Cannabinoids Reduce Melanoma Cell Viability and Do Not Interfere with Commonly Used Targeted Therapy in Metastatic Melanoma In Vivo and In Vitro

 ,
,  , ,
, ,  and
and 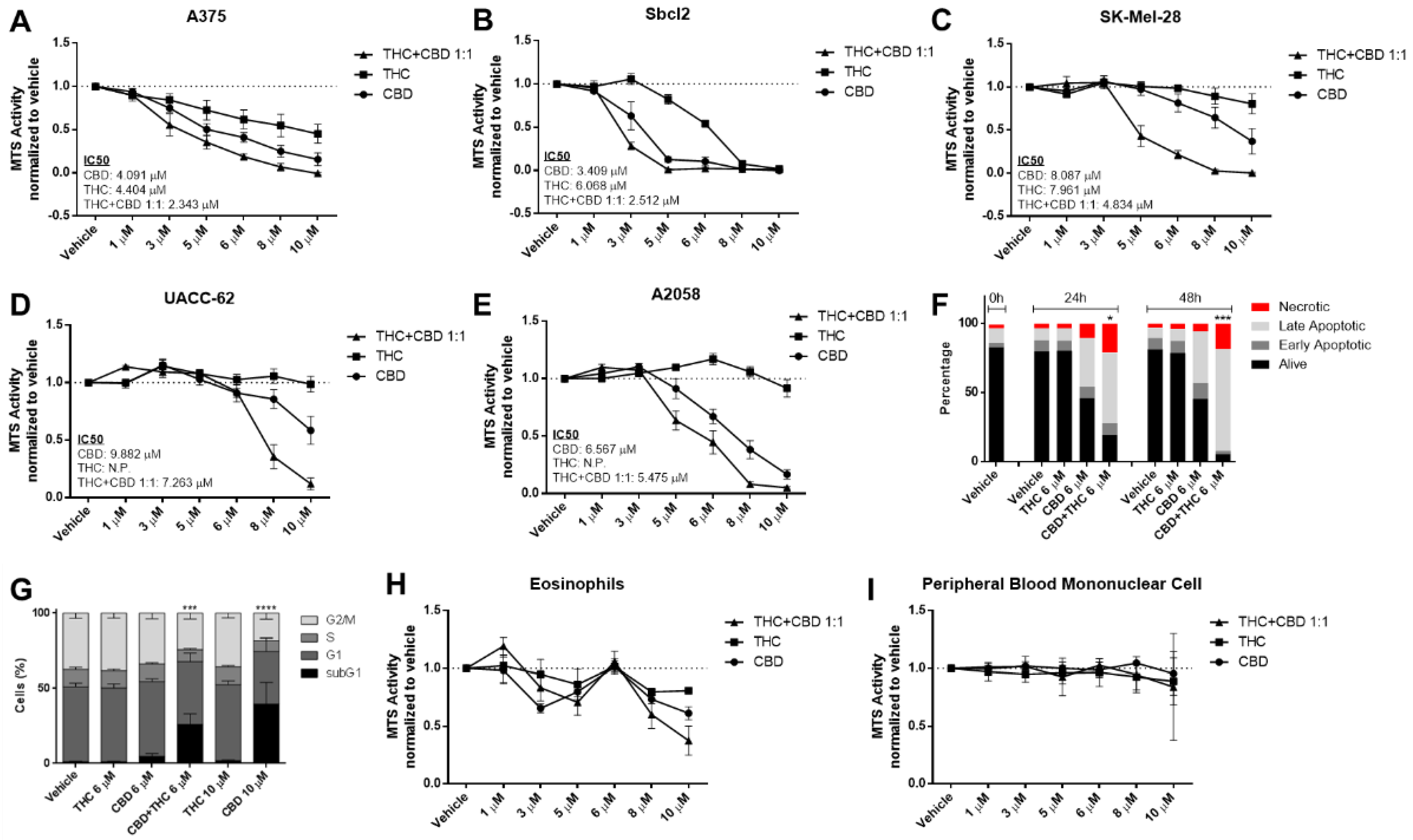{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Lines
2.3. Cell Viability Assay
2.4. Cell Cycle Analysis
2.5. AnnexinV/PI Co-Staining
2.6. Caspase-3/7—Glo Assay
2.7. JC-1 Staining
2.8. Crystal Violet Assay
2.9. Immunohistochemistry
2.10. Cancer Genomics
2.11. In Vivo Experiments
2.12. Statistical Analysis
3. Results
3.1. Cannabinoids Reduce Cell Viability among a Variety of Melanoma Cell Lines in a Concentration-Dependent Manner
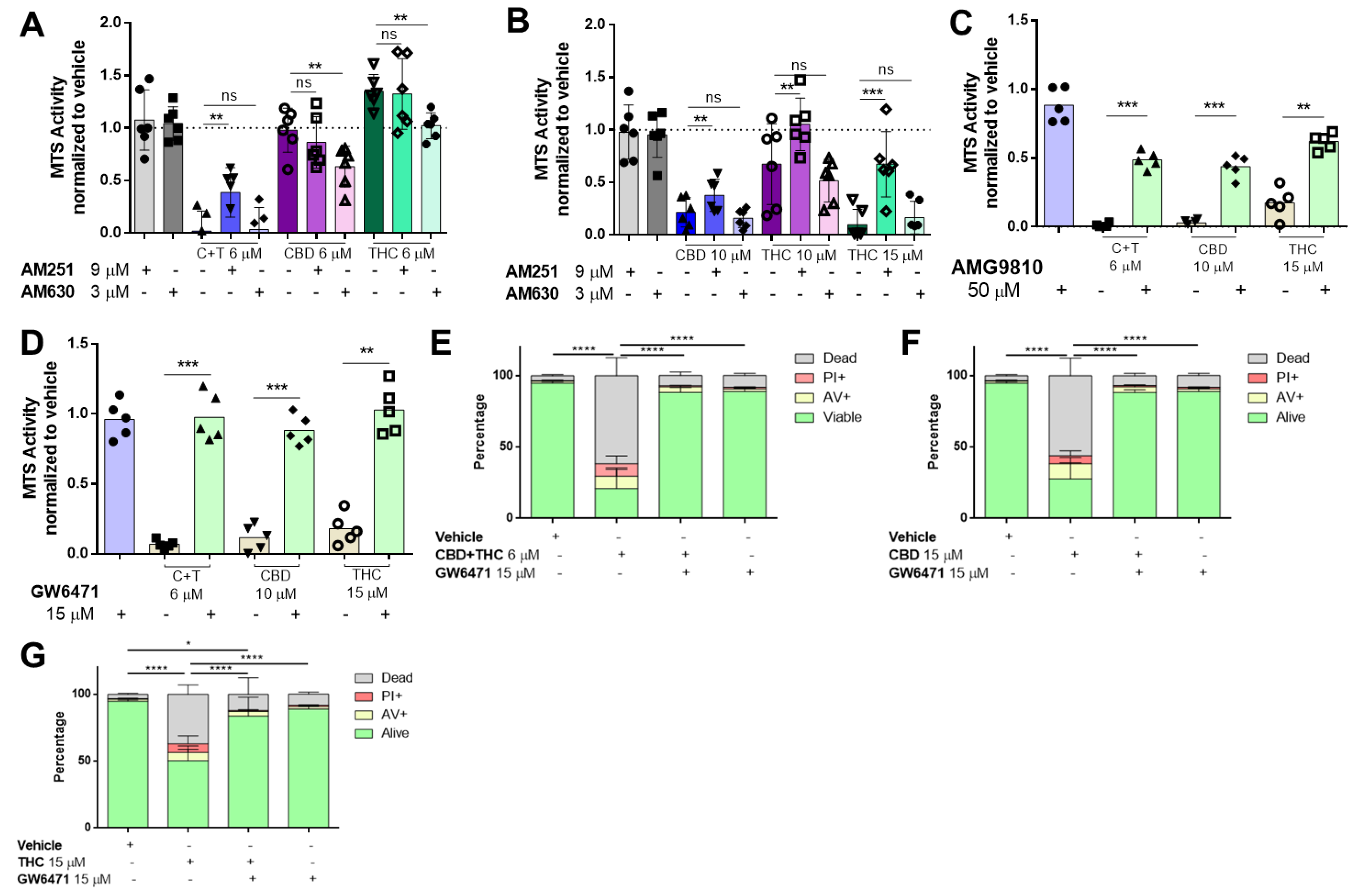
3.2. Cannabinoids Mediate Their Effects by Activation of CB1, TRPV1 and PPARα Receptors
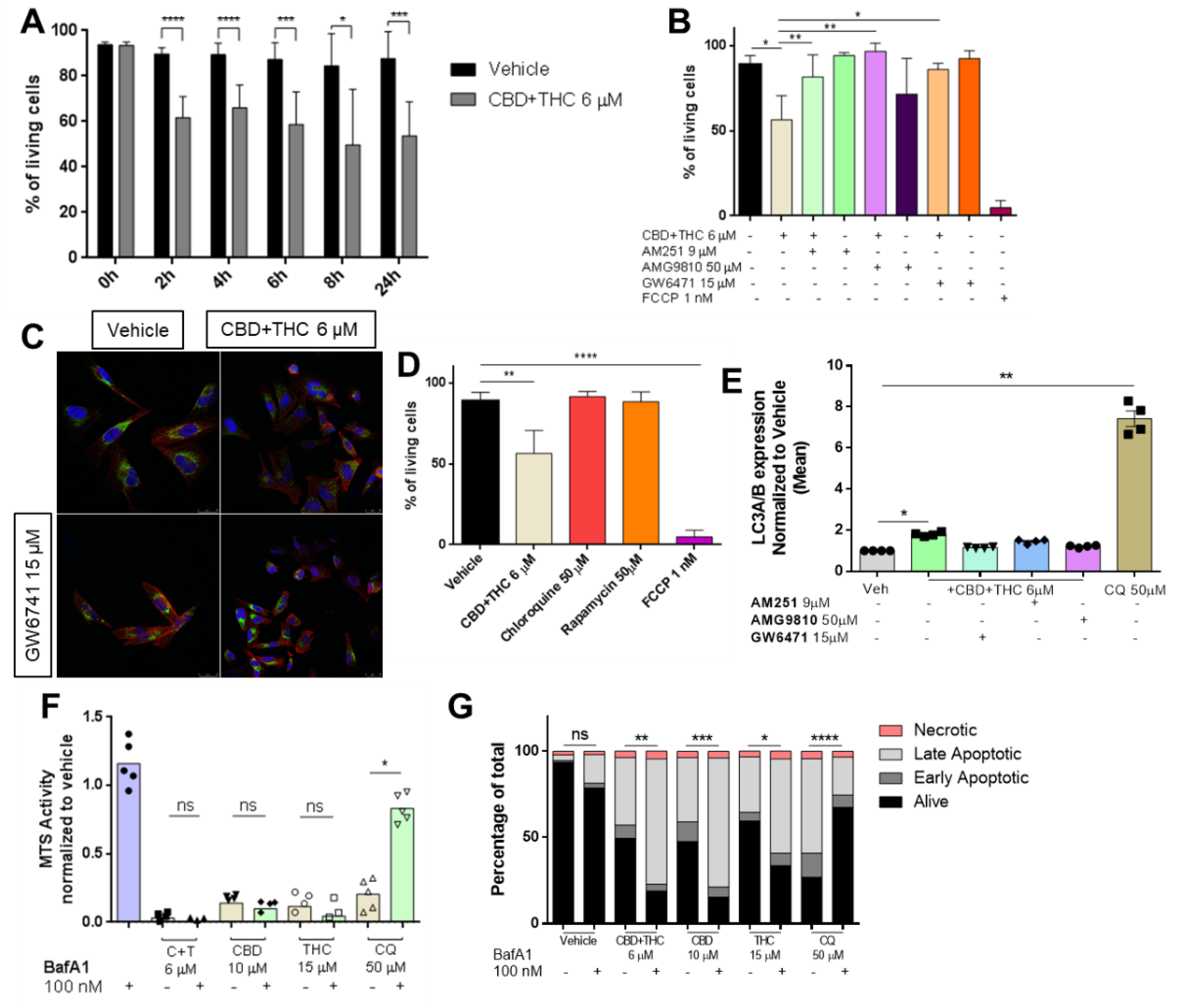
3.3. Cannabinoids Impair Mitochondrial Integrity
3.4. Cannabinoids Do Not Interfere with Commonly Used Targeted Therapy
3.5. Cannabinoids Delay Melanoma Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Wode, K.; Henriksson, R.; Sharp, L.; Stoltenberg, A.; Nordberg, J.H. Cancer patients’ use of complementary and alternative medicine in Sweden: A cross-sectional study. BMC Complement. Altern. Med. 2019, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Buckner, C.A.; Lafrenie, R.M.; Dénommée, J.A.; Caswell, J.M.; Want, D.A. Complementary and alternative medicine use in patients before and after a cancer diagnosis. Curr. Oncol. 2018, 25. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Wagner, A.; Neureiter, D.; Pichler, M.; Jakab, M.; Illig, R.; Berr, F.; Kiesslich, T. The green tea catechin epigallocatechin gallate induces cell cycle arrest and shows potential synergism with cisplatin in biliary tract cancer cells. BMC Complement. Altern. Med. 2015, 15, 194. [Google Scholar] [CrossRef] [PubMed]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Properties of Cannabidiol. Antioxidants 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Atakan, Z. Cannabis, a complex plant: Different compounds and different effects on individuals. Ther. Adv. Psychopharmacol. 2012, 2, 241–254. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Aso, E.; Ferrer, I. Cannabinoids for treatment of alzheimer’s disease: Moving toward the clinic. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef]
