
TAFPred: Torsion Angle Fluctuations Prediction from Protein Sequences



Abstract
:Simple Summary
Abstract
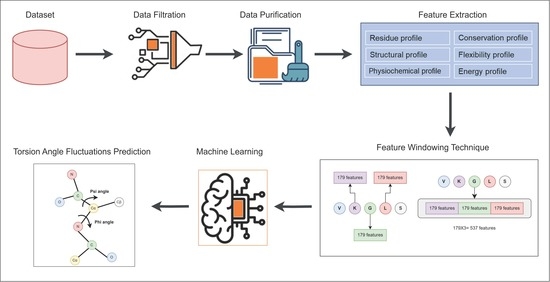
1. Introduction
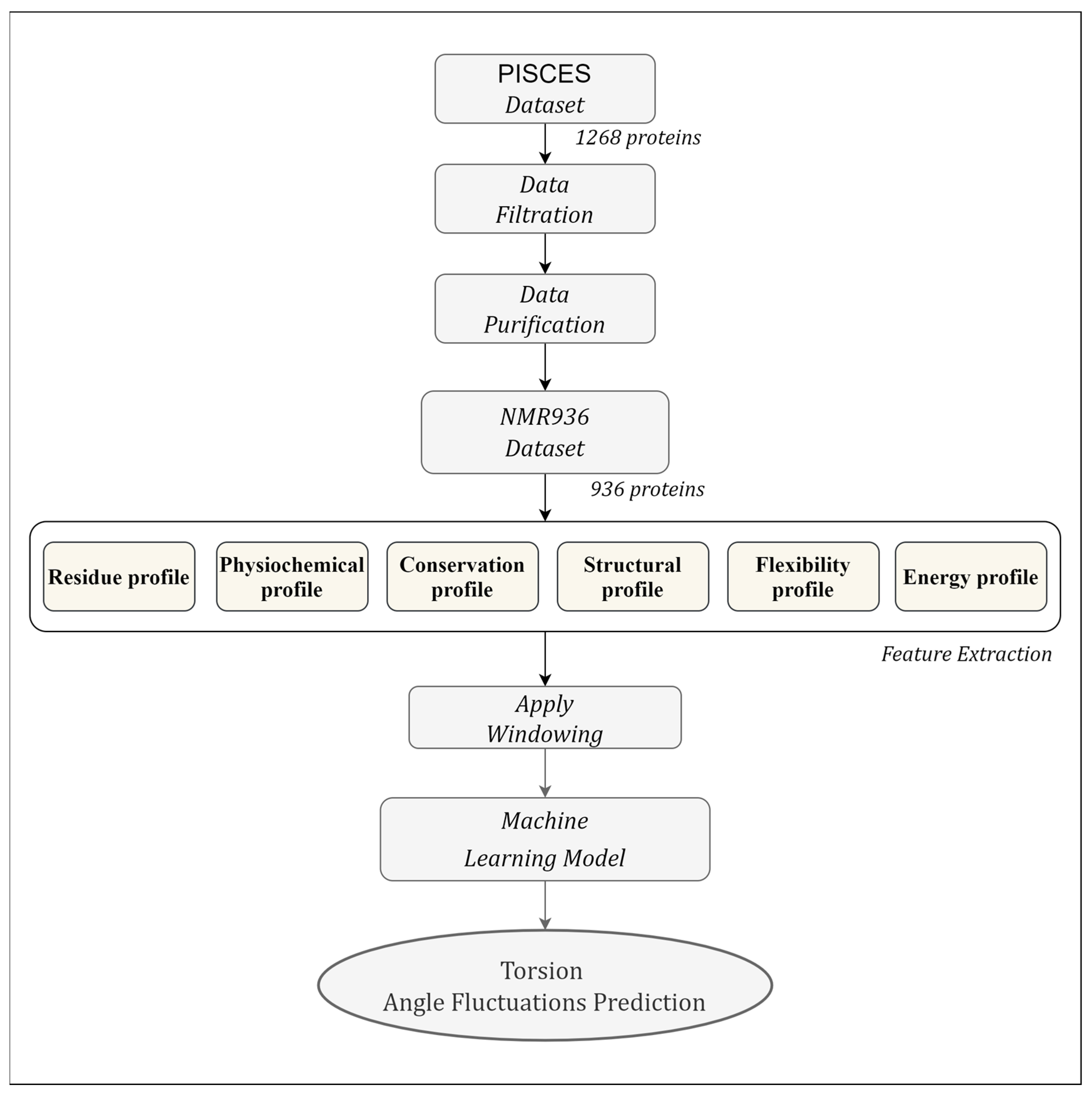
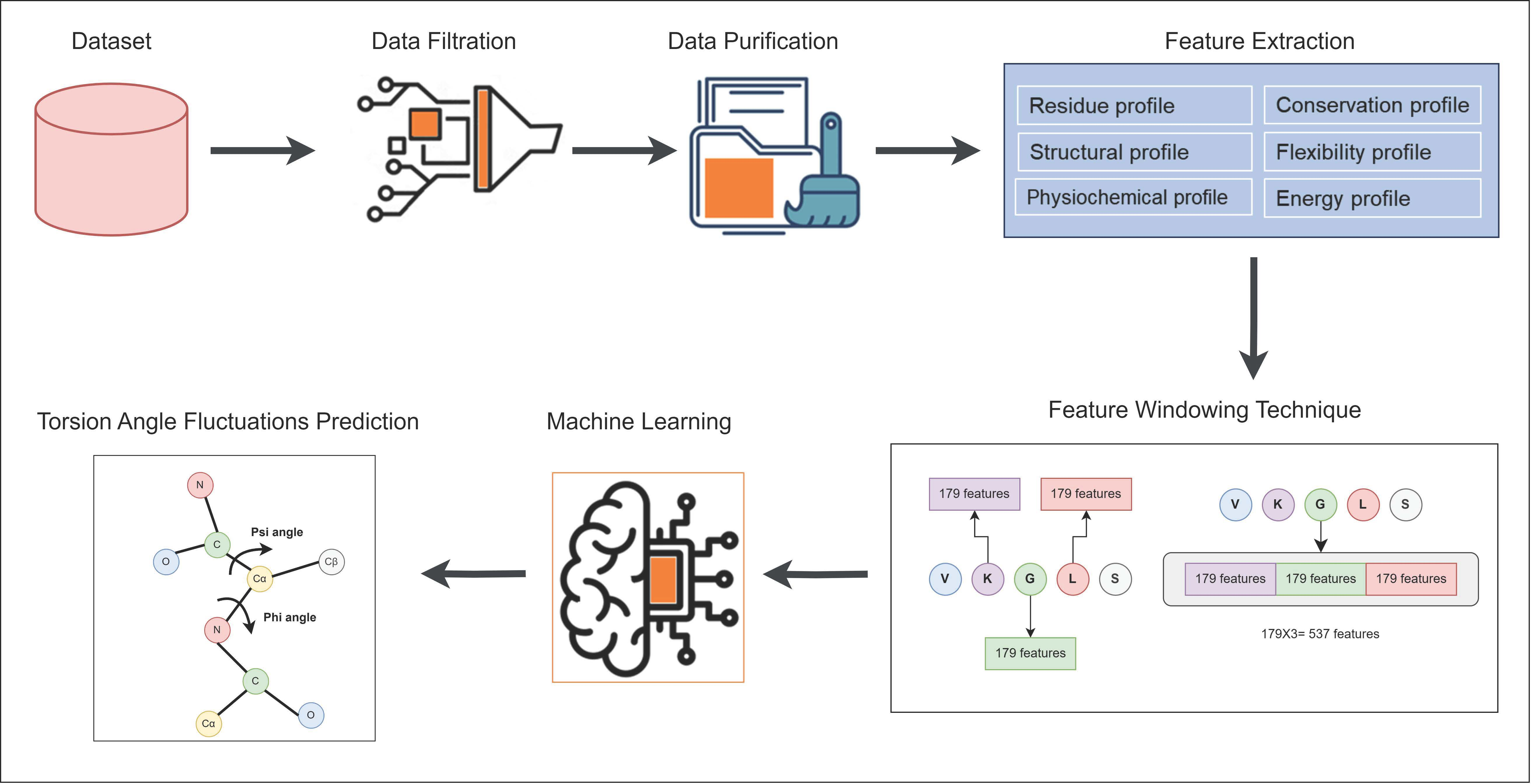
2. Materials and Methods
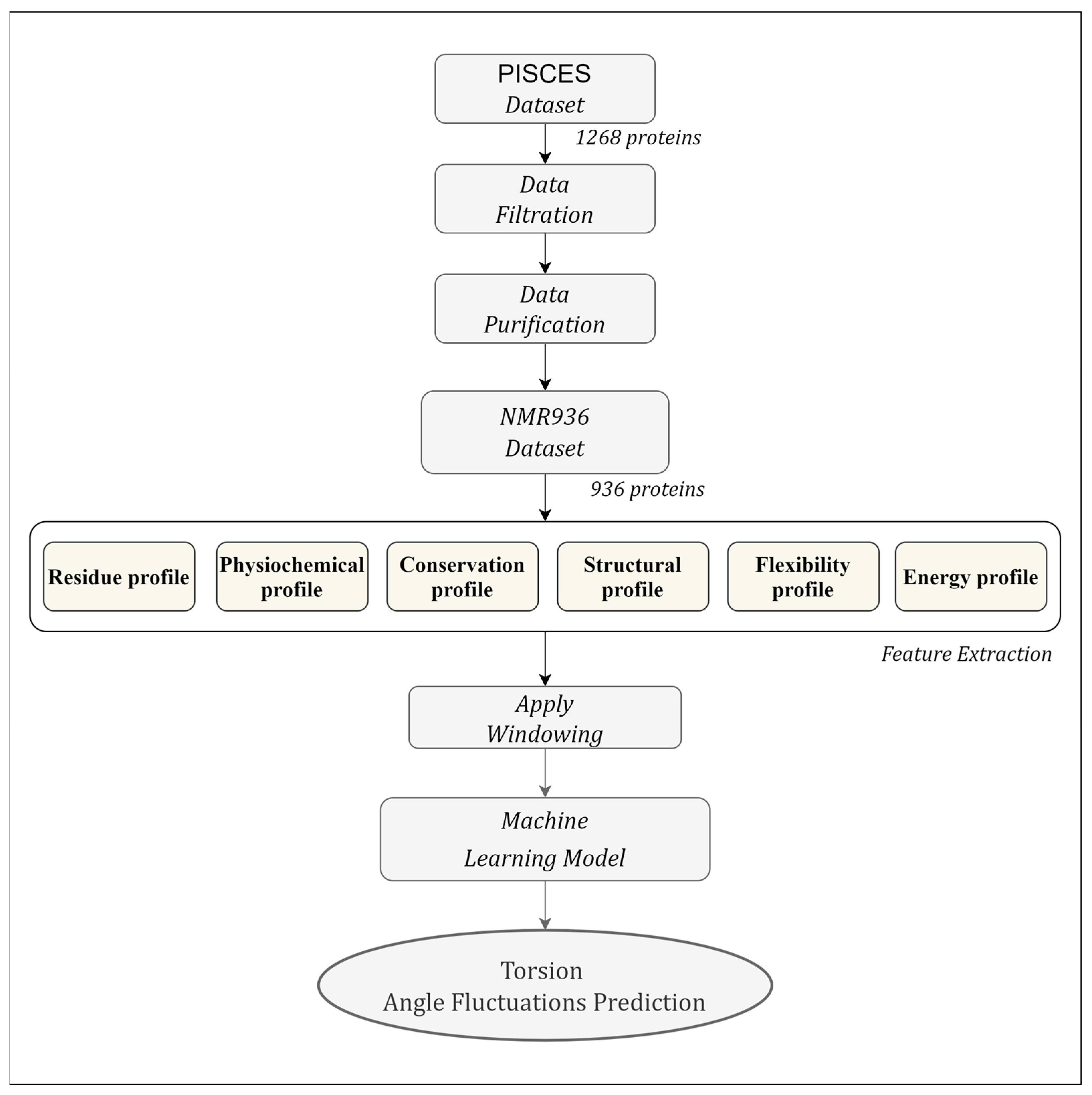
2.1. Dataset
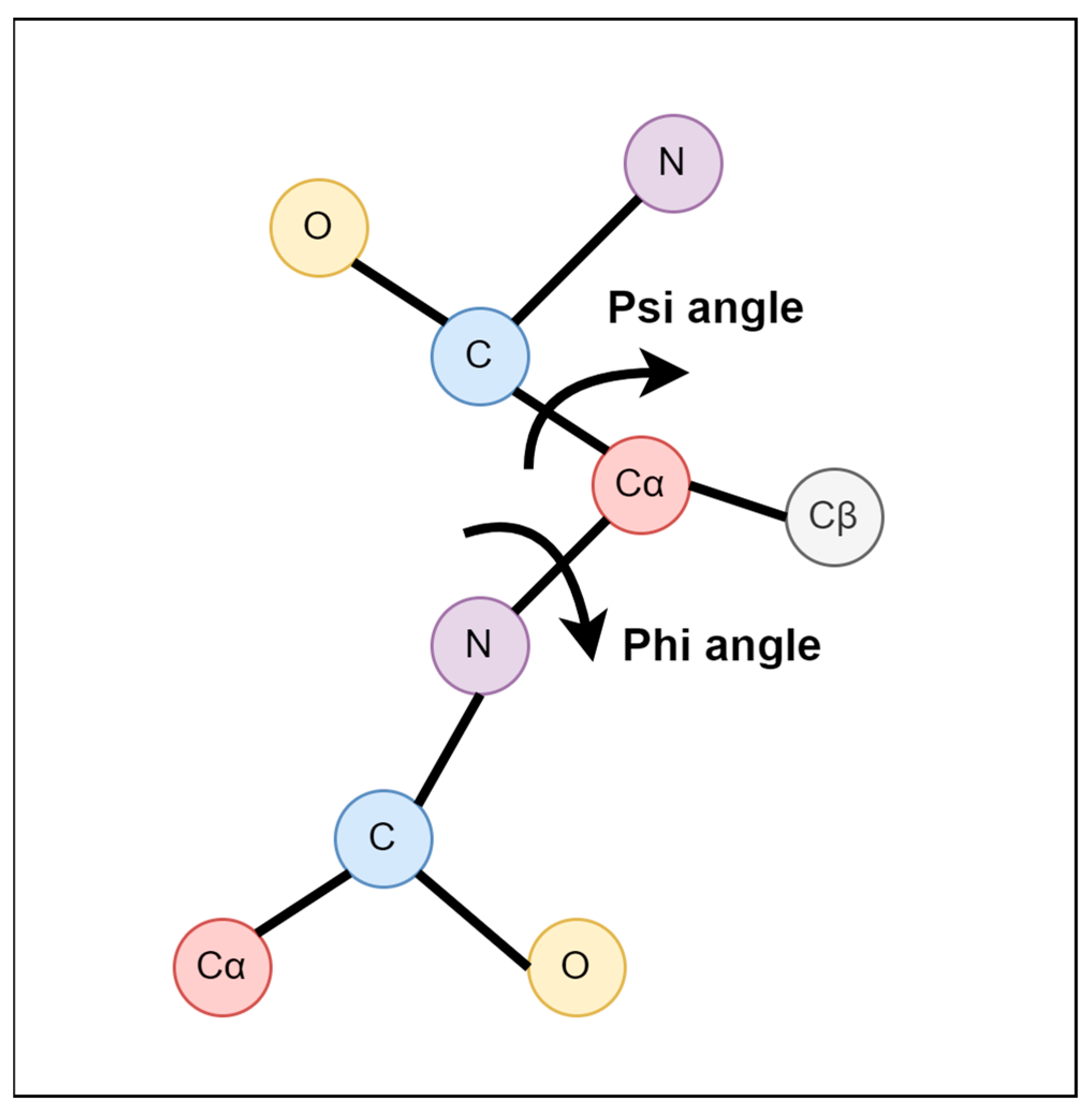
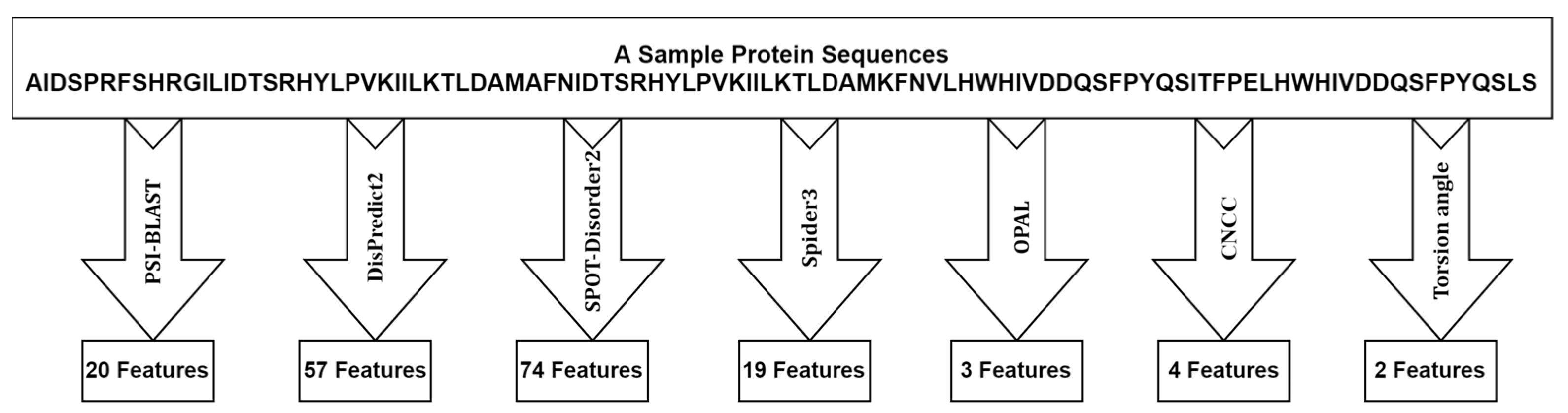
2.2. Feature Extraction
2.3. Machine Learning Methods
2.4. Feature Selection Using Genetic Algorithm (GA)
2.5. Performance Evaluation
3. Results
3.1. Comparison between Different Methods
3.2. Hyperparameters Optimization
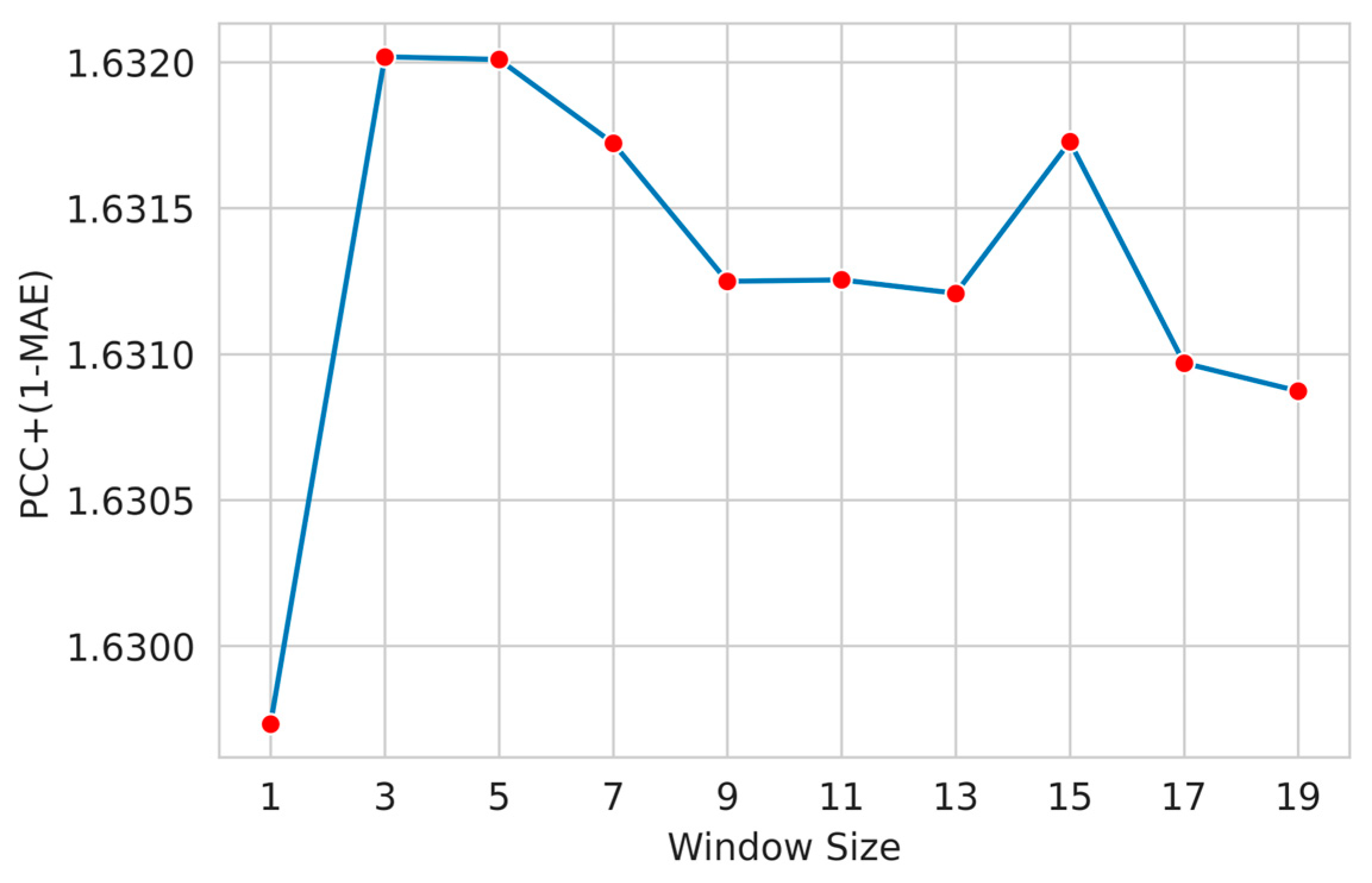
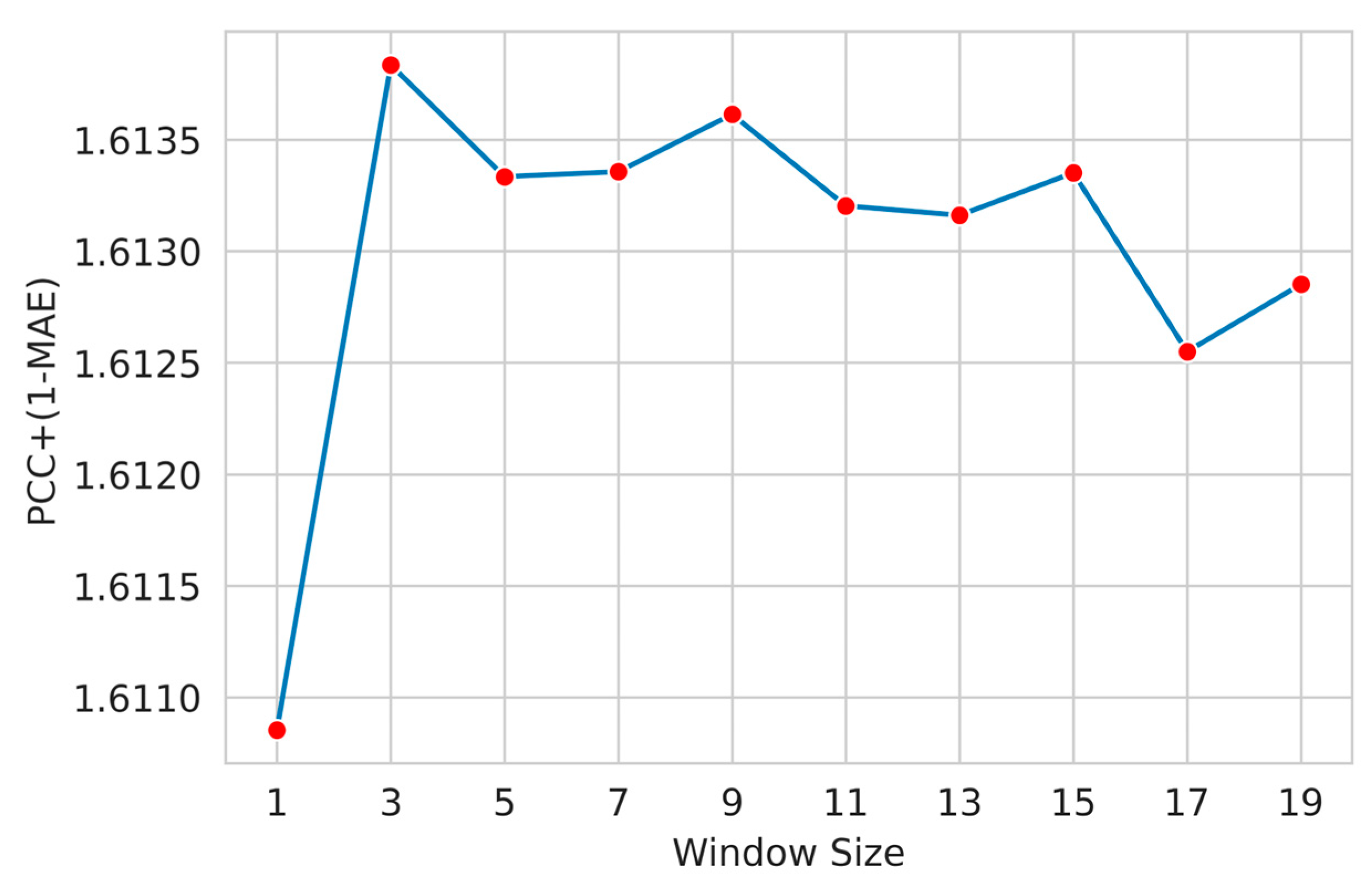
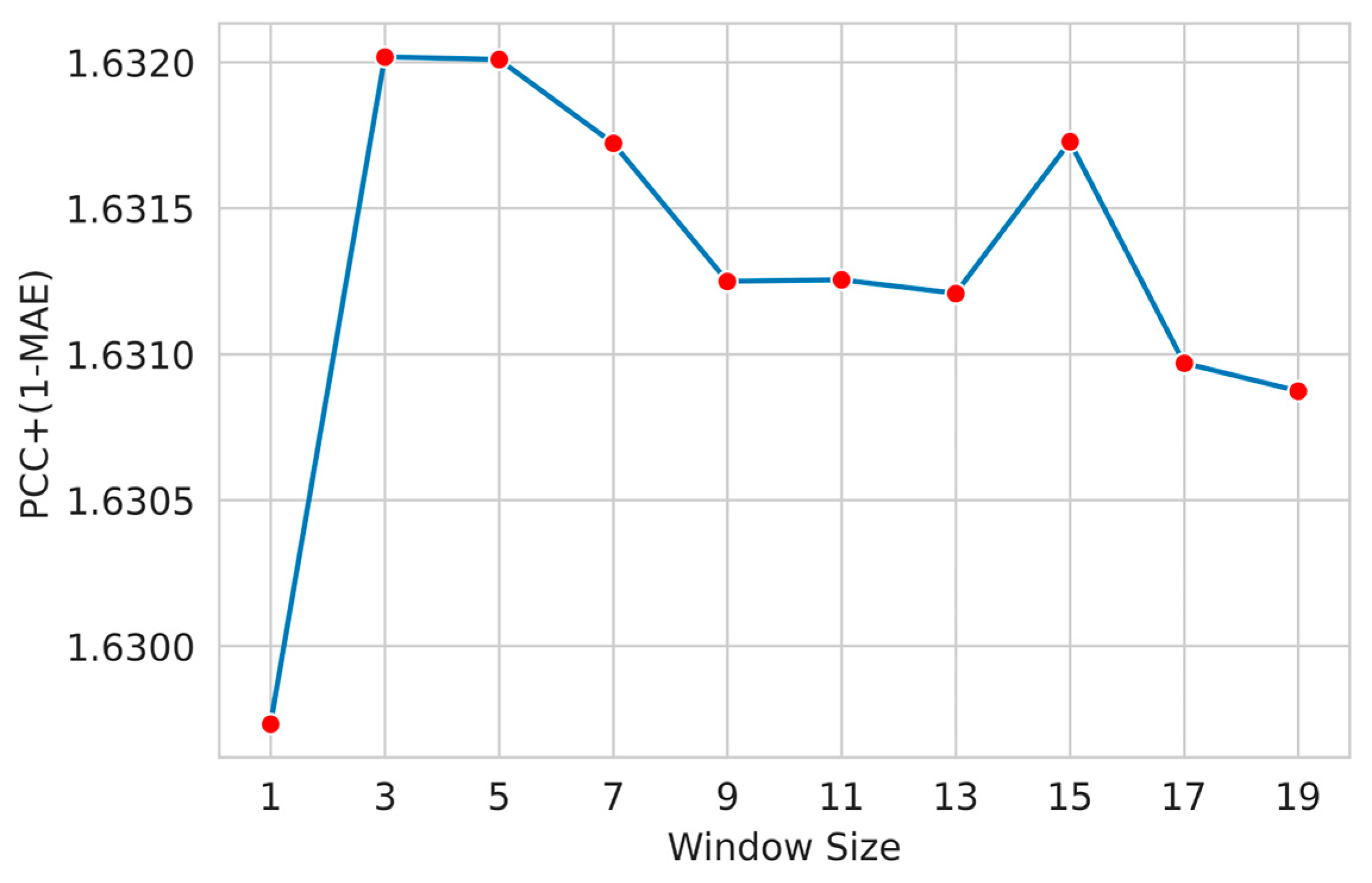
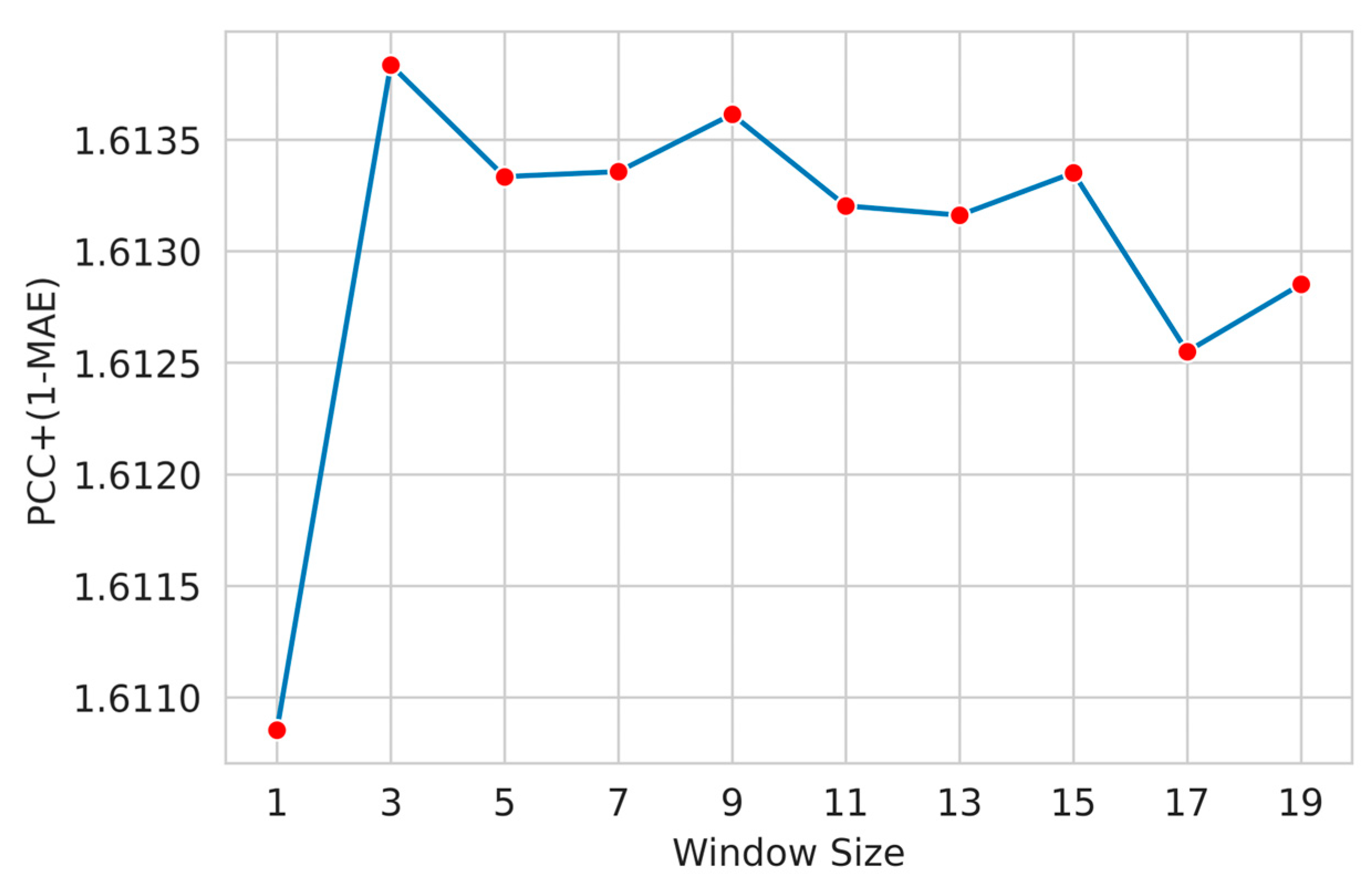
3.3. Feature Window Selection
3.4. Comparison with the State-of-the-Art Method
4. Discussion
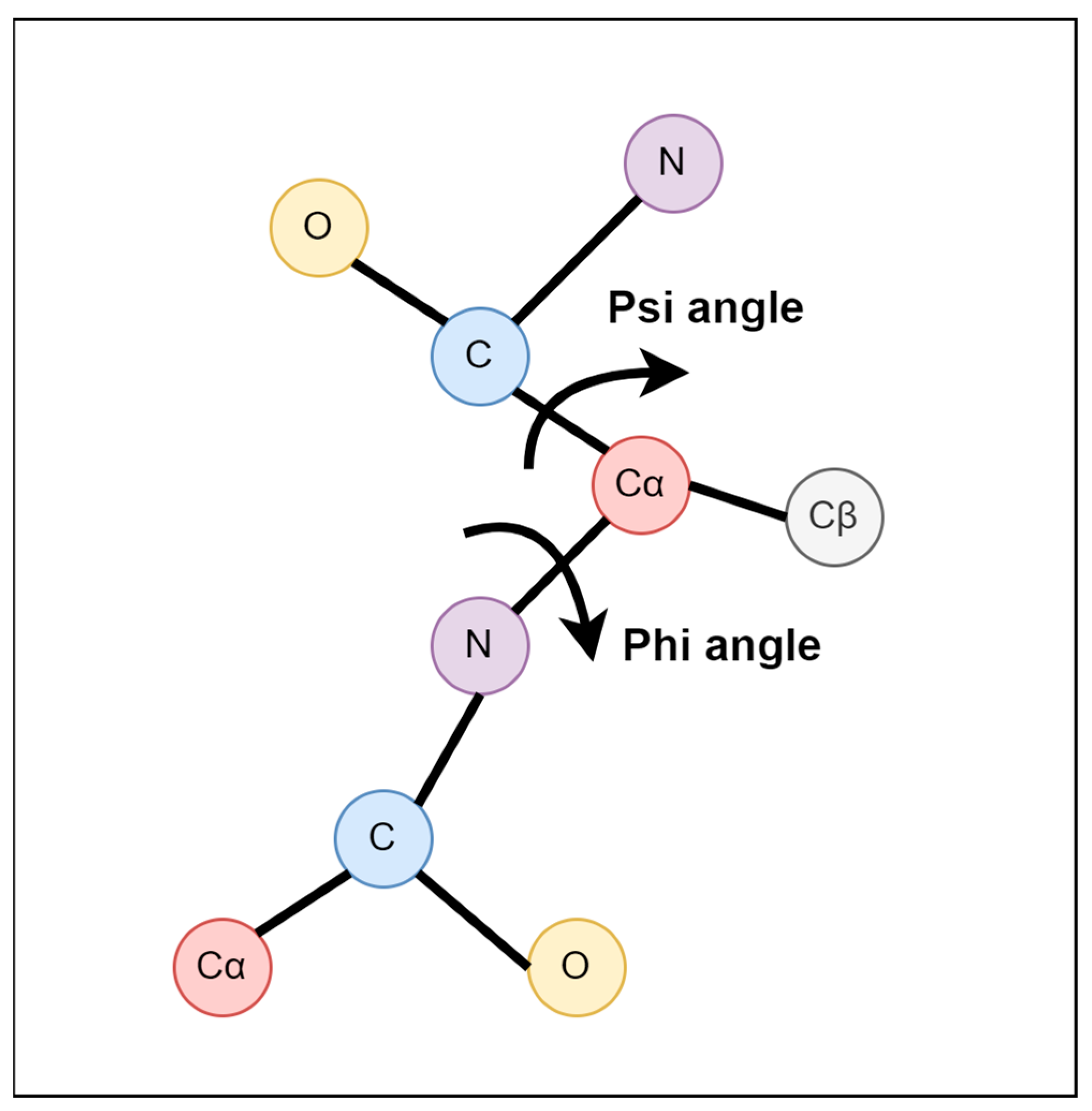
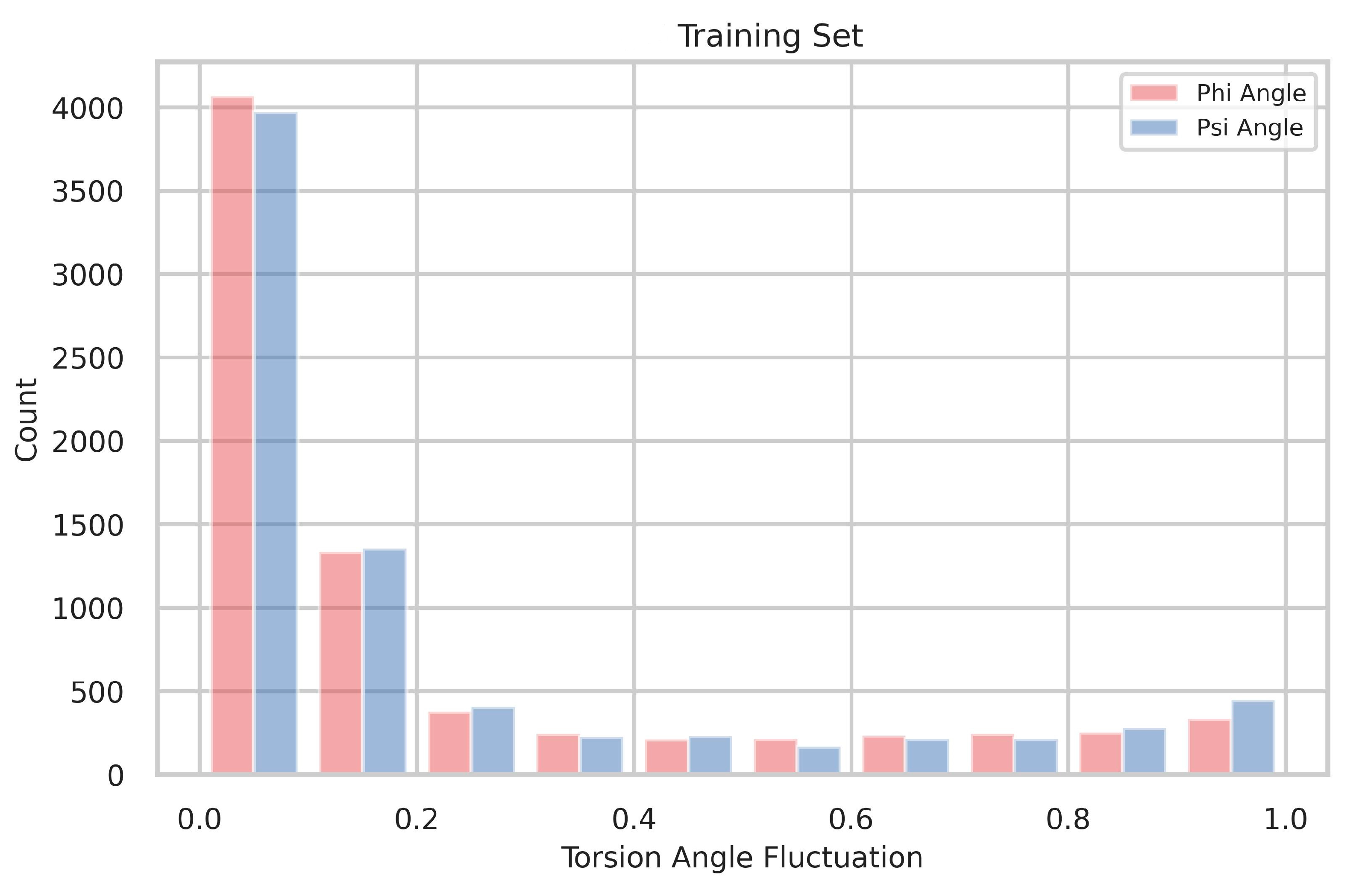
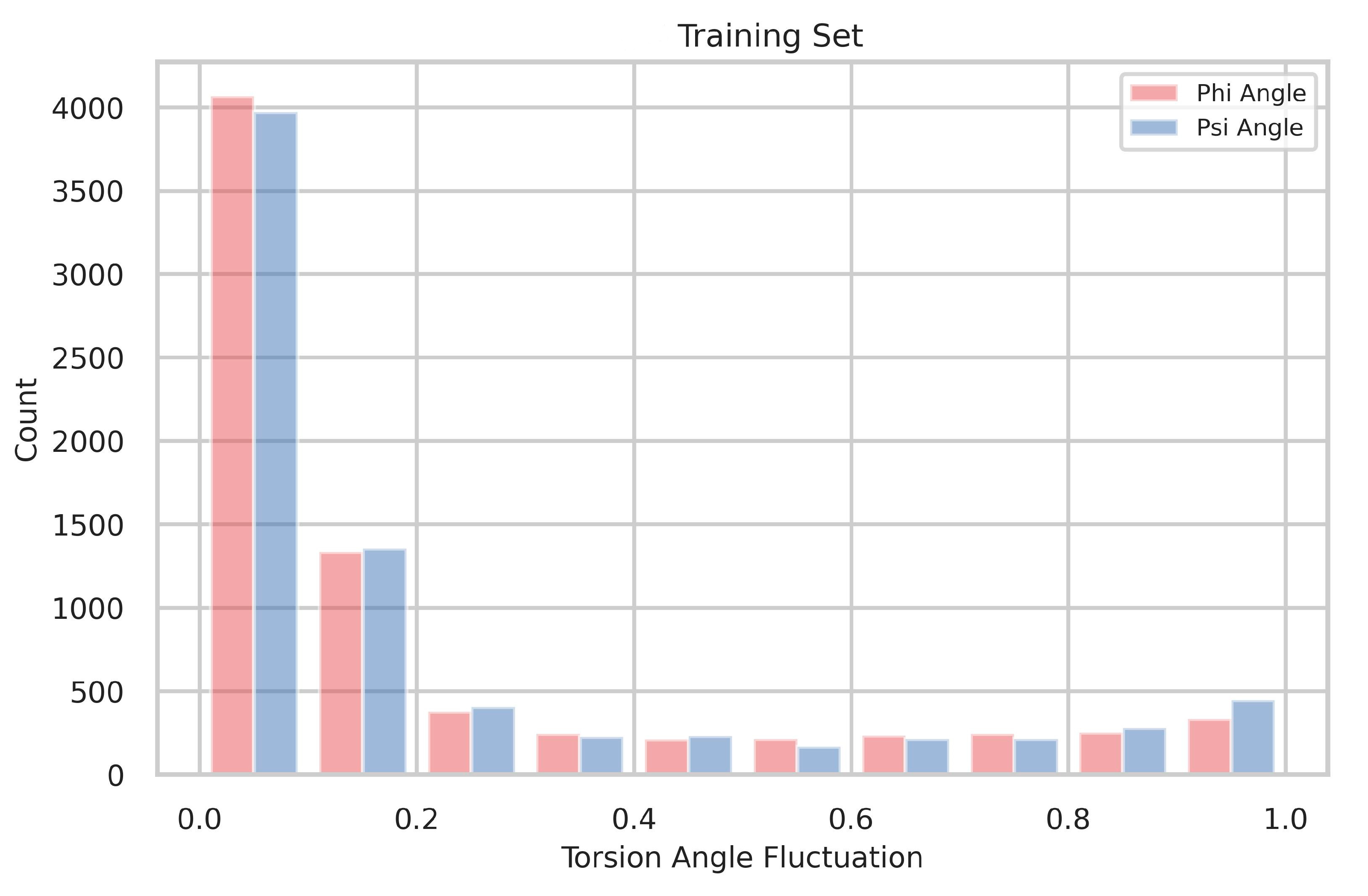
4.1. The Distribution of Torsion-Angle Fluctuation
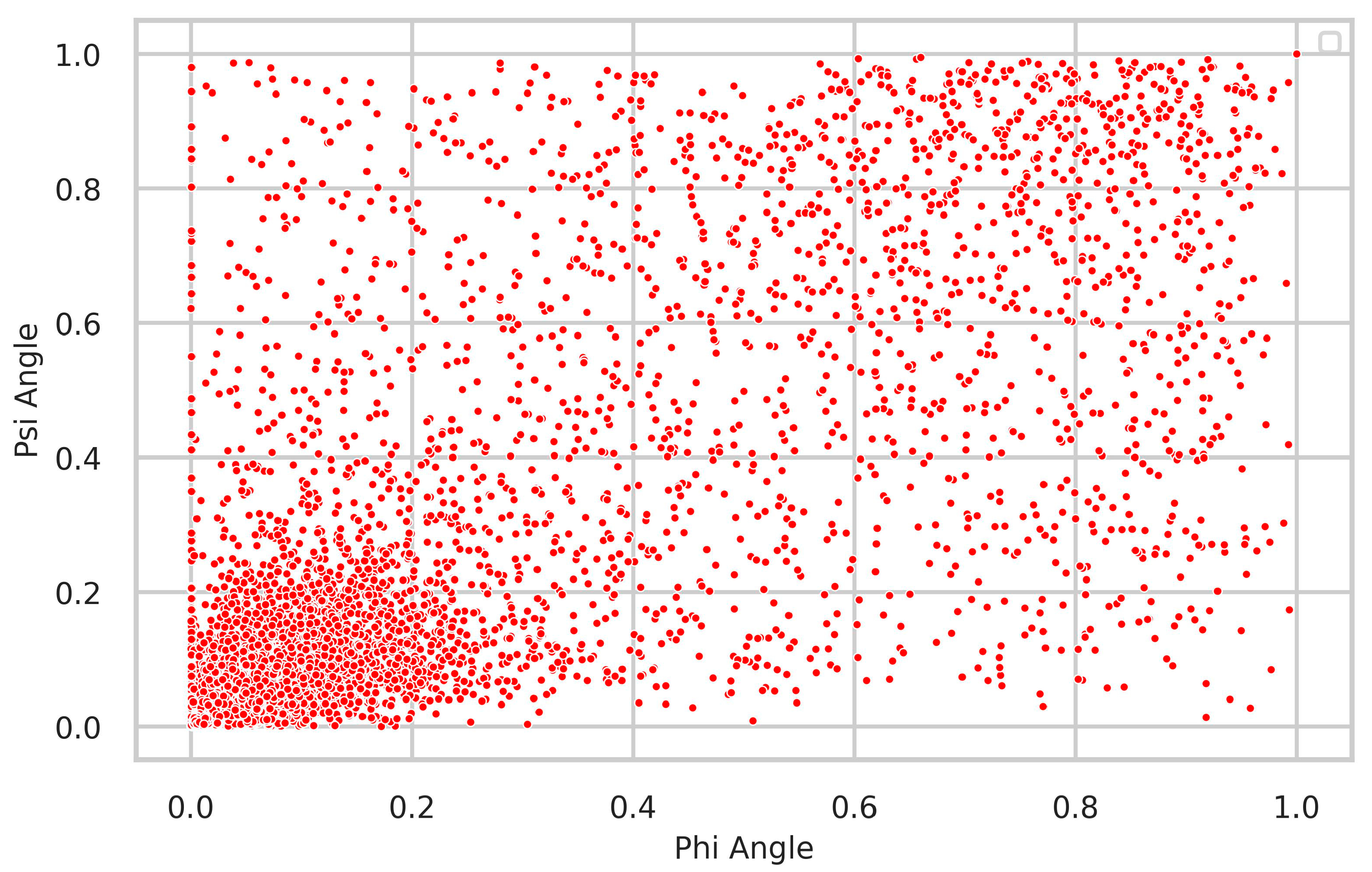
