
Tumor-Suppressive Functions of the Aryl Hydrocarbon Receptor (AhR) and AhR as a Therapeutic Target in Cancer


Abstract
Simple Summary
Abstract
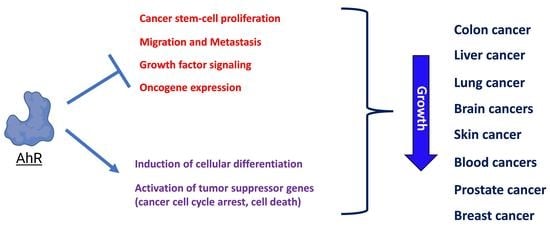
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
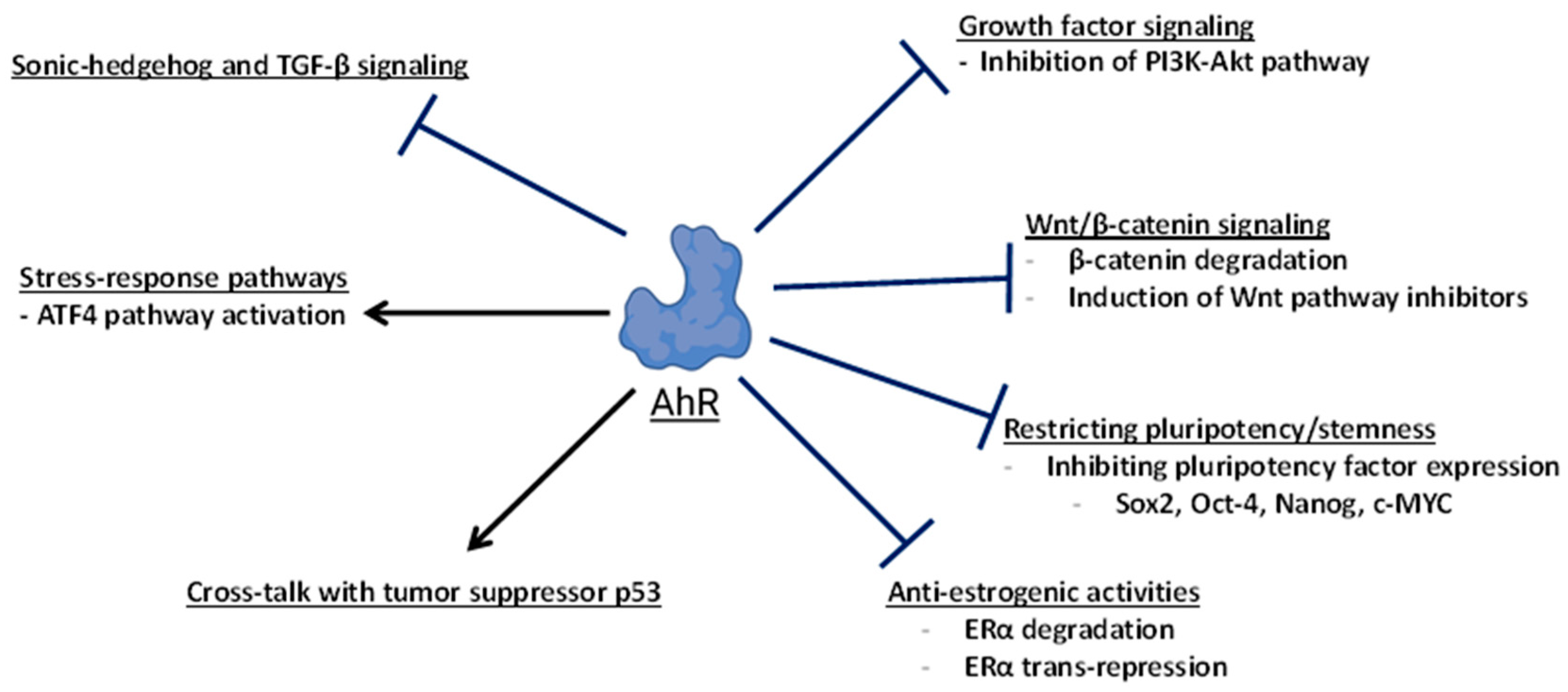{kind=link}
1. Introduction
2. Review of AhR Biology and Signaling
2.1. Role of AhR in Xenobiotic Metabolism
2.2. The AhR Signaling Pathway
2.3. Role of Co-Regulators in AhR Signaling
3. AhR-Driven Tumor Suppression by Cancer Type
3.1. Prostate Cancer
3.2. Lung Cancer
3.3. Intestinal Cancers
3.4. Medulloblastoma/Neuroblastoma/Glioblastoma
3.5. Liver Cancer
3.6. Leukemia
3.7. Melanoma
3.8. Breast Cancer
4. Crosstalk between Tumor Suppressor p53 and AhR in Cancer
5. AhR and Tumor Immunity
6. Tools and Therapies for Modulating the Function of AhR
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hahn, M.E.; Karchner, S.I.; Merson, R.R. Diversity as opportunity: Insights from 600 million years of AHR evolution. Curr. Opin. Toxicol. 2017, 2, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef]
- Ko, C.-I.; Puga, A. Does the aryl hydrocarbon receptor regulate pluripotency? Curr. Opin. Toxicol. 2017, 2, 1–7. [Google Scholar] [CrossRef]
- Rejano-Gordillo, C.; Ordiales-Talavero, A.; Nacarino-Palma, A.; Merino, J.M.; González-Rico, F.J.; Fernández-Salguero, P.M. Aryl Hydrocarbon Receptor: From Homeostasis to Tumor Progression. Front. Cell Dev. Biol. 2022, 10, 884004. [Google Scholar] [CrossRef]
- Lu, P.; Yamaguchi, Y.; Fulton, W.B.; Wang, S.; Zhou, Q.; Jia, H.; Kovler, M.L.; Salazar, A.G.; Sampah, M.; Prindle, T., Jr.; et al. Maternal aryl hydrocarbon receptor activation protects newborns against necrotizing enterocolitis. Nat. Commun. 2021, 12, 1042. [Google Scholar] [CrossRef]
- Barroso, A.; Mahler, J.V.; Fonseca-Castro, P.H.; Quintana, F.J. The aryl hydrocarbon receptor and the gut–brain axis. Cell. Mol. Immunol. 2021, 18, 259–268. [Google Scholar] [CrossRef]
- Safe, S.; Cheng, Y.; Jin, U.-H. The aryl hydrocarbon receptor (AhR) as a drug target for cancer chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.K.; Kerkvliet, N.I. Is chronic AhR activation by rapidly metabolized ligands safe for the treatment of immune-mediated diseases? Curr. Opin. Toxicol. 2017, 2, 72–78. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, E.F.; Koch, D.C.; Bisson, W.H.; Jang, H.S.; Kolluri, S.K. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis. 2014, 5, e1038. [Google Scholar] [CrossRef]
- Díaz-Díaz, C.J.; Ronnekleiv-Kelly, S.; Nukaya, M.; Geiger, P.G.; Balbo, S.; Dator, R.; Megna, B.; Carney, P.; Bradfield, C.A.; Kennedy, G.D. The aryl hydrocarbon receptor mediates the chemopreventive effect of indole-3-carbinol in an inflammation-associated colorectal tumorigenesis model. Ann. Surg. 2016, 264, 429–436. [Google Scholar] [CrossRef]
