Y.; Yamashita, M.; Nakano, M.; Guo, X.; Aikawa, A.; Nakada, S.; Sasagawa, T.; Yamada, S. Adenosquamous carcinoma of the uterine cervix displaying tumor-associated tissue eosinophilia. SAGE Open Med. Case Rep. 2019, 7, 2050313X19828235. [Google Scholar] [CrossRef]
- van Driel, W.J.; Hogendoorn, P.C.W.; Jansen, F.-W.; Zwinderman, A.H.; Trimbos, J.B.; Fleuren, G.J. Tumor-associated eosinophilic infiltrate of cervical cancer is indicative for a less effective immune response. Hum. Pathol. 1996, 27, 904–911. [Google Scholar] [CrossRef]
- Höckel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schäffer, U.; Vaupel, P. Association between Tumor Hypoxia and Malignant Progression in Advanced Cancer of the Uterine Cervix1. Cancer Res. 1996, 56, 4509–4515. [Google Scholar]
- Xie, F.; Meng, Y.-H.; Liu, L.-B.; Chang, K.-K.; Li, H.; Li, M.-Q.; Li, D.-J. Cervical Carcinoma Cells Stimulate the Angiogenesis through TSLP Promoting Growth and Activation of Vascular Endothelial Cells. Am. J. Reprod. Immunol. 2013, 70, 69–79. [Google Scholar] [CrossRef]
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 2020, 20, 594–607. [Google Scholar] [CrossRef]
- Grisaru-Tal, S.; Rothenberg, M.E.; Munitz, A. Eosinophil–lymphocyte interactions in the tumor microenvironment and cancer immunotherapy. Nat. Immunol. 2022, 23, 1309–1316. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Sakref, C.; Bendriss-Vermare, N.; Valladeau-Guilemond, J. Phenotypes and Functions of Human Dendritic Cell Subsets in the Tumor Microenvironment. Methods Mol. Biol. 2023, 2618, 17–35. [Google Scholar]
- Carenza, C.; Calcaterra, F.; Oriolo, F.; Di Vito, C.; Ubezio, M.; Della Porta, M.G.; Mavilio, D.; Della Bella, S. Costimulatory Molecules and Immune Checkpoints Are Differentially Expressed on Different Subsets of Dendritic Cells. Front. Immunol. 2019, 10, 1325. [Google Scholar] [CrossRef]
- Spits, H.; Bernink, J.H.; Lanier, L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat. Immunol. 2016, 17, 758–764. [Google Scholar] [CrossRef]
- Kumar, V.; Bauer, C.; Stewart, J.H. Chasing Uterine Cancer with NK Cell-Based Immunotherapies. Future Pharmacol. 2022, 2, 642–659. [Google Scholar] [CrossRef]
- Wolf, N.K.; Kissiov, D.U.; Raulet, D.H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 2023, 23, 90–105. [Google Scholar] [CrossRef]
- Tu, Y.; Pan, M.; Song, S.; Hua, J.; Liu, R.; Li, L. CD3+CD56+ natural killer T cell infiltration is increased in cervical cancer and negatively correlated with tumour progression. Biotechnol. Biotechnol. Equip. 2019, 33, 1380–1391. [Google Scholar] [CrossRef]
- Céspedes, M.A.; Rodríguez, J.A.; Medina, M.; Bravo, M.; Cómbita, A.L. Analysis of NK Cells in Peripheral Blood and Tumor Infiltrating Lymphocytes in Cervical Cancer Patients. Rev. Colomb. Cancerol. 2012, 16, 16–26. [Google Scholar] [CrossRef]
- Gooden, M.; Lampen, M.; Jordanova, E.S.; Leffers, N.; Trimbos, J.B.; van der Burg, S.H.; Nijman, H.; van Hall, T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8(+) T lymphocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10656–10661. [Google Scholar] [CrossRef]
- Spaans, V.M.; Peters, A.A.W.; Fleuren, G.J.; Jordanova, E.S. HLA-E expression in cervical adenocarcinomas: Association with improved long-term survival. J. Transl. Med. 2012, 10, 184. [Google Scholar] [CrossRef]