4.2. Relationship between Δφ and Δψ
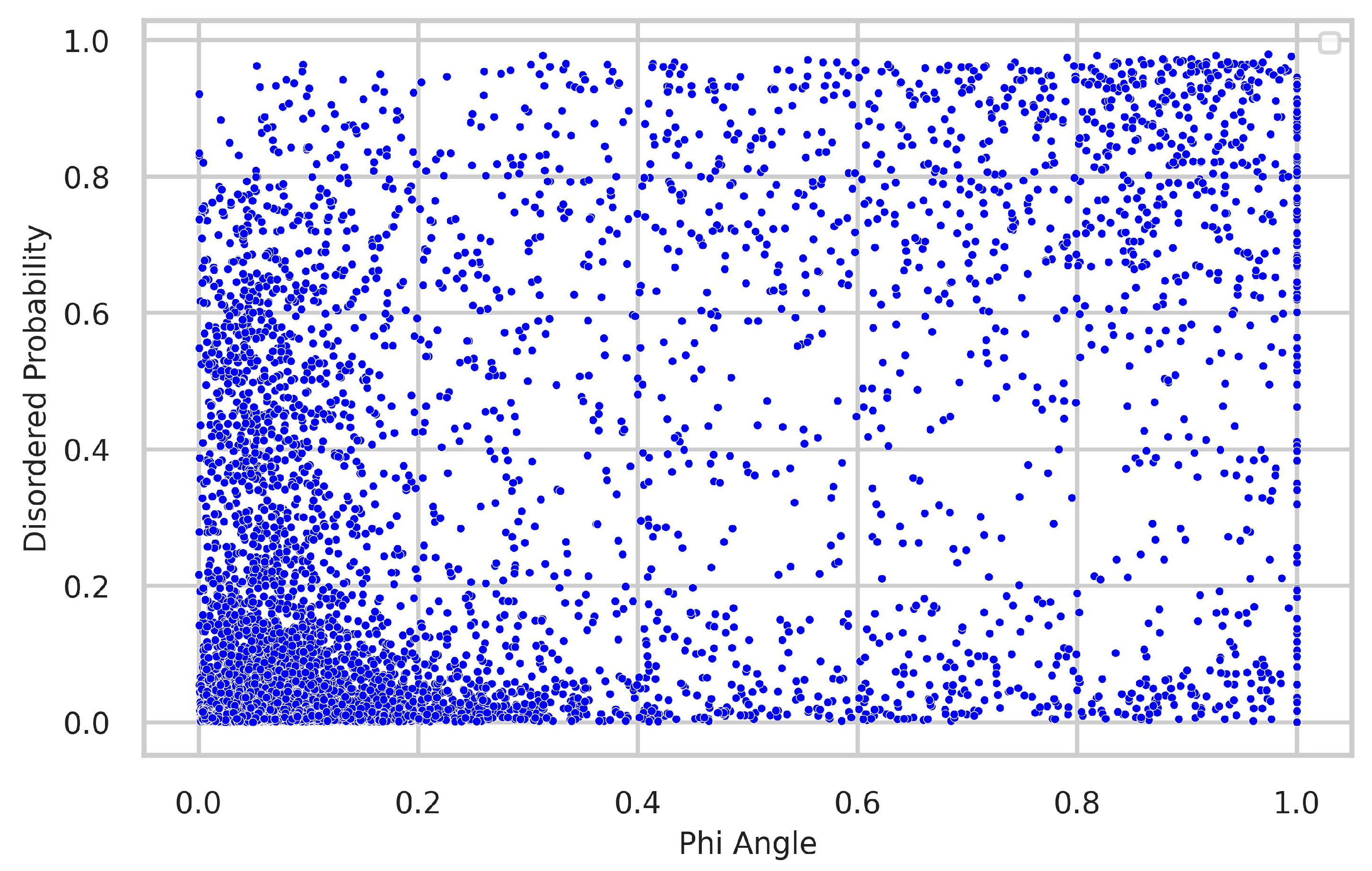
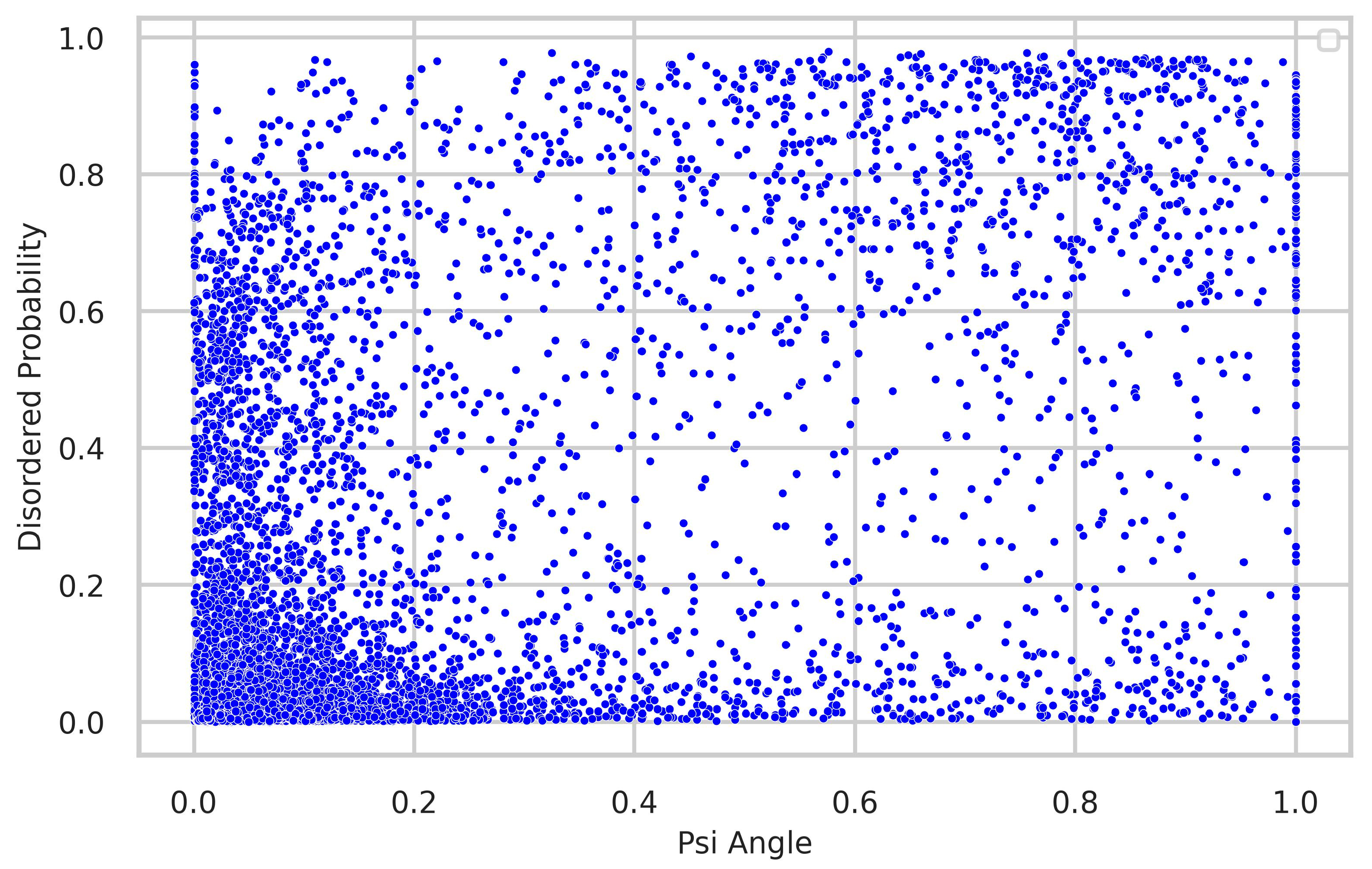
4.3. Relationship between Torsion-Angle Fluctuation and Disordered Regions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biol. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar]
- Jonsson, A.L.; Roberts, M.A.J.; Kiappes, J.L.; Scott, K.A. Essential chemistry for biochemists. Essays Biochem. 2017, 61, 401–427. [Google Scholar] [CrossRef] [PubMed]
- Kulmanov, M.; Hoehndorf, R. DeepGOPlus: Improved protein function prediction from sequence. Bioinformatics 2020, 36, 422–429. [Google Scholar]
- Nechab, M.; Mondal, S.; Bertrand, M.P. 1,n-Hydrogen-Atom Transfer (HAT) Reactions in Which n≠ 5: An Updated Inventory. Chemistry 2014, 20, 16034–16059. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar]
- Quiocho, F.A. Carbohydrate-binding proteins: Tertiary structures and protein-sugar interactions. Annu. Rev. Biochem. 1986, 55, 287–315. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, S.; Meleshko, R.; James, M.N. A critical assessment of comparative molecular modeling of tertiary structures of proteins. Proteins Struct. Funct. Bioinform. 1995, 23, 301–317. [Google Scholar] [CrossRef]