- Grotenhermen, F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin. Pharm. 2003, 42, 327–360. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB 1 and CB 2. Pharm. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.; Fischer, O.M.; Ullrich, A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 2004, 64, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Δ-9-Tetrahydrocannabinol Enhances Breast Cancer Growth and Metastasis by Suppression of the Antitumor Immune Response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Theiler, A.; Bärnthaler, T.; Platzer, W.; Richtig, G.; Peinhaupt, M.; Rittchen, S.; Kargl, J.; Ulven, T.; Marsh, L.M.; Marsche, G.; et al. Butyrate ameliorates allergic airway inflammation by limiting eosinophil trafficking and survival. J. Allergy Clin. Immunol. 2019, 144, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.M.; Jørgensen, J.T.; Binderup, T.; Kjær, A. Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Med. Imaging 2008, 8, 16. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Portenoy, R.K.; Ganae-Motan, E.D.; Allende, S.; Yanagihara, R.; Shaiova, L.; Weinstein, S.; McQuade, R.; Wright, S.; Fallon, M.T. Nabiximols for Opioid-Treated Cancer Patients With Poorly-Controlled Chronic Pain: A Randomized, Placebo-Controlled, Graded-Dose Trial. J. Pain. 2012, 13, 438–449. [Google Scholar] [CrossRef]
- Lynch, M.E.; Cesar-Rittenberg, P.; Hohmann, A.G.; Double-Blind, A. Placebo-Controlled, Crossover Pilot Trial With Extension Using an Oral Mucosal Cannabinoid Extract for Treatment of Chemotherapy-Induced Neuropathic Pain. J. Pain. Symptom Manag. 2014, 47, 166–173. [Google Scholar] [CrossRef]
- López-Valero, I.; Torres, S.; Salazar-Roa, M.; García-Taboada, E.; Hernández-Tiedra, S.; Guzmán, M.; Sepúlveda, J.M.; Velasco, G.; Lorente, M. Optimization of a preclinical therapy of cannabinoids in combination with temozolomide against glioma. Biochem. Pharm. 2018, 157, 275–284. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.P.; Pincus, L.B.; Ruben, B.S.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Fung, C.; Shahheydari, H.; Todd, J.R.; Boyd, S.C.; Irvine, M.; Nagrial, A.M.; Scolyer, R.A.; Kefford, R.F.; Long, G.V.; et al. Preexisting MEK1 P124 Mutations Diminish Response to BRAF Inhibitors in Metastatic Melanoma Patients. Clin. Cancer Res. 2015, 21, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Nagarkatti, P.; Pandey, R.; Rieder, S.A.; Hegde, V.L.; Nagarkatti, M. Cannabinoids as novel anti-inflammatory drugs. Future Med. Chem. 2009, 1, 1333–1349. [Google Scholar] [CrossRef] [PubMed]
