
The Role of Peroxiredoxins in Cancer Development


Abstract
Simple Summary
Abstract
1. Introduction
- Typical 2-Cys Prx. Prx1 to Prx4 are typical 2-Cys Prxs. A catalytic unit consists of a homodimer where both subunits contain CP and CR. Oxidized CP (for example, CPSOH) of one subunit forms disulfide with CRSH of another subunit. This disulfide bond is typically reduced by thioredoxin reductase (TrxR) or GSH–glutaredoxin (Grx) reductase systems. Typical 2-Cys Prxs exist in dimers and decamers (dodecamers for Prx3), with the ratio influenced by oxidation of CP. These Prxs can also function as protein chaperones.
- Atypical 2-Cys Prx. Prx5 is considered atypical 2-Cys Prx. Oxidized CP of a Prx5 molecule forms disulfide with CRSH in the same molecule. This disulfide bond is also reduced by the Trx–TrxR system. Unlike typical 2-Cys Prxs, Prx5 does not form decamers.
- 1-Cys Prx. Prx6 does not contain CR; therefore, disulfide bond formation takes place with other thiol proteins, such as π glutathione S-transferase (πGST), and is reduced by GSH [10]. Unlike other Prxs, Prx6 expresses phospholipase A2 (PLA2) activity.
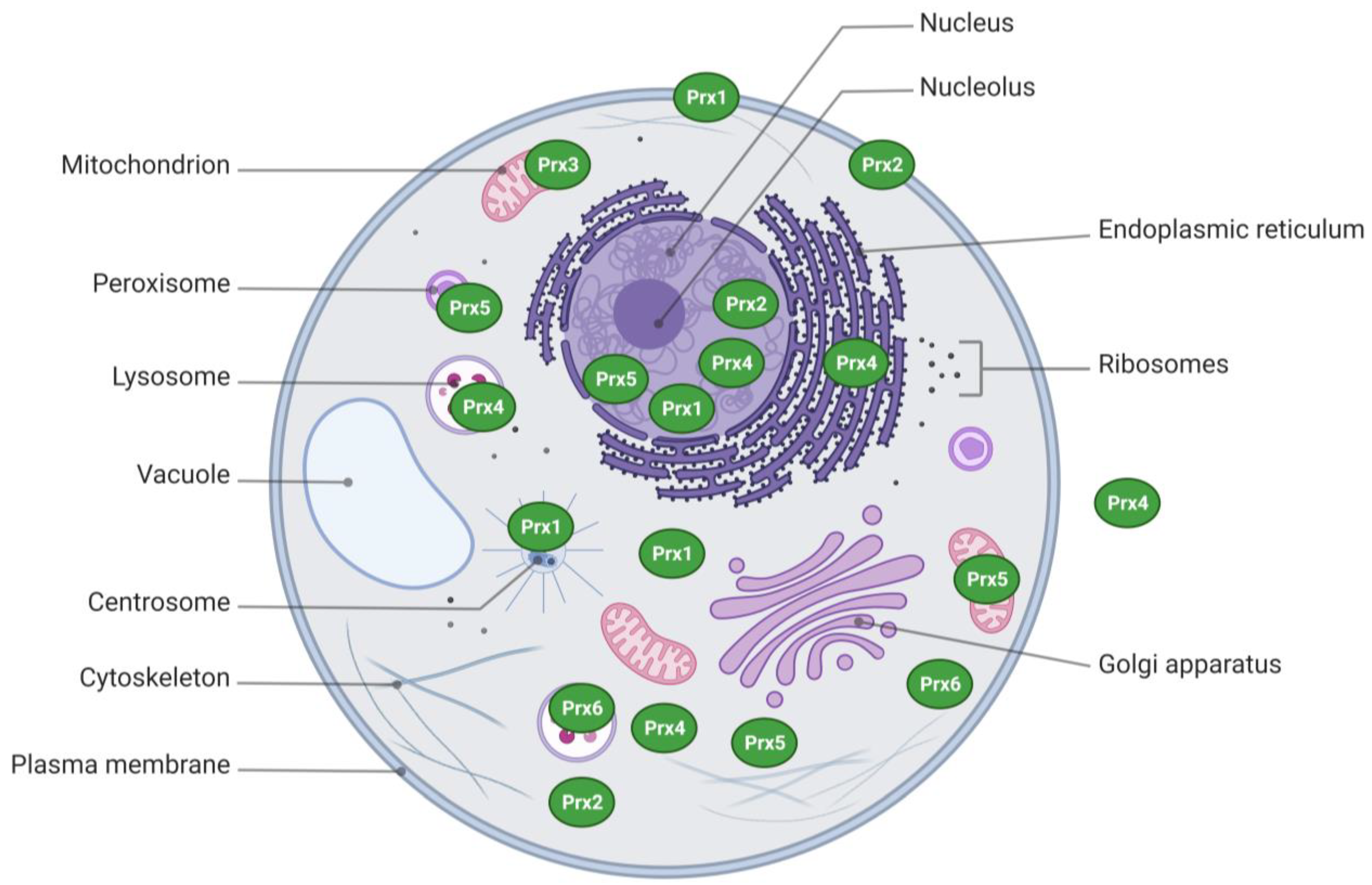
2. Structural Aspects and Subcellular Distribution
3. Peroxidase Mechanism
4. Prxs and Cancer
4.1. Prxs Promote Carcinogenesis
4.2. Prxs Regulate Cancer Progression
4.2.1. Prx1
4.2.2. Prx2
4.2.3. Prx3
4.2.4. Prx4
4.2.5. Prx5
4.2.6. Prx6
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-DE | two-dimensional gel electrophoresis |
| 5-FU | 5-fluorouracil |
| AOM | azoxymethane |
| AR | androgen receptor |
| CRC | colorectal cancer |
| CSC | cancer stem cell |
| DCFDA | 2′,7′-dichlorodihydrofluorescein diacetate |
| DMBA | 7,12-dimethylbenz[a]anthracene |
| DSS | dextran sulfate sodium |
| EMT | epithelial-mesenchymal transition |
| GPx | glutathione peroxidase |
| GSH | glutathione |
| GST | glutathione-s-transferase |
| IHC | immunohistochemistry |
| IP | immunoprecipitation |
| LOX | lysyl oxidase |
| LUAD | lung adenocarcinoma |
| NAC | N-acetyl cysteine |
| NSCLC | non-small cell lung cancer |
| PAM | plasma-activated medium |
| PLA2 | phospholipase A2 |
| Prx | peroxiredoxin |
| PSA | prostate-specific antigen |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| ROS | reactive oxygen species |
| Srx | sulfiredoxin |
| Trx | thioredoxin |
| UC | ulcerative colitis |
| WA | withangulatin A |
References
- Nogoceke, E.; Gommel, D.U.; Kiess, M.; Kalisz, H.M.; Flohé, L. A unique cascade of oxidoreductases catalyses trypanothione-mediated peroxide metabolism in Crithidia fasciculata. Biol. Chem. 1997, 378, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Bryk, R.; Griffin, P.; Nathan, C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 2000, 407, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hillas, P.J.; del Alba, F.S.; Oyarzabal, J.; Wilks, A.; Ortiz De Montellano, P.R. The AhpC and AhpD antioxidant defense system of Mycobacterium tuberculosis. J. Biol. Chem. 2000, 275, 18801–18809. [Google Scholar] [CrossRef] [PubMed]
- Milev, N.B.; Rhee, S.G.; Reddy, A.B. Cellular Timekeeping: It’s Redox o’Clock. Cold Spring Harb. Perspect. Biol. 2018, 10, a027698. [Google Scholar] [CrossRef]
- Troussicot, L.; Burmann, B.M.; Molin, M. Structural determinants of multimerization and dissociation in 2-Cys peroxiredoxin chaperone function. Structure 2021, 29, 640–654. [Google Scholar] [CrossRef]
- Kim, K.; Kim, I.H.; Lee, K.Y.; Rhee, S.G.; Stadtman, E.R. The isolation and purification of a specific “protector” protein which inhibits enzyme inactivation by a thiol/Fe(III)/O2 mixed-function oxidation system. J. Biol. Chem. 1988, 263, 4704–4711. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Storz, G.; Brodsky, M.H.; Lai, A.; Ames, B.N. Alkyl hydroperoxide reductase from Salmonella typhimurium. Sequence and homology to thioredoxin reductase and other flavoprotein disulfide oxidoreductases. J. Biol. Chem. 1990, 265, 10535–10540. [Google Scholar] [CrossRef]