- Das, D.; Sarkar, B.; Mukhopadhyay, S.; Banerjee, C.; Mondal, S.B. An Altered Ratio of CD4+ And CD8+ T Lymphocytes in Cervical Cancer Tissues and Peripheral Blood—A Prognostic Clue? Asian Pac. J. Cancer Prev. 2018, 19, 471–478. [Google Scholar]
- Sheu, B.-C.; Hsu, S.-M.; Ho, H.-N.; Lin, R.-H.; Torng, P.-L.; Huang, S.-C. Reversed CD4/CD8 ratios of tumor-infiltrating lymphocytes are correlated with the progression of human cervical carcinoma. Cancer 1999, 86, 1537–1543. [Google Scholar] [CrossRef]
- Jiang, W.; Kang, L.; Lu, H.-Z.; Pan, X.; Lin, Q.; Pan, Q.; Xue, Y.; Weng, X.; Tang, Y.-W. Normal Values for CD4 and CD8 Lymphocyte Subsets in Healthy Chinese Adults from Shanghai. Clin. Vaccine Immunol. 2004, 11, 811–813. [Google Scholar] [CrossRef]
- Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4+FOXP3+ regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell. Mol. Immunol. 2011, 8, 59–66. [Google Scholar] [CrossRef]
- Zhang, J.; Meng, S.; Zhang, X.; Shao, K.; Lin, C. Infiltration Patterns of Cervical Epithelial Microenvironment Cells During Carcinogenesis. Front. Immunol. 2022, 13, 888176. [Google Scholar] [CrossRef]
- Lin, W.; Niu, Z.; Zhang, H.; Kong, Y.; Wang, Z.; Yang, X.; Yuan, F. Imbalance of Th1/Th2 and Th17/Treg during the development of uterine cervical cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 3604–3612. [Google Scholar]
- Nishimura, T.; Nakui, M.; Sato, M.; Iwakabe, K.; Kitamura, H.; Sekimoto, M.; Ohta, A.; Koda, T.; Nishimura, S. The critical role of Th1-dominant immunity in tumor immunology. Cancer Chemother. Pharmacol. 2000, 46, S52–S61. [Google Scholar] [CrossRef]
- van der Burg, S.H.; Piersma, S.J.; de Jong, A.; van der Hulst, J.M.; Kwappenberg, K.M.C.; van den Hende, M.; Welters, M.J.P.; Van Rood, J.J.; Fleuren, G.J.; Melief, C.J.M.; et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc. Natl. Acad. Sci. USA 2007, 104, 12087–12092. [Google Scholar] [CrossRef]
- Maskey, N.; Thapa, N.; Maharjan, M.; Shrestha, G.; Maharjan, N.; Cai, H.; Liu, S. Infiltrating CD4 and CD8 lymphocytes in HPV infected uterine cervical milieu. Cancer Manag. Res. 2019, 11, 7647–7655. [Google Scholar] [CrossRef]
- Sanif, R.; Nurwany, R. Prognostic significance of CD4/CD8 ratio in patients with advanced cervical cancer. J. Phys. Conf. Ser. 2019, 1246, 012053. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, M.; Jing, Y.; Cheng, J.; Zhang, C.; Cheng, L.; Lu, H.; Cai, M.-C.; Wu, J.; Wang, W.; et al. Baseline immunity and impact of chemotherapy on immune microenvironment in cervical cancer. Br. J. Cancer 2021, 124, 414–424. [Google Scholar] [CrossRef]
- Li, L.; Xu, X.T.; Wang, L.L.; Qin, S.B.; Zhou, J.Y. Expression and clinicopathological significance of Foxp3 and VISTA in cervical cancer. Am. J. Transl. Res. 2021, 13, 10428–10438. [Google Scholar]
- Wu, M.-Y.; Kuo, T.-Y.; Ho, H.-N. Tumor-infiltrating lymphocytes contain a higher proportion of FOXP3+ T lymphocytes in cervical cancer. J. Formos. Med. Assoc. 2011, 110, 580–586. [Google Scholar] [CrossRef]
- Ni, H.; Zhang, H.; Li, L.; Huang, H.; Guo, H.; Zhang, L.; Li, C.; Xu, J.-X.; Nie, C.-P.; Li, K.; et al. T cell-intrinsic STING signaling promotes regulatory T cell induction and immunosuppression by upregulating FOXP3 transcription in cervical cancer. J. ImmunoTherapy Cancer 2022, 10, e005151. [Google Scholar] [CrossRef]