- Delyon, J.; Mateus, C.; Lefeuvre, D.; Lanoy, E.; Zitvogel, L.; Chaput, N.; Roy, S.; Eggermont, A.M.M.; Routier, E.; Robert, C. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: An early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann. Oncol. 2013, 24, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Chen, W. Dual Roles of Immune Cells and Their Factors in Cancer Development and Progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharm. 2016, 173, 1899–1910. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Anagnostou, M.E.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Derm. 2015, 135, 1629–1637. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.-H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell. 2017, 68, 281–292.e5. [Google Scholar] [CrossRef] [PubMed]
- Tsapras, P.; Nezis, I.P. Caspase involvement in autophagy. Cell. Death Differ. 2017, 24, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, K.; Liu, P.; Zhang, X.; Dong, X.; Gao, J.; Liu, Q.; Barr, M.P.; Zhang, Q.; Hou, X.; et al. Bafilomycin A1 induces caspase-independent cell death in hepatocellular carcinoma cells via targeting of autophagy and MAPK pathways. Sci. Rep. 2016, 6, 37052. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Chaiyamongkol, W.; Doolittle, A.C.; Johnson, Z.I.; Gogate, S.S.; Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. COX-2 expression mediated by calcium-TonEBP signaling axis under hyperosmotic conditions serves osmoprotective function in nucleus pulposus cells. J. Biol. Chem. 2018, 293, 8969–8981. [Google Scholar] [CrossRef]
- Xiao, J.; Egger, M.E.; McMasters, K.M.; Hao, H. Differential expression of ABCB5 in BRAF inhibitor-resistant melanoma cell lines. BMC Cancer 2018, 18, 675. [Google Scholar] [CrossRef]
- Ohsie, S.J.; Sarantopoulos, G.P.; Cochran, A.J.; Binder, S.W. Immunohistochemical characteristics of melanoma. J. Cutan. Pathol. 2008, 35, 433–444. [Google Scholar] [CrossRef]