- Chae, H.Z.; Chung, S.J.; Rhee, S.G. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994, 269, 27670–27678. [Google Scholar] [CrossRef]
- Edgar, R.S.; Green, E.W.; Zhao, Y.; van Ooijen, G.; Olmedo, M.; Qin, X.; Xu, Y.; Pan, M.; Valekunja, U.K.; Feeney, K.A.; et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature 2012, 485, 459–464. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Nelson, K.J.; Knutson, S.T.; Soito, L.; Klomsiri, C.; Poole, L.B.; Fetrow, J.S. Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins 2011, 79, 947–964. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Lindsay, J.G. The Peroxiredoxin Family: An Unfolding Story. Subcell Biochem. 2017, 83, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Thapa, P.; Hao, Y.; Ding, N.; Alshahrani, A.; Wei, Q. Protein Disulfide Isomerases Function as the Missing Link Between Diabetes and Cancer. Antioxid. Redox Signal. 2022, 37, 1191–1205. [Google Scholar] [CrossRef]
- Schröder, E.; Ponting, C.P. Evidence that peroxiredoxins are novel members of the thioredoxin fold superfamily. Protein Sci. 1998, 7, 2465–2468. [Google Scholar] [CrossRef]
- Zeida, A.; Reyes, A.M.; Lebrero, M.C.; Radi, R.; Trujillo, M.; Estrin, D.A. The extraordinary catalytic ability of peroxiredoxins: A combined experimental and QM/MM study on the fast thiol oxidation step. Chem. Commun. 2014, 50, 10070–10073. [Google Scholar] [CrossRef]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef]
- Shau, H.; Kim, A. Identification of natural killer enhancing factor as a major antioxidant in human red blood cells. Biochem Biophys. Res. Commun. 1994, 199, 83–88. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef]
- Chang, T.S.; Jeong, W.; Choi, S.Y.; Yu, S.; Kang, S.W.; Rhee, S.G. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J. Biol. Chem. 2002, 277, 25370–25376. [Google Scholar] [CrossRef]
- Goemaere, J.; Knoops, B. Peroxiredoxin distribution in the mouse brain with emphasis on neuronal populations affected in neurodegenerative disorders. J. Comp. Neurol. 2012, 520, 258–280. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Nelson, K.J.; Haynes, A.C.; Lee, J.; Reisz, J.A.; Graff, A.H.; Clodfelter, J.E.; Parsonage, D.; Poole, L.B.; Furdui, C.M.; et al. Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J. Biol. Chem. 2018, 293, 11901–11912. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Okado, A.; Fujii, T.; Fujii, J.; Egashira, M.; Niikawa, N.; Taniguchi, N. Cloning of the peroxiredoxin gene family in rats and characterization of the fourth member. FEBS Lett. 1999, 443, 246–250. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Matsumoto, A.; Fujii, J.; Taniguchi, N. Peroxiredoxin IV is a secretable protein with heparin-binding properties under reduced conditions. J. Biochem. 2000, 127, 493–501. [Google Scholar] [CrossRef]
- Thapa, P.; Ding, N.; Hao, Y.; Alshahrani, A.; Jiang, H.; Wei, Q. Essential Roles of Peroxiredoxin IV in Inflammation and Cancer. Molecules 2022, 27, 6513. [Google Scholar] [CrossRef]
- De Simoni, S.; Goemaere, J.; Knoops, B. Silencing of peroxiredoxin 3 and peroxiredoxin 5 reveals the role of mitochondrial peroxiredoxins in the protection of human neuroblastoma SH-SY5Y cells toward MPP+. Neurosci. Lett. 2008, 433, 219–224. [Google Scholar] [CrossRef]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef]
- Sorokina, E.M.; Feinstein, S.I.; Milovanova, T.N.; Fisher, A.B. Identification of the amino acid sequence that targets peroxiredoxin 6 to lysosome-like structures of lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L871–L880. [Google Scholar] [CrossRef]