- Nedergaard, B.S.; Ladekarl, M.; Thomsen, H.F.; Nyengaard, J.R.; Nielsen, K. Low density of CD3+, CD4+ and CD8+ cells is associated with increased risk of relapse in squamous cell cervical cancer. Br. J. Cancer 2007, 97, 1135–1138. [Google Scholar] [CrossRef]
- Hong, H.S.; Mbah, N.E.; Shan, M.; Loesel, K.; Lin, L.; Sajjakulnukit, P.; Correa, L.O.; Andren, A.; Lin, J.; Hayashi, A.; et al. OXPHOS promotes apoptotic resistance and cellular persistence in TH17 cells in the periphery and tumor microenvironment. Sci. Immunol. 2022, 7, eabm8182. [Google Scholar] [CrossRef]
- Alves, J.J.P.; De Medeiros Fernandes, T.A.A.; De Araújo, J.M.G.; Cobucci, R.N.O.; Lanza, D.C.F.; Bezerra, F.L.; Andrade, V.S.; Fernandes, J.V. Th17 response in patients with cervical cancer. Oncol. Lett. 2018, 16, 6215–6227. [Google Scholar] [CrossRef]
- Oomizu, S.; Arikawa, T.; Niki, T.; Kadowaki, T.; Ueno, M.; Nishi, N.; Yamauchi, A.; Hirashima, M. Galectin-9 suppresses Th17 cell development in an IL-2-dependent but Tim-3-independent manner. Clin. Immunol. 2012, 143, 51–58. [Google Scholar] [CrossRef]
- Punt, S.; van Vliet, M.E.; Spaans, V.M.; de Kroon, C.D.; Fleuren, G.J.; Gorter, A.; Jordanova, E.S. FoxP3(+) and IL-17(+) cells are correlated with improved prognosis in cervical adenocarcinoma. Cancer Immunol. Immunother. 2015, 64, 745–753. [Google Scholar] [CrossRef]
- Yu, Q.; Lou, X.-M.; He, Y. Preferential Recruitment of Th17 Cells to Cervical Cancer via CCR6-CCL20 Pathway. PLoS ONE 2015, 10, e0120855. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune Regulatory Function of B Cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Inoue, S.; Leitner, W.W.; Golding, B.; Scott, D. Inhibitory Effects of B Cells on Antitumor Immunity. Cancer Res. 2006, 66, 7741–7747. [Google Scholar] [CrossRef]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef]
- Kim, S.S.; Shen, S.; Miyauchi, S.; Sanders, P.D.; Franiak-Pietryga, I.; Mell, L.; Gutkind, J.S.; Cohen, E.E.W.; Califano, J.A.; Sharabi, A.B. B Cells Improve Overall Survival in HPV-Associated Squamous Cell Carcinomas and Are Activated by Radiation and PD-1 Blockade. Clin. Cancer Res. 2020, 26, 3345–3359. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, Y.; Du, R.; Pang, N.; Zhang, F.; Dong, D.; Ding, J.; Ding, Y. Role of Regulatory B Cells in the Progression of Cervical Cancer. Mediat. Inflamm. 2019, 2019, 6519427. [Google Scholar] [CrossRef]
- Vora, C.; Gupta, S. Targeted therapy in cervical cancer. ESMO Open. 2018, 3, e000462. [Google Scholar] [CrossRef]
- Harper, D.M.; Vierthaler, S.L.; Santee, J.A. Review of Gardasil. J. Vaccines Vaccin. 2010, 1, 1000107. [Google Scholar] [CrossRef]
- Monie, A.; Hung, C.F.; Roden, R.; Wu, T.C. Cervarix: A vaccine for the prevention of HPV 16, 18-associated cervical cancer. Biologics 2008, 2, 97–105. [Google Scholar]
- Grau-Bejar, J.F.; Garcia-Duran, C.; Garcia-Illescas, D.; Mirallas, O.; Oaknin, A. Advances in immunotherapy for cervical cancer. Ther. Adv. Med. Oncol. 2023, 15, 17588359231163836. [Google Scholar] [CrossRef]