- Casanova, M.L.; Blázquez, C.; Martínez-Palacio, J.; Villanueva, C.; Fernández-Aceñerp, M.J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Investig. 2003, 111, 43–50. [Google Scholar] [CrossRef]
- Liu, C.; Sadat, S.H.; Ebisumoto, K.; Sakai, A.; Panuganti, B.A.; Ren, S.; Goto, Y.; Haft, S.; Fukusumi, T.; Ando, M.; et al. Cannabinoids Promote Progression of HPV-Positive Head and Neck Squamous Cell Carcinoma via p38 MAPK Activation. Clin. Cancer Res. 2020, 26, 2693–2703. [Google Scholar] [CrossRef]
- Blázquez, C.; Salazar, M.; Carracedo, A.; Lorente, M.; Egia, A.; González-Feria, L.; Haro, A.; Velasco, G.; Guzmán, M. Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res. 2008, 68, 1945–1952. [Google Scholar] [CrossRef]
- Preet, A.; Ganju, R.; Groopman, J. Δ9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene 2008, 27, 339–346. [Google Scholar] [CrossRef]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117–3129. [Google Scholar] [CrossRef]
- Ramer, R.; Hinz, B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 2008, 100, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Frei, R.B.; Luschnig, P.; Parzmair, G.P.; Peinhaupt, M.; Schranz, S.; Fauland, A.; Wheelock, C.E.; Heinemann, A.; Sturm, E.M. Cannabinoid receptor 2 augments eosinophil responsiveness and aggravates allergen-induced pulmonary inflammation in mice. Allergy 2016, 71, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Abood, M.E. CB 1 and CB 2 Receptor Pharmacology. Adv. Pharmacol. 2017, 80, 169–206. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Basu, S.; Maresz, K.; Bifulco, M.; Dittel, B.N. What we know and do not know about the cannabinoid receptor 2 (CB2). Semin. Immunol. 2014, 26, 369–379. [Google Scholar] [CrossRef]
- Hillard, C.J. Stress regulates endocannabinoid-CB1 receptor signaling. Semin. Immunol. 2014, 26, 380–388. [Google Scholar] [CrossRef]
- Moldrich, G.; Wenger, T. Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study✩. Peptides 2000, 21, 1735–1742. [Google Scholar] [CrossRef]