- Wu, Y.; Feinstein, S.I.; Manevich, Y.; Chowdhury, I.; Pak, J.H.; Kazi, A.; Dodia, C.; Speicher, D.W.; Fisher, A.B. Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipase A2 activity. Biochem. J. 2009, 419, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Haridas, V.; Ni, J.; Meager, A.; Su, J.; Yu, G.L.; Zhai, Y.; Kyaw, H.; Akama, K.T.; Hu, J.; Van Eldik, L.J.; et al. TRANK, a novel cytokine that activates NF-kappa B and c-Jun N-terminal kinase. J. Immunol. 1998, 161, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Roussel, X.; Béchade, G.; Kriznik, A.; Van Dorsselaer, A.; Sanglier-Cianferani, S.; Branlant, G.; Rahuel-Clermont, S. Evidence for the formation of a covalent thiosulfinate intermediate with peroxiredoxin in the catalytic mechanism of sulfiredoxin. J. Biol. Chem. 2008, 283, 22371–22382. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Jeong, W.; Chang, T.S.; Park, K.J.; Park, S.J.; Yang, J.S.; Rhee, S.G. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 2005, 280, 3125–3128. [Google Scholar] [CrossRef]
- Horie, K.; Mikami, T.; Yoshida, T.; Sato, Y.; Okayasu, I. Peroxiredoxin 1 expression in active ulcerative colitis mucosa identified by proteome analysis and involvement of thioredoxin based on immunohistochemistry. Oncol. Lett. 2018, 15, 2364–2372. [Google Scholar] [CrossRef]
- Bostanci, Z.; Mack, R.P., Jr.; Enomoto, L.M.; Alam, S.; Brown, A.; Neumann, C.; Soybel, D.I.; Kelleher, S.L. Marginal zinc intake reduces the protective effect of lactation on mammary gland carcinogenesis in a DMBA-induced tumor model in mice. Oncol. Rep. 2016, 35, 1409–1416. [Google Scholar] [CrossRef]
- Jezierska-Drutel, A.; Attaran, S.; Hopkins, B.L.; Skoko, J.J.; Rosenzweig, S.A.; Neumann, C.A. The peroxidase PRDX1 inhibits the activated phenotype in mammary fibroblasts through regulating c-Jun N-terminal kinases. BMC Cancer 2019, 19, 812. [Google Scholar] [CrossRef]
- Kang, D.H.; Lee, D.J.; Lee, S.; Lee, S.Y.; Jun, Y.; Kim, Y.; Kim, Y.; Lee, J.S.; Lee, D.K.; Lee, S.; et al. Interaction of tankyrase and peroxiredoxin II is indispensable for the survival of colorectal cancer cells. Nat. Commun. 2017, 8, 40. [Google Scholar] [CrossRef]
- Zheng, J.; Guo, X.; Nakamura, Y.; Zhou, X.; Yamaguchi, R.; Zhang, J.; Ishigaki, Y.; Uramoto, H.; Yamada, S. Overexpression of PRDX4 Modulates Tumor Microenvironment and Promotes Urethane-Induced Lung Tumorigenesis. Oxid. Med. Cell. Longev. 2020, 2020, 8262730. [Google Scholar] [CrossRef]
- Hao, Y.; Jiang, H.; Thapa, P.; Ding, N.; Alshahrani, A.; Fujii, J.; Toledano, M.B.; Wei, Q. Critical Role of the Sulfiredoxin-Peroxiredoxin IV Axis in Urethane-Induced Non-Small Cell Lung Cancer. Antioxidants 2023, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Jiang, H.; Ding, N.; Hao, Y.; Alshahrani, A.; Lee, E.Y.; Fujii, J.; Wei, Q. Loss of Peroxiredoxin IV Protects Mice from Azoxymethane/Dextran Sulfate Sodium-Induced Colorectal Cancer Development. Antioxidants 2023, 12, 677. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Yun, H.M.; Hwang, C.J.; Park, S.I.; Han, S.B.; Hwang, D.Y.; Yoon, D.Y.; Kim, S.; Hong, J.T. Presenilin Mutation Suppresses Lung Tumorigenesis via Inhibition of Peroxiredoxin 6 Activity and Expression. Theranostics 2017, 7, 3624–3637. [Google Scholar] [CrossRef]
- Ha, B.; Kim, E.K.; Kim, J.H.; Lee, H.N.; Lee, K.O.; Lee, S.Y.; Jang, H.H. Human peroxiredoxin 1 modulates TGF-β1-induced epithelial-mesenchymal transition through its peroxidase activity. Biochem. Biophys. Res. Commun. 2012, 421, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wu, L.; Mishra, M.; Chawsheen, H.A.; Wei, Q. Expression of peroxiredoxin 1 and 4 promotes human lung cancer malignancy. Am. J. Cancer Res. 2014, 4, 445–460. [Google Scholar]
- Yang, Y.J.; Baek, J.Y.; Goo, J.; Shin, Y.; Park, J.K.; Jang, J.Y.; Wang, S.B.; Jeong, W.; Lee, H.J.; Um, H.D.; et al. Effective Killing of Cancer Cells through ROS-Mediated Mechanisms by AMRI-59 Targeting Peroxiredoxin I. Antioxid. Redox Signal. 2016, 24, 453–469. [Google Scholar] [CrossRef]