- Kaye, J.; Piryatinsky, V.; Birnberg, T.; Hingaly, T.; Raymond, E.; Kashi, R.; Amit-Romach, E.; Caballero, I.S.; Towfic, F.; Ator, M.A.; et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E6145–E6152. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Zulick, E.S.; Novikov, O.; Parks, A.J.; Schlezinger, J.J.; Wang, Z.; Laroche, F.; Feng, H.; Mulas, F.; Monti, S.; et al. Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2018, 19, 1388. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, B.E.; Hogenesch, J.B.; Bradfield, C.A. Mammalian Per-Arnt-Sim Proteins in Environmental Adaptation. Annu. Rev. Physiol. 2010, 72, 625–645. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Bin Zhao, B. Exactly the Same but Different: Promiscuity and Diversity in the Molecular Mechanisms of Action of the Aryl Hydrocarbon (Dioxin) Receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ciolino, H.P.; Macdonald, C.J.; Memon, O.S.; Bass, S.E.; Yeh, G.C. Sulindac regulates the aryl hydrocarbon receptor-mediated expression of Phase 1 metabolic enzymes in vivo and in vitro. Carcinogenesis 2005, 27, 1586–1592. [Google Scholar] [CrossRef]
- Poland, A.P.; Glover, E.; Robinson, J.R.; Nebert, D.W. Genetic expression of aryl hydrocarbon hydroxylase activity. Induction of monooxygenase activities and cytochrome P1-450 formation by 2,3,7,8 tetrachlorodibenzo p dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J. Biol. Chem. 1974, 249, 5599–5606. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. Veterans and Agent Orange: Update 2014; The National Academies Press: Washington, DC, USA, 2016. [Google Scholar] [CrossRef]
- Antonsson, C.; Whitelaw, M.L.; McGuire, J.; Gustafsson, J.A.; Poellinger, L. Distinct roles of the molecular chaperone hsp90 in modulating dioxin receptor function via the basic helix-loop-helix and PAS domains. Mol. Cell. Biol. 1995, 15, 756–765. [Google Scholar] [CrossRef]
- Tsuji, N.; Fukuda, K.; Nagata, Y.; Okada, H.; Haga, A.; Hatakeyama, S.; Yoshida, S.; Okamoto, T.; Hosaka, M.; Sekine, K.; et al. The activation mechanism of the aryl hydrocarbon receptor (AhR) by molecular chaperone HSP90. FEBS Open Bio 2014, 4, 796–803. [Google Scholar] [CrossRef]
- Pappas, B.; Yang, Y.; Wang, Y.; Kim, K.; Chung, H.J.; Cheung, M.; Ngo, K.; Shinn, A.; Chan, W.K. p23 protects the human aryl hydrocarbon receptor from degradation via a heat shock protein 90-independent mechanism. Biochem. Pharmacol. 2018, 152, 34–44. [Google Scholar] [CrossRef]
- Meyer, B.K.; Pray-Grant, M.G.; Heuvel, J.P.V.; Perdew, G.H. Hepatitis B Virus X-Associated Protein 2 Is a Subunit of the Unliganded Aryl Hydrocarbon Receptor Core Complex and Exhibits Transcriptional Enhancer Activity. Mol. Cell. Biol. 1998, 18, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Enan, E.; Matsumura, F. Identification of c-Src as the integral component of the cytosolic Ah receptor complex, transducing the signal of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through the protein phosphorylation pathway. Biochem. Pharmacol. 1996, 52, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Petrulis, J.R.; Perdew, G.H. The role of chaperone proteins in the aryl hydrocarbon receptor core complex. Chem. Biol. Interact. 2002, 141, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Yao, E.F.; Denison, M.S. DNA sequence determinants for binding of transformed Ah receptor to a dioxin-responsive enhancer. Biochemistry 1992, 31, 5060–5067. [Google Scholar] [CrossRef]