- Shakhova, O. Neural crest stem cells in melanoma development. Curr. Opin. Oncol. 2014, 26, 215–221. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Q.; Xu, Y.; Gong, X.; Zhang, R.; Zhou, C.; Su, Z.; Jin, J.; Shi, H.; Shi, J.; et al. PPARα induces cell apoptosis by destructing Bcl2. Oncotarget 2015, 6, 44635–44642. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Q.; Zhang, J.; Yang, G.; Shao, Z.; Luo, J.; Fan, M.; Ni, C.; Wu, Z.; Hu, X. Fenofibrate induces apoptosis of triple-negative breast cancer cells via activation of NF-κB pathway. BMC Cancer 2014, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.G.; Ball, G.R.; Rakha, E.A.; Nolan, C.C.; Caldas, C.; Ellis, I.O.; Green, A.R. Lack of expression of the proteins GMPR2 and PPARα are associated with the basal phenotype and patient outcome in breast cancer. Breast Cancer Res. Treat. 2013, 137, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Plonka, P.M.; Urbanska, K.; Reiss, K. Peroxisome Proliferator–Activated Receptor α Activation Decreases Metastatic Potential of Melanoma Cells In vitro via Down-Regulation of Akt. Clin. Cancer Res. 2006, 12, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) Apoptosis Systems. Exp. Cell. Res. 2000, 256, 58–66. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell. Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell. Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Marsicano, G.; Desprez, T. Cannabinoids and Mitochondria. In Endocannabinoids and Lipid Mediators in Brain Functions; Springer International Publishing: Cham, Switzerland, 2017; pp. 211–235. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, S.U.; Oh, U.; Jin, B.K. Transient Receptor Potential Vanilloid Subtype 1 Mediates Microglial Cell Death In Vivo and In Vitro via Ca 2+ -Mediated Mitochondrial Damage and Cytochrome c Release. J. Immunol. 2006, 177, 4322–4329. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, S.U.; Oh, U.; Jin, B.K. Transient Receptor Potential Vanilloid 1 Expression Mediates Capsaicin-Induced Cell Death. Front. Physiol. 2018, 9, 682. [Google Scholar] [CrossRef]
- Pellerito, O.; Notaro, A.; Sabella, S.; De Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. WIN induces apoptotic cell death in human colon cancer cells through a block of autophagic flux dependent on PPARγ down-regulation. Apoptosis 2014, 19, 1029–1042. [Google Scholar] [CrossRef]
- Dando, I.; Donadelli, M.; Costanzo, C.; Dalla Pozza, E.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell. Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Fröhlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef]