- Shao, D.-D.; Meng, F.-Z.; Liu, Y.; Xu, X.-Q.; Wang, X.; Hu, W.-H.; Hou, W.; Ho, W.-Z. Poly(dA:dT) Suppresses HSV-2 Infection of Human Cervical Epithelial Cells Through RIG-I Activation. Front. Immunol. 2021, 11, 598884. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Shi, F.; Su, J.; Wang, J.; Liu, Z.; Wang, T. Activation of STING inhibits cervical cancer tumor growth through enhancing the anti-tumor immune response. Mol. Cell. Biochem. 2021, 476, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kim, S.; Herndon, J.M.; Goedegebuure, P.; Belt, B.A.; Satpathy, A.T.; Fleming, T.P.; Hansen, T.H.; Murphy, K.M.; Gillanders, W.E. Cross-dressed CD8α+/CD103+ dendritic cells prime CD8+ T cells following vaccination. Proc. Natl. Acad. Sci. USA 2012, 109, 12716–12721. [Google Scholar] [CrossRef]
- Williford, J.-M.; Ishihara, J.; Ishihara, A.; Mansurov, A.; Hosseinchi, P.; Marchell, T.M.; Potin, L.; Swartz, M.A.; Hubbell, J.A. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 2019, 5, eaay1357. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Langers, I.; Renoux, V.; Reschner, A.; Touze, A.; Coursaget, P.; Boniver, J.; Koch, J.; Delvenne, P.; Jacobs, N. Natural killer and dendritic cells collaborate in the immune response induced by the vaccine against uterine cervical cancer. Eur. J. Immunol. 2014, 44, 3585–3595. [Google Scholar] [CrossRef]
- Soca Gallego, L.; Dominguez, A.; Parmar, M. Human Papilloma Virus Vaccine. In StatPearls; Statpearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chan, J.K.; Deng, W.; Higgins, R.V.; Tewari, K.S.; Bonebrake, A.J.; Hicks, M.; Gaillard, S.; Ramirez, P.T.; Chafe, W.; Monk, B.J.; et al. A phase II evaluation of brivanib in the treatment of persistent or recurrent carcinoma of the cervix: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 146, 554–559. [Google Scholar] [CrossRef]
- Santin, A.D.; Deng, W.; Frumovitz, M.; Buza, N.; Bellone, S.; Huh, W.; Khleif, S.; Lankes, H.A.; Ratner, E.S.; O’Cearbhaill, R.E.; et al. Phase II evaluation of nivolumab in the treatment of persistent or recurrent cervical cancer (NCT02257528/NRG-GY002). Gynecol. Oncol. 2020, 157, 161–166. [Google Scholar] [CrossRef]
- Naumann, R.W.; Oaknin, A.; Meyer, T.; Lopez-Picazo, J.M.; Lao, C.; Bang, Y.J.; Boni, V.; Sharfman, W.H.; Park, J.C.; Devriese, L.A.; et al. Efficacy and safety of nivolumab (Nivo) + ipilimumab (Ipi) in patients (pts) with recurrent/metastatic (R/M) cervical cancer: Results from CheckMate 358. Ann. Oncol. 2019, 30, v898–v899. [Google Scholar] [CrossRef]
- Liu, K.; Zhu, Y.; Zhou, Y.; Zhu, H. Cemiplimab as Second-Line Therapy for Patients with Recurrent Cervical Cancer: A United States-based Cost-effectiveness Analysis. Adv. Ther. 2023, 40, 1838–1849. [Google Scholar] [CrossRef]
- Zheng, X.; Gu, H.; Cao, X.; Pan, B.; Xiang, H.; Ju, M.; Xu, S.; Zheng, M. Tislelizumab for cervical cancer: A retrospective study and analysis of correlative blood biomarkers. Front. Immunol. 2023, 14, 1113369. [Google Scholar] [CrossRef]
- Basu, P.; Mehta, A.; Jain, M.; Gupta, S.; Nagarkar, R.V.; John, S.; Petit, R. A Randomized Phase 2 Study of ADXS11-001 Listeria monocytogenes-Listeriolysin O Immunotherapy with or without Cisplatin in Treatment of Advanced Cervical Cancer. Int. J. Gynecol. Cancer 2018, 28, 764–772. [Google Scholar] [CrossRef]