- Gao, J.; Yang, Y.; Zhou, Y. Grid-based prediction of torsion angle probabilities of protein backbone and its application to discrimination of protein intrinsic disorder regions and selection of model structures. BMC Bioinform. 2018, 19, 29. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, R.; Yang, Y.; Paliwal, K.; Zhou, Y. Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics 2017, 33, 2842–2849. [Google Scholar] [CrossRef] [Green Version]
- Lyons, J.; Dehzangi, A.; Heffernan, R.; Sharma, A.; Paliwal, K.; Sattar, A.; Zhou, Y.; Yang, Y. Predicting backbone Cα angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network. J. Comput. Chem. 2014, 35, 2040–2046. [Google Scholar] [CrossRef]
- Iqbal, S.; Hoque, M.T. DisPredict: A Predictor of Disordered Protein Using Optimized RBF Kernel. PLoS ONE 2015, 10, e0141551. [Google Scholar] [CrossRef]
- Konrat, R. NMR contributions to structural dynamics studies of intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Katuwawala, A.; Wang, K.; Wu, Z.; Ghadermarzi, S.; Gao, J.; Kurgan, L. flDPnn: Accurate intrinsic disorder prediction with putative propensities of disorder functions. Nat. Commun. 2021, 12, 4438. [Google Scholar] [CrossRef]
- Zhang, T.; Faraggi, E.; Zhou, Y. Fluctuations of backbone torsion angles obtained from NMR-determined structures and their prediction. Proteins 2010, 78, 3353–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Bung, N.; Vangala, S.R.; Srinivasan, R.; Bulusu, G.; Roy, A. De Novo Structure-Based Drug Design Using Deep Learning. J. Chem. Inf. Model. 2022, 62, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.M.; Laskowski, R.A.; Borkakoti, N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat. Med. 2021, 27, 1666–1669. [Google Scholar] [CrossRef]
- Bulacu, M.; Goga, N.; Zhao, W.; Rossi, G.; Monticelli, L.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Angle Potentials for Coarse-Grained Molecular Dynamics Simulations. J. Chem. Theory Comput. 2013, 9, 3282–3292. [Google Scholar] [CrossRef] [Green Version]