- Sreevalsan, S.; Joseph, S.; Jutooru, I.; Chadalapaka, G.; Safe, S.H. Induction of apoptosis by cannabinoids in prostate and colon cancer cells is phosphatase dependent. Anticancer. Res. 2011, 31, 3799–3807. [Google Scholar] [PubMed]
- Berthenet, K.; Ferrer, C.C.; Fanfone, D.; Popgeorgiev, N.; Neves, D.; Bertolino, P.; Gibert, B.; Hernandez-Vargas, H.; Ichim, G. Failed Apoptosis Enhances Melanoma Cancer Cell Aggressiveness. Cell. Rep. 2020, 31, 107731. [Google Scholar] [CrossRef] [PubMed]
- Cortellini, A.; Porzio, G.; Cofini, V.; Necozione, S.; Giusti, R.; Marchetti, P.; Spiriti, M.A.A.; Costanzi, A.; Peris, F.; Ravoni, G.; et al. What cancer patients actually know regarding medical cannabis? A cross-sectional survey with a critical analysis of the current attitudes. J. Oncol. Pharm. Pract. 2019, 25, 1439–1444. [Google Scholar] [CrossRef]
- Johns, A. Psychiatric effects of cannabis. Br. J. Psychiatry 2001, 178, 116–122. [Google Scholar] [CrossRef]
- Large, M.; Sharma, S.; Compton, M.T.; Slade, T.; Nielssen, O. Cannabis Use and Earlier Onset of Psychosis. Arch. Gen. Psychiatry 2011, 68, 555. [Google Scholar] [CrossRef]
- Price, C.; Hemmingsson, T.; Lewis, G.; Zammit, S.; Allebeck, P. Cannabis and suicide: Longitudinal study. Br. J. Psychiatry 2009, 195, 492–497. [Google Scholar] [CrossRef]
- Serafini, G.; Pompili, M.; Innamorati, M.; Rihmer, Z.; Sher, L.; Girardi, P. Can Cannabis Increase the Suicide Risk in Psychosis? A Critical Review. Curr. Pharm. Des. 2012, 18, 5165–5187. [Google Scholar] [CrossRef]
- Pergam, S.A.; Bs, M.C.W.; Lee, C.M.; Cheng, G.-S.; Baker, K.K.; Marquis, S.R.; Fann, J.R. Cannabis use among patients at a comprehensive cancer center in a state with legalized medicinal and recreational use. Cancer 2017, 123, 4488–4497. [Google Scholar] [CrossRef] [PubMed]
- Taha, T.; Meiri, D.; Talhamy, S.; Wollner, M.; Peer, A.; Bar-Sela, G. Cannabis Impacts Tumor Response Rate to Nivolumab in Patients with Advanced Malignancies. Oncologist 2019, 24, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef]