- Galicia-Carmona, T.; Arango-Bravo, E.; Serrano-Olvera, J.A.; Flores-de La Torre, C.; Cruz-Esquivel, I.; Villalobos-Valencia, R.; Morán-Mendoza, A.; Castro-Eguiluz, D.; Cetina-Pérez, L. ADXS11-001 LM-LLO as specific immunotherapy in cervical cancer. Hum. Vaccin. Immunother. 2021, 17, 2617–2625. [Google Scholar] [CrossRef]
- Hong, D.S.; Concin, N.; Vergote, I.; de Bono, J.S.; Slomovitz, B.M.; Drew, Y.; Arkenau, H.T.; Machiels, J.P.; Spicer, J.F.; Jones, R.; et al. Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clin. Cancer Res. 2020, 26, 1220–1228. [Google Scholar] [CrossRef]
- Song, X.; Li, R.; Wang, H.; Song, P.; Guo, W.; Chen, Z.S. Tisotumab vedotin for the treatment of cervical carcinoma. Drugs Today 2022, 58, 213–222. [Google Scholar] [CrossRef]
- Sherer, M.V.; Kotha, N.V.; Williamson, C.; Mayadev, J. Advances in immunotherapy for cervical cancer: Recent developments and future directions. Int. J. Gynecol. Cancer 2022, 32, 281–287. [Google Scholar] [CrossRef]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos Usta, E.; Yañez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. N. Engl. J. Med. 2021, 385, 1856–1867. [Google Scholar] [CrossRef]
- Youn, J.W.; Hur, S.Y.; Woo, J.W.; Kim, Y.M.; Lim, M.C.; Park, S.Y.; Seo, S.S.; No, J.H.; Kim, B.G.; Lee, J.K.; et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: Interim results of a single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1653–1660. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, K.; Huang, Y.; Zhang, H.; Zhou, L.; Li, A.; Sun, Y. Photosensitizer-induced HPV16 E7 nanovaccines for cervical cancer immunotherapy. Biomaterials 2022, 282, 121411. [Google Scholar] [CrossRef]
- van Meir, H.; Nout, R.A.; Welters, M.J.P.; Loof, N.M.; de Kam, M.L.; van Ham, J.J.; Samuels, S.; Kenter, G.G.; Cohen, A.F.; Melief, C.J.M.; et al. Impact of (chemo)radiotherapy on immune cell composition and function in cervical cancer patients. OncoImmunology 2017, 6, e1267095. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Bonini, C.; Mondino, A. Adoptive T-cell therapy for cancer: The era of engineered T cells. Eur. J. Immunol. 2015, 45, 2457–2469. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Ellis, G.I.; Sheppard, N.C.; Riley, J.L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 2021, 22, 427–447. [Google Scholar] [CrossRef]
- Etxeberria, I.; Olivera, I.; Bolaños, E.; Cirella, A.; Teijeira, Á.; Berraondo, P.; Melero, I. Engineering bionic T cells: Signal 1, signal 2, signal 3, reprogramming and the removal of inhibitory mechanisms. Cell. Mol. Immunol. 2020, 17, 576–586. [Google Scholar] [CrossRef]
- Jazaeri, A.A.; Zsiros, E.; Amaria, R.N.; Artz, A.S.; Edwards, R.P.; Wenham, R.M.; Slomovitz, B.M.; Walther, A.; Thomas, S.S.; Chesney, J.A.; et al. Safety and efficacy of adoptive cell transfer using autologous tumor infiltrating lymphocytes (LN-145) for treatment of recurrent, metastatic, or persistent cervical carcinoma. J. Clin. Oncol. 2019, 37, 2538. [Google Scholar] [CrossRef]
- Radvanyi, L.G. Tumor-Infiltrating Lymphocyte Therapy: Addressing Prevailing Questions. Cancer J. 2015, 21, 450–464. [Google Scholar] [CrossRef]