- Hong, W.G.; Kim, J.Y.; Cho, J.H.; Hwang, S.G.; Song, J.Y.; Lee, E.; Chang, T.S.; Um, H.D.; Park, J.K. AMRI-59 functions as a radiosensitizer via peroxiredoxin I-targeted ROS accumulation and apoptotic cell death induction. Oncotarget 2017, 8, 114050–114064. [Google Scholar] [CrossRef]
- Sun, H.H.; Li, Y.L.; Jiang, H.; Yin, X.H.; Jin, X.L. PRDX1 Influences The Occurrence and Progression of Liver Cancer by Inhibiting Mitochondrial Apoptosis Pathway. Cell J. 2022, 24, 657–664. [Google Scholar] [CrossRef]
- Jiang, Y.; Cao, W.; Wu, K.; Qin, X.; Wang, X.; Li, Y.; Yu, B.; Zhang, Z.; Wang, X.; Yan, M.; et al. LncRNA LINC00460 promotes EMT in head and neck squamous cell carcinoma by facilitating peroxiredoxin-1 into the nucleus. J. Exp. Clin. Cancer Res. 2019, 38, 365. [Google Scholar] [CrossRef]
- Dasari, C.; Reddy, K.R.K.; Natani, S.; Murthy, T.R.L.; Bhukya, S.; Ummanni, R. Tumor protein D52 (isoform 3) interacts with and promotes peroxidase activity of Peroxiredoxin 1 in prostate cancer cells implicated in cell growth and migration. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1298–1309. [Google Scholar] [CrossRef]
- Feng, T.; Zhao, R.; Sun, F.; Lu, Q.; Wang, X.; Hu, J.; Wang, S.; Gao, L.; Zhou, Q.; Xiong, X.; et al. TXNDC9 regulates oxidative stress-induced androgen receptor signaling to promote prostate cancer progression. Oncogene 2020, 39, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Sun, X.Y.; Yang, S.M.; Wang, Q.; Wang, Z.Y. Peroxiredoxin 1 promoted tumor metastasis and angiogenesis in colorectal cancer. Pathol. Res. Pract. 2018, 214, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Li, J.; Hong, Z.; Jia, F.; He, Y.; Yuan, L. The role of human umbilical cord mesenchymal stem cells-derived exosomal microRNA-431-5p in survival and prognosis of colorectal cancer patients. Mutagenesis 2022, 37, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ma, Y.; Tong, Q.; Yang, J.; Liu, J.; Wang, Y.; Li, G.; Zeng, J.; Fang, S.; Li, F.; et al. Cullin-5 neddylation-mediated NOXA degradation is enhanced by PRDX1 oligomers in colorectal cancer. Cell Death Dis. 2021, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Bajor, M.; Zych, A.O.; Graczyk-Jarzynka, A.; Muchowicz, A.; Firczuk, M.; Trzeciak, L.; Gaj, P.; Domagala, A.; Siernicka, M.; Zagozdzon, A.; et al. Targeting peroxiredoxin 1 impairs growth of breast cancer cells and potently sensitises these cells to prooxidant agents. Br. J. Cancer 2018, 119, 873–884. [Google Scholar] [CrossRef]
- Bajor, M.; Graczyk-Jarzynka, A.; Marhelava, K.; Kurkowiak, M.; Rahman, A.; Aura, C.; Russell, N.; Zych, A.O.; Firczuk, M.; Winiarska, M.; et al. Triple Combination of Ascorbate, Menadione and the Inhibition of Peroxiredoxin-1 Produces Synergistic Cytotoxic Effects in Triple-Negative Breast Cancer Cells. Antioxidants 2020, 9, 320. [Google Scholar] [CrossRef]
- Fiskus, W.; Coothankandaswamy, V.; Chen, J.; Ma, H.; Ha, K.; Saenz, D.T.; Krieger, S.S.; Mill, C.P.; Sun, B.; Huang, P.; et al. SIRT2 Deacetylates and Inhibits the Peroxidase Activity of Peroxiredoxin-1 to Sensitize Breast Cancer Cells to Oxidant Stress-Inducing Agents. Cancer Res. 2016, 76, 5467–5478. [Google Scholar] [CrossRef]
- Skoko, J.J.; Cao, J.; Gaboriau, D.; Attar, M.; Asan, A.; Hong, L.; Paulsen, C.E.; Ma, H.; Liu, Y.; Wu, H.; et al. Redox regulation of RAD51 Cys319 and homologous recombination by peroxiredoxin 1. Redox Biol. 2022, 56, 102443. [Google Scholar] [CrossRef]
- Attaran, S.; Skoko, J.J.; Hopkins, B.L.; Wright, M.K.; Wood, L.E.; Asan, A.; Woo, H.A.; Feinberg, A.; Neumann, C.A. Peroxiredoxin-1 Tyr194 phosphorylation regulates LOX-dependent extracellular matrix remodelling in breast cancer. Br. J. Cancer 2021, 125, 1146–1157. [Google Scholar] [CrossRef]
- Yang, L.; Hong, Q.; Xu, S.G.; Kuang, X.Y.; Di, G.H.; Liu, G.Y.; Wu, J.; Shao, Z.M.; Yu, S.J. Downregulation of transgelin 2 promotes breast cancer metastasis by activating the reactive oxygen species/nuclear factor-κB signaling pathway. Mol. Med. Rep. 2019, 20, 4045–4258. [Google Scholar] [CrossRef]