- Yee, A.A.; Savchenko, A.; Ignachenko, A.; Lukin, J.; Xu, X.; Skarina, T.; Evdokimova, E.; Liu, C.S.; Semesi, A.; Guido, V.; et al. NMR and X-ray Crystallography, Complementary Tools in Structural Proteomics of Small Proteins. J. Am. Chem. Soc. 2005, 127, 16512–16517. [Google Scholar] [CrossRef]
- Bryant, R.G. The NMR time scale. J. Chem. Educ. 1983, 60, 933. [Google Scholar] [CrossRef]
- Camacho-Zarco, A.R.; Schnapka, V.; Guseva, S.; Abyzov, A.; Adamski, W.; Milles, S.; Jensen, M.R.; Zidek, L.; Salvi, N.; Blackledge, M. NMR Provides Unique Insight into the Functional Dynamics and Interactions of Intrinsically Disordered Proteins. Chem. Rev. 2022, 122, 9331–9356. [Google Scholar] [CrossRef] [PubMed]
- Adamski, W.; Salvi, N.; Maurin, D.; Magnat, J.; Milles, S.; Jensen, M.R.; Abyzov, A.; Moreau, C.J.; Blackledge, M. A Unified Description of Intrinsically Disordered Protein Dynamics under Physiological Conditions Using NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141, 17817–17829. [Google Scholar] [CrossRef] [PubMed]