- Kazemi, M.H.; Sadri, M.; Najafi, A.; Rahimi, A.; Baghernejadan, Z.; Khorramdelazad, H.; Falak, R. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front. Immunol. 2022, 13, 1018962. [Google Scholar] [CrossRef]
- Li, B. Why do tumor-infiltrating lymphocytes have variable efficacy in the treatment of solid tumors? Front. Immunol. 2022, 13, 973881. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, J.; Ma, W.; Yang, W.; Hu, B.A.-O. The Antitumor Activity of CAR-T-PD1 Cells Enhanced by HPV16mE7-Pulsed and SOCS1-Silenced DCs in Cervical Cancer Models. Cancer Manag. Res. 2021, 13, 6045–6053. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, X.-M.; Yin, C.-H.; Wu, Y.-M. Killing cervical cancer cells by specific chimeric antigen receptor-modified T cells. J. Reprod. Immunol. 2020, 139, 103115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Zhang, J.; Mao, L. Novel cellular immunotherapy using NKG2D CAR-T for the treatment of cervical cancer. Biomed. Pharmacother. 2020, 131, 110562. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.-M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.-F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 102. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal. Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef]
- Ghanaat, M.; Goradel, N.H.; Arashkia, A.; Ebrahimi, N.; Ghorghanlu, S.; Malekshahi, Z.V.; Fattahi, E.; Negahdari, B.; Kaboosi, H. Virus against virus: Strategies for using adenovirus vectors in the treatment of HPV-induced cervical cancer. Acta Pharmacol. Sin. 2021, 42, 1981–1990. [Google Scholar] [CrossRef]
- Petry, K.U. HPV and cervical cancer. Scand. J. Clin. Lab. Investig. 2014, 74, 59–62. [Google Scholar] [CrossRef]
- Castellsagué, X. Natural history and epidemiology of HPV infection and cervical cancer. Gynecol. Oncol. 2008, 110, S4–S7. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Das, S.S.; Biswal, S.S.; Nath, A.; Das, D.; Basu, A.; Malik, S.; Kumar, L.; Kar, S.; Singh, S.K.; et al. Mechanistic role of HPV-associated early proteins in cervical cancer: Molecular pathways and targeted therapeutic strategies. Crit. Rev. Oncol./Hematol. 2022, 174, 103675. [Google Scholar] [CrossRef]
- Yu, S.; Li, X.; Zhang, J.; Wu, S. Development of a Novel Immune Infiltration-Based Gene Signature to Predict Prognosis and Immunotherapy Response of Patients With Cervical Cancer. Front. Immunol. 2021, 12, 709493. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Cai, X.; Xie, Z.; Zhou, L.; Cheng, B.; Zhong, R.; Xiong, S.; Li, J.; Chen, Z.; et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. eClinicalMedicine 2021, 41, 101134. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, C.; Murakami, Y.; Ishii, E.; Fujita, H.; Wakao, H. Reprogramming and redifferentiation of mucosal-associated invariant T cells reveal tumor inhibitory activity. eLife 2022, 11, e70848. [Google Scholar] [CrossRef] [PubMed]
- Stoitzner, P.; Sparber, F.; Tripp, C.H. Langerhans cells as targets for immunotherapy against skin cancer. Immunol. Cell Biol. 2010, 88, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Bellone, S.; El-Sahwi, K.; Cocco, E.; Casagrande, F.; Cargnelutti, M.; Palmieri, M.; Bignotti, E.; Romani, C.; Silasi, D.A.; Azodi, M.; et al. Human papillomavirus type 16 (HPV-16) virus-like particle L1-specific CD8+ cytotoxic T lymphocytes (CTLs) are equally effective as E7-specific CD8+ CTLs in killing autologous HPV-16-positive tumor cells in cervical cancer patients: Implications for L1 dendritic cell-based therapeutic vaccines. J. Virol. 2009, 83, 6779–6789. [Google Scholar]