- Wang, R.; Liu, Y.; Liu, L.; Chen, M.; Wang, X.; Yang, J.; Gong, Y.; Ding, B.S.; Wei, Y.; Wei, X. Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. eBioMedicine 2019, 40, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Klopotowska, M.; Bajor, M.; Graczyk-Jarzynka, A.; Kraft, A.; Pilch, Z.; Zhylko, A.; Firczuk, M.; Baranowska, I.; Lazniewski, M.; Plewczynski, D.; et al. PRDX-1 Supports the Survival and Antitumor Activity of Primary and CAR-Modified NK Cells under Oxidative Stress. Cancer Immunol. Res. 2022, 10, 228–244. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Zhou, H.; Su, D. PRDX2 Promotes the Proliferation and Metastasis of Non-Small Cell Lung Cancer In Vitro and In Vivo. Biomed. Res. Int. 2020, 2020, 8359860. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Du, L.; Niu, A.; Wang, Y.; Wang, Y.; Wang, C. Silencing of PRDX2 Inhibits the Proliferation and Invasion of Non-Small Cell Lung Cancer Cells. Biomed. Res. Int. 2020, 2020, 1276328. [Google Scholar] [CrossRef] [PubMed]
- Chandimali, N.; Huynh, D.L.; Zhang, J.J.; Lee, J.C.; Yu, D.Y.; Jeong, D.K.; Kwon, T. MicroRNA-122 negatively associates with peroxiredoxin-II expression in human gefitinib-resistant lung cancer stem cells. Cancer Gene Ther. 2019, 26, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, C.; Xiao, G.; Shan, H.; Tang, L.; Yi, Y.; Yu, W.; Gu, Y. S-nitrosylation of the Peroxiredoxin-2 promotes S-nitrosoglutathione-mediated lung cancer cells apoptosis via AMPK-SIRT1 pathway. Cell Death Dis. 2019, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, J.; Zhang, H.; Zheng, X.; Zhou, H.; Luo, Y.; Yang, J.; Deng, Q.; Huang, S.; Fu, Z. PRDX2 promotes the proliferation of colorectal cancer cells by increasing the ubiquitinated degradation of p53. Cell Death Dis. 2021, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Xiong, Y.; Wang, R.; Xiang, L.; Zhou, H.; Fu, Z. The critical role of peroxiredoxin-2 in colon cancer stem cells. Aging 2021, 13, 11170–11187. [Google Scholar] [CrossRef]
- Wang, R.; Wei, J.; Zhang, S.; Wu, X.; Guo, J.; Liu, M.; Du, K.; Xu, J.; Peng, L.; Lv, Z.; et al. Peroxiredoxin 2 is essential for maintaining cancer stem cell-like phenotype through activation of Hedgehog signaling pathway in colon cancer. Oncotarget 2016, 7, 86816–86828. [Google Scholar] [CrossRef]
- Cerda, M.B.; Lloyd, R.; Batalla, M.; Giannoni, F.; Casal, M.; Policastro, L. Silencing peroxiredoxin-2 sensitizes human colorectal cancer cells to ionizing radiation and oxaliplatin. Cancer Lett. 2017, 388, 312–319. [Google Scholar] [CrossRef]
- Peng, L.; Jiang, J.; Chen, H.N.; Zhou, L.; Huang, Z.; Qin, S.; Jin, P.; Luo, M.; Li, B.; Shi, J.; et al. Redox-sensitive cyclophilin A elicits chemoresistance through realigning cellular oxidative status in colorectal cancer. Cell Rep. 2021, 37, 110069. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wei, J.; You, W.; Wang, R.; Shang, J.; Xiong, Y.; Yang, H.; Yang, X.; Fu, Z. Disruption of the c-Myc/miR-200b-3p/PRDX2 regulatory loop enhances tumor metastasis and chemotherapeutic resistance in colorectal cancer. J. Transl. Med. 2017, 15, 257. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, S.; Wang, R.; Wu, X.; Zeng, L.; Fu, Z. Knockdown of PRDX2 sensitizes colon cancer cells to 5-FU by suppressing the PI3K/AKT signaling pathway. Biosci. Rep. 2017, 37, BSR20160447. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chen, D.; Wu, T.; Lin, H.; Ni, L.; Sui, H.; Xiao, S.; Wang, C.; Jiang, S.; Pan, H.; et al. Dihydroartemisinin enhances the anti-tumor activity of oxaliplatin in colorectal cancer cells by altering PRDX2-reactive oxygen species-mediated multiple signaling pathways. Phytomedicine 2022, 98, 153932. [Google Scholar] [CrossRef]