- Kosol, S.; Contreras-Martos, S.; Cedeño, C.; Tompa, P. Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules 2013, 18, 10802–10828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graether, S.P. Troubleshooting Guide to Expressing Intrinsically Disordered Proteins for Use in NMR Experiments. Front. Mol. Biosci. 2019, 5, 118. [Google Scholar] [CrossRef]
- Yang, Y.; Faraggi, E.; Zhao, H.; Zhou, Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics 2011, 27, 2076–2082. [Google Scholar] [CrossRef] [Green Version]
- Karchin, R.; Cline, M.; Mandel-Gutfreund, Y.; Karplus, K. Hidden Markov models that use predicted local structure for fold recognition: Alphabets of backbone geometry. Proteins Struct. Funct. Bioinform. 2003, 51, 504–514. [Google Scholar] [CrossRef]
- Rohl, C.A.; Strauss, C.E.; Misura, K.M.; Baker, D. Protein Structure Prediction Using Rosetta. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2004; pp. 66–93. [Google Scholar]
- Faraggi, E.; Yang, Y.; Zhang, S.; Zhou, Y. Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction. Structure 2009, 17, 1515–1527. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; and Zhang, Y. ANGLOR: A composite machine-learning algorithm for protein backbone torsion angle prediction. PLoS ONE 2008, 3, e3400. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, J.; Wang, J.; Heffernan, R.; Hanson, J.; Paliwal, K.; Zhou, Y. Sixty-five years of the long march in protein secondary structure prediction: The final stretch? Brief. Bioinform. 2018, 19, 482–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Hou, J.; Adhikari, B.; Lyu, Q.; Cheng, J. Deep learning methods for protein torsion angle prediction. BMC Bioinform. 2017, 18, 417. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kabir, M.W.U.; Alawad, D.M.; Mishra, A.; Hoque, M.T. Prediction of Phi and Psi Angle Fluctuations from Protein Sequences. In Proceedings of the 20th IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology, Eindhoven, The Netherlands, 29–31 August 2023. [Google Scholar]
- Md Kauser, A.; Avdesh, M.; Md Tamjidul, H. TAFPred: An Efficient Torsion Angle Fluctuation Predictor of a Protein from Its Sequence, Baton Rouge, LA, USA, 6–7 April 2018.