- Lv, Y.; Ma, X.; Ma, Y.; Du, Y.; Feng, J. A new emerging target in cancer immunotherapy: Galectin-9 (LGALS9). Genes. Dis. 2022. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Wang, Y.; Wu, X. The Role of Galectins in Cervical Cancer Biology and Progression. BioMed. Res. Int. 2018, 2018, 2175927. [Google Scholar] [CrossRef]
- Kandel, S.; Adhikary, P.; Li, G.; Cheng, K. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett. 2021, 510, 67–78. [Google Scholar] [CrossRef]
- Kaesler, S.; Wölbing, F.; Kempf, W.E.; Skabytska, Y.; Köberle, M.; Volz, T.; Sinnberg, T.; Amaral, T.; Möckel, S.; Yazdi, A.; et al. Targeting tumor-resident mast cells for effective anti-melanoma immune responses. JCI Insight 2019, 4, e125057. [Google Scholar] [CrossRef]
- Oldford, S.A.; Marshall, J.S. Mast cells as targets for immunotherapy of solid tumors. Mol. Immunol. 2015, 63, 113–124. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| CC Prevention Strategies | Target |
|---|---|
| Prevents HPV-16 and -18-associated CC via inducing immunity, including anti-HPV-16 and -18 antibodies (IgG1) |
| Gardasil protects against low-risk HPV-6 and -11, which cause most genital warts, and against HR-HPV-16 and -18 for at least five years [243]. Gardasil 9 protects against infection with low-risk HPV types 6 and 11, which cause most genital warts, and against HR-HPV types 16, 18, 31, 33, 45, 52, and 58, responsible for different cancers by inducing the humoral antiviral immunity [253]. |
| Targeted drug therapies for CC | |
| Targets human vascular endothelial growth factor (VEGF) to inhibit angiogenesis or neoangiogenesis in CC |
| Targets VEGF and fibroblast growth factor receptor (FGFR) |
| Targets PD-1 on T cells to prevent their exhaustion that increases anticancer immunity to clear CC cells |
| Targets human PD-1 |
| Blocks PD-1–PD-L1 interaction to enhance the antitumor immunity |
| Targets human PD-1 to block PD-1–PD-L1 interaction to prevent immune exhaustion |
| Targets human PD-1 to inhibit PD-1–PD-L1 interaction and minimizes binding to Fcγ receptors to serve as an ICI. |
| Raises anti-HPV-16 cellular immunity, including cytotoxic T cell-mediated immune response |
| This ADC delivers cytotoxic agent monomethyl auristatin E (MMAE) directly into tumor cells to target highly expressed tissue factor (TF) or conjugation factor III in CC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; Bauer, C.; Stewart, J.H., IV. TIME Is Ticking for Cervical Cancer. Biology 2023, 12, 941. https://doi.org/10.3390/biology12070941
Kumar V, Bauer C, Stewart JH IV. TIME Is Ticking for Cervical Cancer. Biology. 2023; 12(7):941. https://doi.org/10.3390/biology12070941
Chicago/Turabian StyleKumar, Vijay, Caitlin Bauer, and John H. Stewart, IV. 2023. "TIME Is Ticking for Cervical Cancer" Biology 12, no. 7: 941. https://doi.org/10.3390/biology12070941
APA StyleKumar, V., Bauer, C., & Stewart, J. H., IV. (2023). TIME Is Ticking for Cervical Cancer. Biology, 12(7), 941. https://doi.org/10.3390/biology12070941