- Ahmad, A.; Prakash, R.; Khan, M.S.; Altwaijry, N.; Asghar, M.N.; Raza, S.S.; Khan, R. N-Carbamoyl Alanine-Mediated Selective Targeting for CHEK2-Null Colorectal Cancer. ACS Omega 2022, 7, 13095–13101. [Google Scholar] [CrossRef]
- Zhang, S.; Fu, Z.; Wei, J.; Guo, J.; Liu, M.; Du, K. Peroxiredoxin 2 is involved in vasculogenic mimicry formation by targeting VEGFR2 activation in colorectal cancer. Med. Oncol. 2015, 32, 414. [Google Scholar] [CrossRef]
- Zheng, X.; Wei, J.; Li, W.; Li, X.; Wang, W.; Guo, J.; Fu, Z. PRDX2 removal inhibits the cell cycle and autophagy in colorectal cancer cells. Aging 2020, 12, 16390–16409. [Google Scholar] [CrossRef]
- Shi, J.; Zhou, L.; Huang, H.S.; Peng, L.; Xie, N.; Nice, E.; Fu, L.; Jiang, C.; Huang, C. Repurposing Oxiconazole against Colorectal Cancer via PRDX2-mediated Autophagy Arrest. Int. J. Biol. Sci. 2022, 18, 3747–3761. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, C.; Li, H.; Yuan, Y.; Wang, X.; Zhao, T.; Xu, J. DACH1 inhibits the proliferation and invasion of lung adenocarcinoma through the downregulation of peroxiredoxin 3. Tumour Biol. 2016, 37, 9781–9788. [Google Scholar] [CrossRef]
- Hao, C.C.; Luo, J.N.; Xu, C.Y.; Zhao, X.Y.; Zhong, Z.B.; Hu, X.N.; Jin, X.M.; Ge, X. TRIAP1 knockdown sensitizes non-small cell lung cancer to ionizing radiation by disrupting redox homeostasis. Thorac. Cancer 2020, 11, 1015–1025. [Google Scholar] [CrossRef]
- Myers, C.R. Enhanced targeting of mitochondrial peroxide defense by the combined use of thiosemicarbazones and inhibitors of thioredoxin reductase. Free Radic. Biol. Med. 2016, 91, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Long, M.; Gao, Z.W.; Liu, C.; Wu, X.N.; Yang, L.; Dong, K.; Zhang, H.Z. Silencing of B7-H4 induces intracellular oxidative stress and inhibits cell viability of breast cancer cells via downregulating PRDX3. Neoplasma 2022, 69, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Jeong, Y.J.; Jeong, S.H.; Heo, H.J.; Kim, H.K.; Bae, K.B.; Park, Y.H.; Kim, S.U.; Kim, J.M.; Kim, N.; et al. FOXM1-Induced PRX3 Regulates Stemness and Survival of Colon Cancer Cells via Maintenance of Mitochondrial Function. Gastroenterology 2015, 149, 1006–1016.e1009. [Google Scholar] [CrossRef]
- Ding, N.; Jiang, H.; Thapa, P.; Hao, Y.; Alshahrani, A.; Allison, D.; Izumi, T.; Rangnekar, V.M.; Liu, X.; Wei, Q. Peroxiredoxin IV plays a critical role in cancer cell growth and radioresistance through the activation of the Akt/GSK3 signaling pathways. J. Biol. Chem. 2022, 298, 102123. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, S.; Tiedemann, K.; Tabaries, S.; Siegel, P.M.; Komarova, S.V. Peroxiredoxin 4: A novel secreted mediator of cancer induced osteoclastogenesis. Cancer Lett. 2015, 361, 262–270. [Google Scholar] [CrossRef]
- Yi, N.; Xiao, M.B.; Ni, W.K.; Jiang, F.; Lu, C.H.; Ni, R.Z. High expression of peroxiredoxin 4 affects the survival time of colorectal cancer patients, but is not an independent unfavorable prognostic factor. Mol. Clin. Oncol. 2014, 2, 767–772. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lee, K.C.; Tung, S.Y.; Huang, W.S.; Teng, C.C.; Lee, K.F.; Hsieh, M.C.; Kuo, H.C. 2D-DIGE-MS Proteomics Approaches for Identification of Gelsolin and Peroxiredoxin 4 with Lymph Node Metastasis in Colorectal Cancer. Cancers 2022, 14, 3189. [Google Scholar] [CrossRef]
- Kim, B.; Kim, Y.S.; Ahn, H.M.; Lee, H.J.; Jung, M.K.; Jeong, H.Y.; Choi, D.K.; Lee, J.H.; Lee, S.R.; Kim, J.M.; et al. Peroxiredoxin 5 overexpression enhances tumorigenicity and correlates with poor prognosis in gastric cancer. Int. J. Oncol. 2017, 51, 298–306. [Google Scholar] [CrossRef]
- Sato, K.; Shi, L.; Ito, F.; Ohara, Y.; Motooka, Y.; Tanaka, H.; Mizuno, M.; Hori, M.; Hirayama, T.; Hibi, H.; et al. Non-thermal plasma specifically kills oral squamous cell carcinoma cells in a catalytic Fe(II)-dependent manner. J. Clin. Biochem. Nutr. 2019, 65, 8–15. [Google Scholar] [CrossRef]