- Iqbal, S.; Mishra, A.; Hoque, T. Improved Prediction of Accessible Surface Area Results in Efficient Energy Function Application. J. Theor. Biol. 2015, 380, 380–391. [Google Scholar] [CrossRef]
- Iqbal, S.; Hoque, M.T. PBRpredict-Suite: A suite of models to predict peptide-recognition domain residues from protein sequence. Bioinformatics 2018, 34, 3289–3299. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.; Hoque, M.T. Estimation of position specific energy as a feature of protein residues from sequence alone for structural classification. PLoS ONE 2016, 11, e0161452. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yang, J.; Song, J.N.; Chou, K.C.; Shen, H.B. Improving the accuracy of predicting disulfide connectivity by feature selection. Comput. Chem. 2010, 31, 1478–1485. [Google Scholar] [CrossRef]
- Islam, M.N.; Iqbal, S.; Katebi, A.R.; Hoque, M.T. A balanced secondary structure predictor. J. Theor. Biol. 2016, 389, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Tan, H.; Rost, B. Loopy proteins appear conserved in evolution. J. Mol. Biol. 2002, 322, 53–64. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. LightGBM: A highly efficient gradient boosting decision tree. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (ACM, 2016), San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning, 2nd ed.; Springer: New York, NY, USA, 2009. [Google Scholar]
- Altman, N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am. Stat. 1992, 46, 175–185. [Google Scholar]
- Arik, S.O.; Pfister, T. TabNet: Attentive Interpretable Tabular Learning. arXiv 2019, arXiv:1908.07442. [Google Scholar] [CrossRef]
- Hoque, M.T.; Iqbal, S. Genetic algorithm-based improved sampling for protein structure prediction. Int. J. Bio-Inspired Comput. 2017, 9, 129–141. [Google Scholar] [CrossRef]
- Hoque, M.T.; Chetty, M.; Sattar, A. Protein Folding Prediction in 3D FCC HP Lattice Model using Genetic Algorithm. In Proceedings of the IEEE Congress on Evolutionary Computation (CEC), Singapore, 25–28 September 2007. [Google Scholar]
- Hoque, M.T.; Chetty, M.; Lewis, A.; Sattar, A.; Avery, V.M. DFS Generated Pathways in GA Crossover for Protein Structure Prediction. Neurocomputing 2010, 73, 2308–2316. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Metric | Definition |
|---|---|
| Pearson Correlation Coefficient (PCC) = | |
| Mean Absolute Error (MAE) = |
| Methods/Metric | MAE | PCC | MAE (% imp.) | PCC (% imp.) | Average (% imp.) |
|---|---|---|---|---|---|
| State-of-the-art Method [16] | 0.126 | 0.598 | - | - | - |
| Extra Trees Regressor | 0.122 | 0.741 | 3.57% | 23.88% | 13.73% |
| XGB Regressor | 0.123 | 0.727 | 2.67% | 21.57% | 12.12% |
| KNN Regressor | 0.129 | 0.681 | −2.30% | 13.89% | 5.79% |
| Decision Tree Regressor | 0.167 | 0.527 | −24.38% | −11.84% | −18.11% |
| LSTM | 0.125 | 0.678 | 1.13% | 13.35% | 7.24% |
| CNN | 0.166 | 0.608 | −24.21% | 1.68% | −11.27% |
| Tabnet | 0.117 | 0.736 | 7.26% | 23.09% | 15.18% |
| LightGBM Regressor | 0.118 | 0.745 | 6.59% | 24.50% | 15.54% |
| Methods/Metric | MAE | PCC | MAE (% imp.) | PCC (% imp.) | Average (% imp.) |
|---|---|---|---|---|---|
| State-of-the-art Method [16] | 0.135 | 0.602 | - | - | - |
| Extra Trees Regressor | 0.131 | 0.729 | 2.77% | 21.10% | 11.94% |
| XGB Regressor | 0.132 | 0.715 | 2.22% | 18.73% | 10.48% |
| KNN Regressor | 0.139 | 0.670 | −2.63% | 11.24% | 4.31% |
| Decision Tree Regressor | 0.179 | 0.511 | −24.65% | −15.11% | −19.88% |
| LSTM | 0.132 | 0.665 | 2.29% | 10.48% | 6.38% |
| CNN | 0.144 | 0.702 | −6.46% | 16.61% | 5.07% |
| Tabnet | 0.126 | 0.724 | 7.24% | 20.28% | 13.76% |
| LightGBM Regressor | 0.127 | 0.733 | 6.09% | 21.84% | 13.96% |
| Methods/Metric | MAE | PCC | MAE (% imp.) | PCC (% imp.) | Average (% imp.) |
|---|---|---|---|---|---|
| State-of-the-art Method [16] | 0.126 | 0.598 | - | - | - |
| TAFPred | 0.114 | 0.746 | 10.08% | 24.83% | 17.45% |
| Methods/Metric | MAE | PCC | MAE (% imp.) | PCC (% imp.) | Average (% imp.) |
|---|---|---|---|---|---|
| State-of-the-art Method [16] | 0.135 | 0.602 | - | - | - |
| TAFPred | 0.123 | 0.737 | 9.93% | 22.37% | 16.15% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabir, M.W.U.; Alawad, D.M.; Mishra, A.; Hoque, M.T. TAFPred: Torsion Angle Fluctuations Prediction from Protein Sequences. Biology 2023, 12, 1020. https://doi.org/10.3390/biology12071020
Kabir MWU, Alawad DM, Mishra A, Hoque MT. TAFPred: Torsion Angle Fluctuations Prediction from Protein Sequences. Biology. 2023; 12(7):1020. https://doi.org/10.3390/biology12071020
Chicago/Turabian StyleKabir, Md Wasi Ul, Duaa Mohammad Alawad, Avdesh Mishra, and Md Tamjidul Hoque. 2023. "TAFPred: Torsion Angle Fluctuations Prediction from Protein Sequences" Biology 12, no. 7: 1020. https://doi.org/10.3390/biology12071020
APA StyleKabir, M. W. U., Alawad, D. M., Mishra, A., & Hoque, M. T. (2023). TAFPred: Torsion Angle Fluctuations Prediction from Protein Sequences. Biology, 12(7), 1020. https://doi.org/10.3390/biology12071020