- Simmerman, E.; Qin, X.; Yu, J.C.; Baban, B. Cannabinoids as a Potential New and Novel Treatment for Melanoma: A Pilot Study in a Murine Model. J. Surg. Res. 2019, 235, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Glodde, N.; Jakobs, M.; Bald, T.; Tüting, T.; Gaffal, E. Differential role of cannabinoids in the pathogenesis of skin cancer. Life Sci. 2015, 138, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Valjent, E.; Pagès, C.; Rogard, M.; Besson, M.-J.; Maldonado, R.; Caboche, J. Δ9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur. J. Neurosci. 2001, 14, 342–352. [Google Scholar] [CrossRef]
- Vrechi, T.A.M.; Leão, A.H.F.F.; Morais, I.B.M.; Abílio, V.C.; Zuardi, A.W.; Hallak, J.E.C.; Crippa, J.A.; Bincoletto, C.; Ureshino, R.P.; Smaili, S.S.; et al. Cannabidiol induces autophagy via ERK1/2 activation in neural cells. Sci. Rep. 2021, 11, 5434. [Google Scholar] [CrossRef]
- Greenhough, A.; Patsos, H.A.; Williams, A.C.; Paraskeva, C. The cannabinoid δ9-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int. J. Cancer 2007, 121, 2172–2180. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richtig, G.; Kienzl, M.; Rittchen, S.; Roula, D.; Eberle, J.; Sarif, Z.; Pichler, M.; Hoefler, G.; Heinemann, A. Cannabinoids Reduce Melanoma Cell Viability and Do Not Interfere with Commonly Used Targeted Therapy in Metastatic Melanoma In Vivo and In Vitro. Biology 2023, 12, 706. https://doi.org/10.3390/biology12050706
Richtig G, Kienzl M, Rittchen S, Roula D, Eberle J, Sarif Z, Pichler M, Hoefler G, Heinemann A. Cannabinoids Reduce Melanoma Cell Viability and Do Not Interfere with Commonly Used Targeted Therapy in Metastatic Melanoma In Vivo and In Vitro. Biology. 2023; 12(5):706. https://doi.org/10.3390/biology12050706
Chicago/Turabian StyleRichtig, Georg, Melanie Kienzl, Sonja Rittchen, David Roula, Jürgen Eberle, Zina Sarif, Martin Pichler, Gerald Hoefler, and Akos Heinemann. 2023. "Cannabinoids Reduce Melanoma Cell Viability and Do Not Interfere with Commonly Used Targeted Therapy in Metastatic Melanoma In Vivo and In Vitro" Biology 12, no. 5: 706. https://doi.org/10.3390/biology12050706
APA StyleRichtig, G., Kienzl, M., Rittchen, S., Roula, D., Eberle, J., Sarif, Z., Pichler, M., Hoefler, G., & Heinemann, A. (2023). Cannabinoids Reduce Melanoma Cell Viability and Do Not Interfere with Commonly Used Targeted Therapy in Metastatic Melanoma In Vivo and In Vitro. Biology, 12(5), 706. https://doi.org/10.3390/biology12050706