- Sun, H.N.; Guo, X.Y.; Xie, D.P.; Wang, X.M.; Ren, C.X.; Han, Y.H.; Yu, N.N.; Huang, Y.L.; Kwon, T. Knockdown of Peroxiredoxin V increased the cytotoxicity of non-thermal plasma-treated culture medium to A549 cells. Aging 2022, 14, 4000–4013. [Google Scholar] [CrossRef]
- Chen, X.; Cao, X.; Xiao, W.; Li, B.; Xue, Q. PRDX5 as a novel binding partner in Nrf2-mediated NSCLC progression under oxidative stress. Aging 2020, 12, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Chen, X.M.; Xiao, W.Z.; Li, B.; Zhang, B.; Wu, Q.; Xue, Q. ROS-mediated hypomethylation of PRDX5 promotes STAT3 binding and activates the Nrf2 signaling pathway in NSCLC. Int. J. Mol. Med. 2021, 47, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.M.; Yoo, J.W.; Lee, S.; Lee, H.J.; Lee, H.S.; Lee, D.S. Peroxiredoxin 5 promotes the epithelial-mesenchymal transition in colon cancer. Biochem. Biophys. Res. Commun. 2017, 487, 580–586. [Google Scholar] [CrossRef]
- Liu, Y.; Kwon, T.; Kim, J.S.; Chandimali, N.; Jin, Y.H.; Gong, Y.X.; Xie, D.P.; Han, Y.H.; Jin, M.H.; Shen, G.N.; et al. Peroxiredoxin V Reduces β-Lapachone-induced Apoptosis of Colon Cancer Cells. Anticancer Res. 2019, 39, 3677–3686. [Google Scholar] [CrossRef]
- Chandimali, N.; Sun, H.N.; Kong, L.Z.; Zhen, X.; Liu, R.; Kwon, T.; Lee, D.S. Shikonin-induced Apoptosis of Colon Cancer Cells Is Reduced by Peroxiredoxin V Expression. Anticancer Res. 2019, 39, 6115–6123. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, D.; Li, B.; Zhen, H.; Chen, W.; Men, Q. PRDX6 Overexpression Promotes Proliferation, Invasion, and Migration of A549 Cells in vitro and in vivo. Cancer Manag. Res. 2021, 13, 1245–1255. [Google Scholar] [CrossRef]
- Xu, J.; Su, Q.; Gao, M.; Liang, Q.; Li, J.; Chen, X. Differential Expression And Effects Of Peroxiredoxin-6 On Drug Resistance And Cancer Stem Cell-Like Properties In Non-Small Cell Lung Cancer. Onco Targets Ther. 2019, 12, 10477–10486. [Google Scholar] [CrossRef]
- Li, B.Z.; Bai, H.H.; Tan, F.W.; Gao, Y.B.; He, J. Identification of peroxiredoxin 6 as a potential lung-adenocarcinoma biomarker for predicting chemotherapy response by proteomic analysis. J. Biol. Regul. Homeost. Agents 2021, 35, 537–546. [Google Scholar] [CrossRef]
- Chen, C.; Gong, L.; Liu, X.; Zhu, T.; Zhou, W.; Kong, L.; Luo, J. Identification of peroxiredoxin 6 as a direct target of withangulatin A by quantitative chemical proteomics in non-small cell lung cancer. Redox Biol. 2021, 46, 102130. [Google Scholar] [CrossRef]
- Huang, W.S.; Huang, C.Y.; Hsieh, M.C.; Kuo, Y.H.; Tung, S.Y.; Shen, C.H.; Hsieh, Y.Y.; Teng, C.C.; Lee, K.C.; Lee, K.F.; et al. Expression of PRDX6 Correlates with Migration and Invasiveness of Colorectal Cancer Cells. Cell. Physiol. Biochem. 2018, 51, 2616–2630. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Percent Sequence Similarity to Human Prxs | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prx1 | Prx2 | Prx3 | Prx4 | Prx5 | Prx6 | |||||||
| Species | DNA | Protein | DNA | Protein | DNA | Protein | DNA | Protein | DNA | Protein | DNA | Protein |
| Macaca mulatta | 98.5 | 99.5 | 98.1 | 100 | 97.5 | 97.7 | 98.4 | 98.5 | 97.8 | 99.2 | 98.4 | 98.7 |
| Mus musculus | 90.8 | 95.5 | 88.2 | 93.4 | 84.2 | 86.3 | 89.1 | 95 | 86.1 | 87.2 | 87.4 | 89.7 |
| Rattus norvegicus | 91.3 | 97.5 | 87 | 93.4 | 83.2 | 85.2 | 90.3 | 94.5 | 85.3 | 88.1 | 88.8 | 91.5 |
| Danio rerio | 73.9 | 81.3 | 72.8 | 76.6 | 66.8 | 75.4 | 74.8 | 88.7 | 64 | 61.3 | 67.3 | 73.6 |
| Prxs | Cancers | Pro- or Anti-tumor | Mechanism/Pathway | References |
|---|---|---|---|---|
| Prx1 | Lung | Pro-tumor | Downregulation of E-cadherin | [44] |
| Pro-tumor | Protection against oxidative stress-induced cell death and inhibition of ASK1-JNK | [46,47] | ||
| Prostate | Pro-tumor | Protection against oxidative stress-induced cell death and interaction with TPD52 | [50] | |
| Pro-tumor | Activation of AR signaling | [51] | ||