- Swanson, H.; Tullis, K.; Denison, M.S. Binding of transformed Ah receptor complex to a dioxin responsive transcriptional enhancer: Evidence for two distinct heteromeric DNA-binding forms. Biochemistry 1993, 32, 12841–12849. [Google Scholar] [CrossRef]
- Tanos, R.; Patel, R.D.; Murray, I.A.; Smith, P.B.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology 2012, 55, 1994–2004. [Google Scholar] [CrossRef]
- Jackson, D.P.; Li, H.; Mitchell, K.A.; Joshi, A.D.; Elferink, C.J. Ah Receptor–Mediated Suppression of Liver Regeneration through NC-XRE–Driven p21Cip1Expression. Mol. Pharmacol. 2014, 85, 533–541. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The Tumor Suppressor Kruppel-Like Factor 6 Is a Novel Aryl Hydrocarbon Receptor DNA Binding Partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Matsumura, F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-κB family. Biochem. Pharmacol. 2009, 77, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Reisz-Porszasz, S.; Probst, M.R.; Fukunaga, B.N.; Hankinson, O. Identification of functional domains of the aryl hydrocarbon receptor nuclear translocator protein (ARNT). Mol. Cell. Biol. 1994, 14, 6075–6086. [Google Scholar] [PubMed]
- Crews, S.T.; Fan, C.-M. Remembrance of things PAS: Regulation of development by bHLH–PAS proteins. Curr. Opin. Genet. Dev. 1999, 9, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Lindebro, M.; Poellinger, L.; Whitelaw, M. Protein-protein interaction via PAS domains: Role of the PAS domain in positive and negative regulation of the bHLH/PAS dioxin receptor-Arnt transcription factor complex. EMBO J. 1995, 14, 3528–3539. [Google Scholar] [CrossRef] [PubMed]
- Pongratz, I.; Antonsson, C.; Whitelaw, M.L.; Poellinger, L. Role of the PAS Domain in Regulation of Dimerization and DNA Binding Specificity of the Dioxin Receptor. Mol. Cell. Biol. 1998, 18, 4079–4088. [Google Scholar] [CrossRef]
- Andersson, P.; McGuire, J.; Rubio, C.; Gradin, K.; Whitelaw, M.L.; Pettersson, S.; Hanberg, A.; Poellinger, L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 9990–9995. [Google Scholar] [CrossRef]
- Schulte, K.W.; Green, E.; Wilz, A.; Platten, M.; Daumke, O. Structural Basis for Aryl Hydrocarbon Receptor-Mediated Gene Activation. Structure 2017, 25, 1025–1033.e3. [Google Scholar] [CrossRef]
- Huang, X.; Powell-Coffman, J.A.; Jin, Y. The AHR-1 aryl hydrocarbon receptor and its co-factor the AHA-1 aryl hydrocarbon receptor nuclear translocator specify GABAergic neuron cell fate in C. elegans. Development 2004, 131, 819–828. [Google Scholar] [CrossRef]
- Qin, H.; Powell-Coffman, J.A. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev. Biol. 2004, 270, 64–75. [Google Scholar] [CrossRef]