| Colorectal | Pro-tumor | Cullin-5 neddylation-mediated NOXA degradation | [54] | |
| Pro-tumor | Increased inflammation | [36] | ||
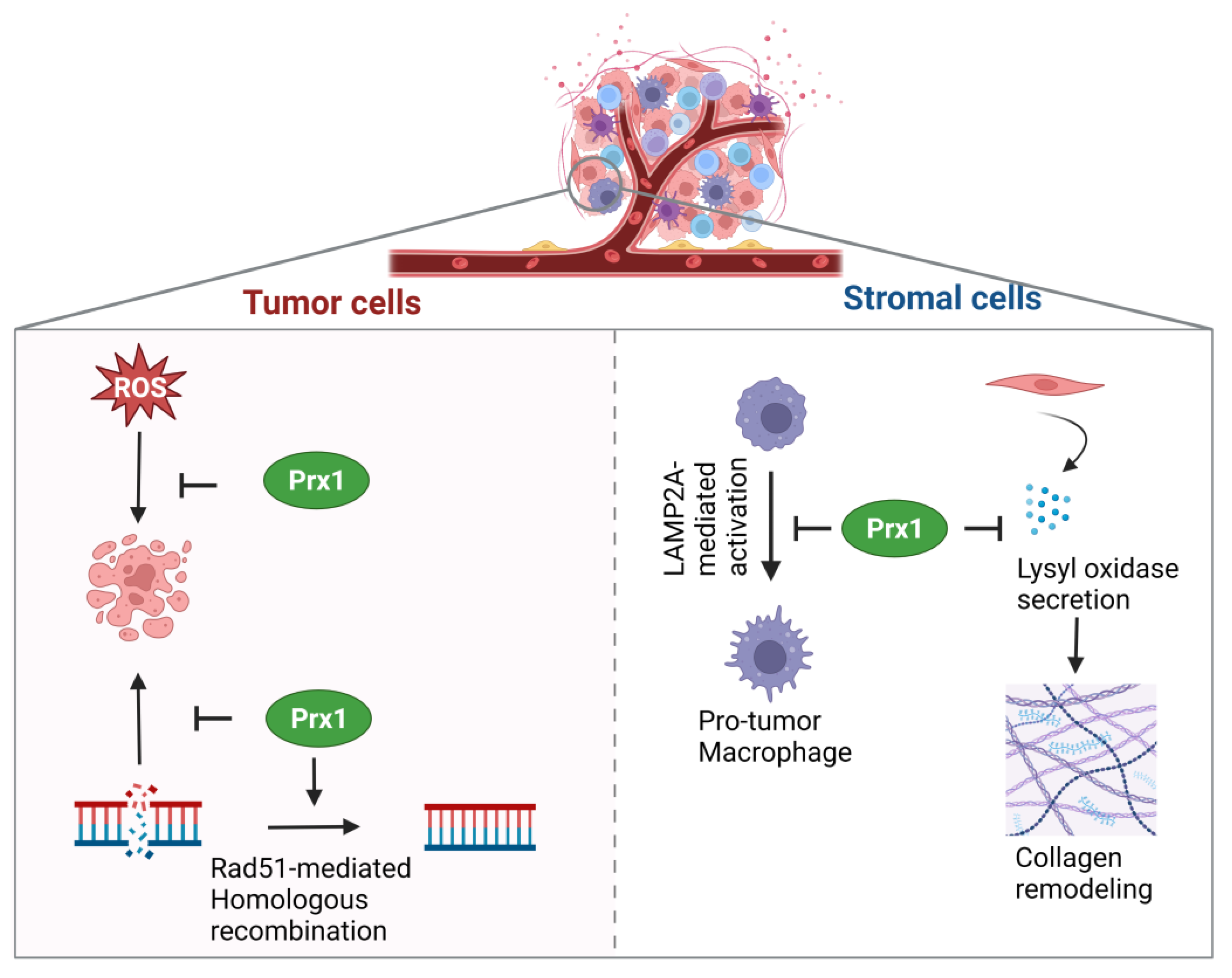
| Breast | Pro-tumor | Prevention of Rad51 oxidation to promote homologous recombination | [58] | |
| Anti-tumor | Mediation of TAGLN2 activity | [60] | ||
| Anti-tumor | Maintenance of redox homeostasis | [37] | ||
| Anti-tumor | Inhibition of LOX secretion and extracellular matrix remodeling | [59] | ||
| Anti-tumor | Inhibition of pro-tumorigenic macrophage differentiation | [61] | ||
| Anti-tumor | Inhibition of pro-tumorigenic fibroblast differentiation | [38] | ||
| Anti-tumor | Increased survival of Natural killer cells | [62] | ||
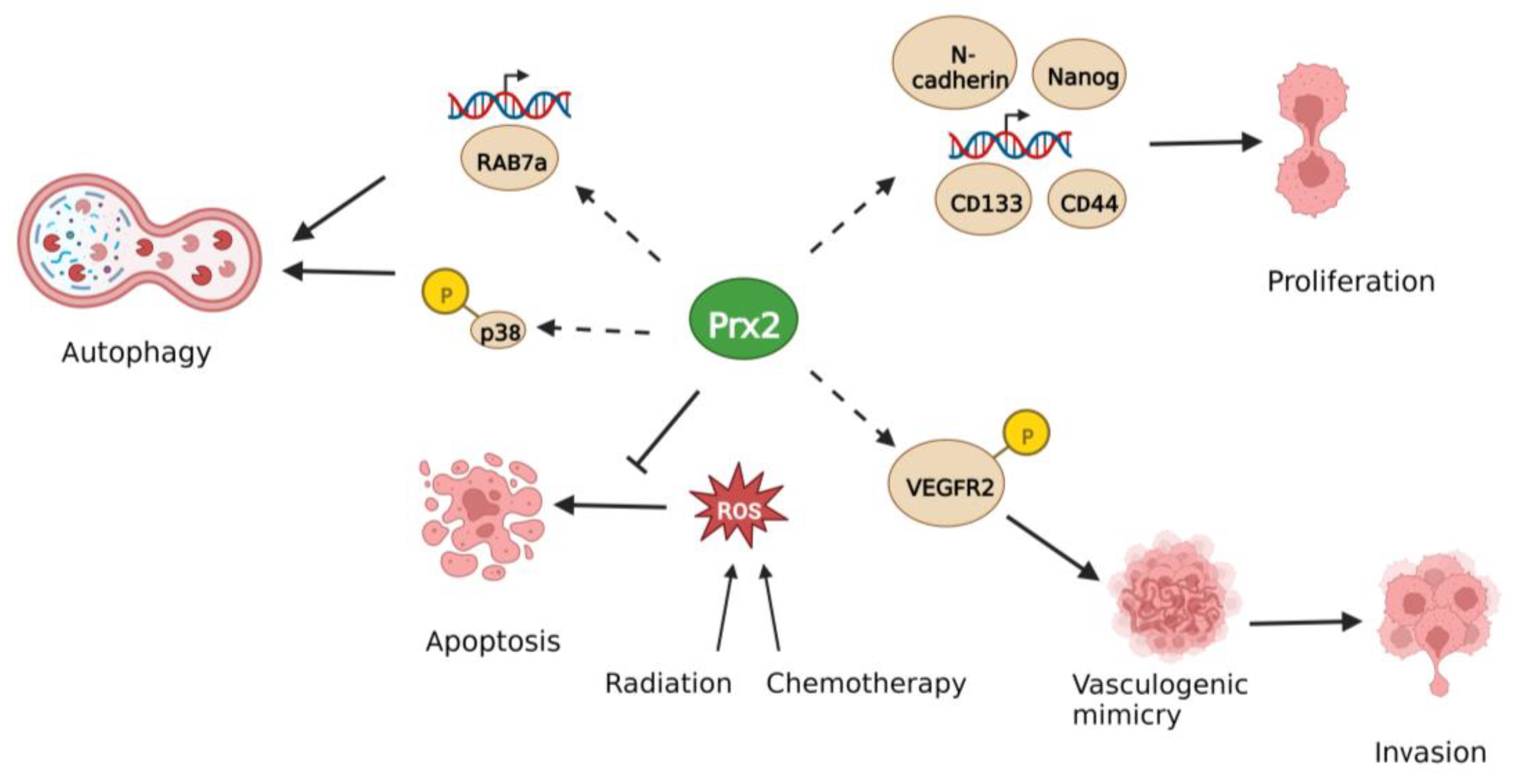
| Prx2 | Lung | Pro-tumor | Upregulation of Vimentin, Slug and, activation of AKT/mTOR | [63,64] |
| Pro-tumor | Activation of Hedgehog, Notch, Wnt/β-catenin | [65] | ||
| Pro-tumor | Maintenance of SIRT1 activity through inhibition of AMPK | [66] | ||
| Colorectal | Pro-tumor | Degradation of p53 | [67] | |
| Pro-tumor | Increased stemness, radioresistance and chemoresistance | [68,69,70,71,72,73] | ||
| Pro-tumor | Stabilization of β-catenin in APC mutant cells | [39] | ||
| Pro-tumor | Protection against oxidative stress-induced cell death | [74,75] | ||
| Pro-tumor | Formation of vasculogenic mimicry | [76] | ||
| Pro-tumor | Inhibition of autophagy | [77,78] | ||
| Prx3 | Lung | Pro-tumor | Radioresistance and chemoresistance | [80,81] |
| Breast | Pro-tumor | Protection against oxidative stress-induced cell death | [82] | |
| Colorectal | Pro-tumor | Increased stemness and chemoresistance | [83] | |
| Prx4 | Prostate | Pro-tumor | AR activation and radioresistance | [84] |
| Prostate, Breast | Pro-tumor | Activation of ERK/NFATc1 | [85] | |
| Lung | Pro-tumor | Activation of c-jun/AP-1 | [45] | |
| Pro-tumor | Protection against oxidative stress-induced cell death and increased activation of NF-κB and AP-1 signaling | [40] | ||
| Pro-tumor | Increased cell transformation, proliferation and increase in intra-tumoral M2 macrophage infiltration | [41] | ||
| Colorectal | Pro-tumor | Activation of EGFR, RhoA, PKCα, ERK | [87] | |
| Pro-tumor | Protection against oxidative stress-induced cell death and increase in intra-tumoral immune infiltration | [42] | ||
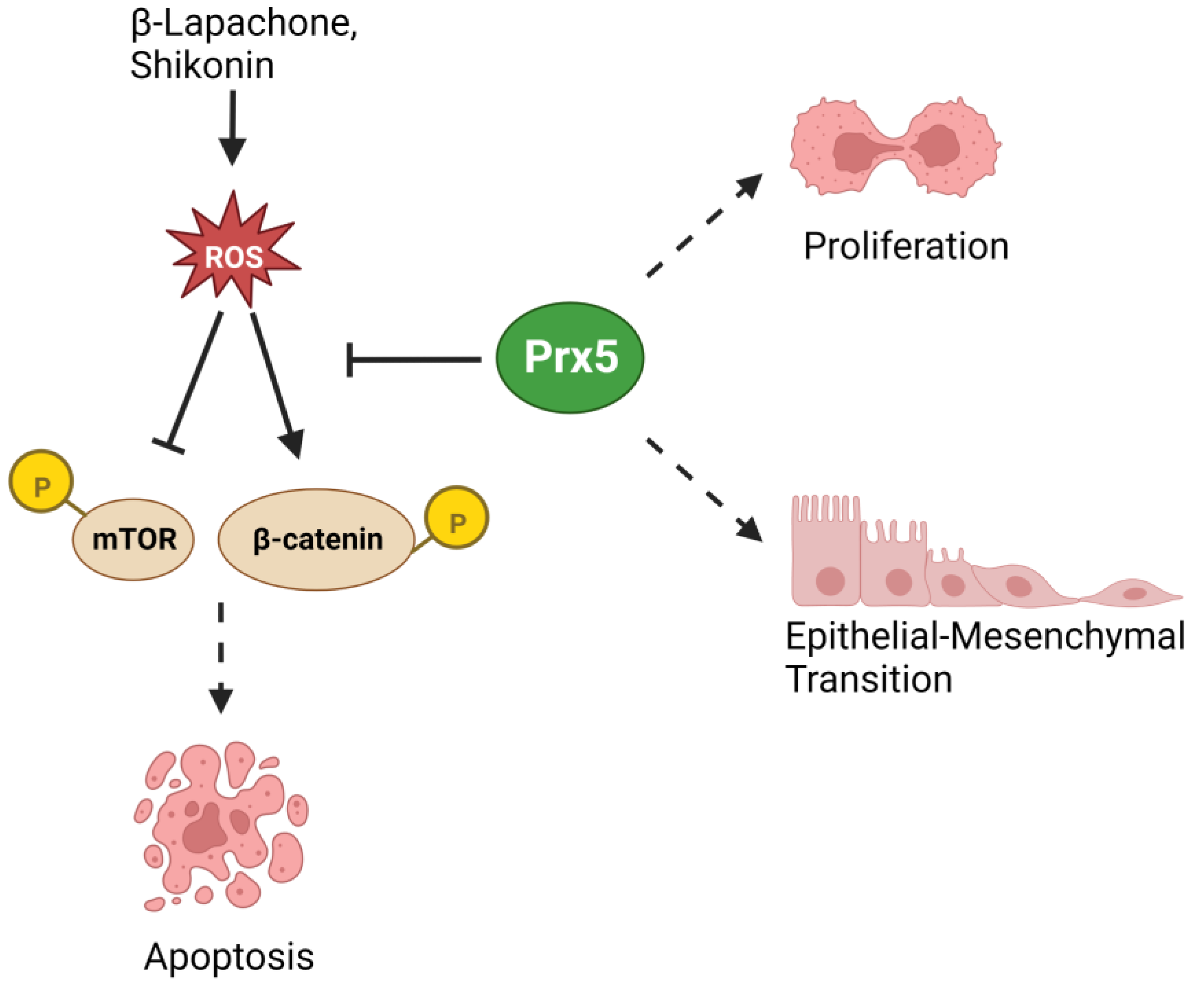
| Prx5 | Lung | Pro-tumor | Protection against oxidative stress-induced cell death | [90] |
| Pro-tumor | Upregulation of Vimentin, Nrf2, NQO1 | [91,92] | ||
| Colorectal | Pro-tumor | Upregulation of Vimentin, Slug | [93] | |
| Pro-tumor | Protection against oxidative stress-induced cell death | [94,95] | ||
| Prx6 | Lung | Pro-tumor | Upregulation of Vimentin, Twist, β-catenin | [96] |
| Pro-tumor | Protection against oxidative stress-induced cell death | [99] | ||
| Pro-tumor | Increased stemness and chemoresistance | [97,98] | ||
| Pro-tumor | Increased PLA2 activity | [43] | ||
| Colorectal | Pro-tumor | Activation of PI3K/AKT and upregulation of N-cadherin | [100] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa, P.; Jiang, H.; Ding, N.; Hao, Y.; Alshahrani, A.; Wei, Q. The Role of Peroxiredoxins in Cancer Development. Biology 2023, 12, 666. https://doi.org/10.3390/biology12050666
Thapa P, Jiang H, Ding N, Hao Y, Alshahrani A, Wei Q. The Role of Peroxiredoxins in Cancer Development. Biology. 2023; 12(5):666. https://doi.org/10.3390/biology12050666
Chicago/Turabian StyleThapa, Pratik, Hong Jiang, Na Ding, Yanning Hao, Aziza Alshahrani, and Qiou Wei. 2023. "The Role of Peroxiredoxins in Cancer Development" Biology 12, no. 5: 666. https://doi.org/10.3390/biology12050666
APA StyleThapa, P., Jiang, H., Ding, N., Hao, Y., Alshahrani, A., & Wei, Q. (2023). The Role of Peroxiredoxins in Cancer Development. Biology, 12(5), 666. https://doi.org/10.3390/biology12050666