- Sonnenfeld, M.; Ward, M.; Nystrom, G.; Mosher, J.; Stahl, S.; Crews, S. The Drosophila tango gene encodes a bHLH-PAS protein that is orthologous to mammalian Arnt and controls CNS midline and tracheal development. Development 1997, 124, 4571–4582. [Google Scholar] [CrossRef]
- Di Giaimo, R.; Durovic, T.; Barquin, P.; Kociaj, A.; Lepko, T.; Aschenbroich, S.; Breunig, C.T.; Irmler, M.; Cernilogar, F.M.; Schotta, G.; et al. The Aryl Hydrocarbon Receptor Pathway Defines the Time Frame for Restorative Neurogenesis. Cell Rep. 2018, 25, 3241–3251.e5. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Kenison, J.E.; Hewson, P.; Wang, Z.; Bakshi, R.; Sherr, D.H.; Quintana, F.J. Detection of aryl hydrocarbon receptor agonists in human samples. Sci. Rep. 2018, 8, 4970. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.L.H.; Martin, K.C.; Resseguie, E.; Lawrence, B.P. Differential Consequences of Two Distinct AhR Ligands on Innate and Adaptive Immune Responses to Influenza A Virus. Toxicol. Sci. 2014, 137, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Bohonowych, J.E.; Denison, M.S. Persistent Binding of Ligands to the Aryl Hydrocarbon Receptor. Toxicol. Sci. 2007, 98, 99–109. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Pennington, J.M.; Bisson, W.H.; Kolluri, S.K.; Kerkvliet, N.I. TCDD, FICZ, and Other High Affinity AhR Ligands Dose-Dependently Determine the Fate of CD4+ T Cell Differentiation. Toxicol. Sci. 2018, 161, 310–320. [Google Scholar] [CrossRef]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between Aryl Hydrocarbon Receptor and the Inflammatory Response: A role for nuclear factor-κB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef]
- DiNatale, B.C.; Smith, K.; John, K.; Krishnegowda, G.; Amin, S.G.; Perdew, G.H. Ah Receptor Antagonism Represses Head and Neck Tumor Cell Aggressive Phenotype. Mol. Cancer Res. 2012, 10, 1369–1379. [Google Scholar] [CrossRef]
- Fletcher, N.; Hanberg, A.; Håkansson, H. Hepatic Vitamin A Depletion Is a Sensitive Marker of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) Exposure in Four Rodent Species. Toxicol. Sci. 2001, 62, 166–175. [Google Scholar] [CrossRef]
- Korkalainen, M.; Tuomisto, J.; Pohjanvirta, R. The AH Receptor of the Most Dioxin-Sensitive Species, Guinea Pig, Is Highly Homologous to the Human AH Receptor. Biochem. Biophys. Res. Commun. 2001, 285, 1121–1129. [Google Scholar] [CrossRef]
- Ramadoss, P.; Perdew, G.H. Use of 2-Azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a Probe to Determine the Relative Ligand Affinity of Human versus Mouse Aryl Hydrocarbon Receptor in Cultured Cells. Mol. Pharmacol. 2004, 66, 129–136. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Phillips, J.L.; Kerkvliet, N.I.; Tanguay, R.L.; Perdew, G.H.; Kolluri, S.K.; Bisson, W.H. A Structural Switch between Agonist and Antagonist Bound Conformations for a Ligand-Optimized Model of the Human Aryl Hydrocarbon Receptor Ligand Binding Domain. Biology 2014, 3, 645–669. [Google Scholar] [CrossRef] [PubMed]
- Bisson, W.H.; Koch, D.C.; O’Donnell, E.F.; Khalil, S.M.; Kerkvliet, N.I.; Tanguay, R.L.; Abagyan, R.; Kolluri, S.K. Modeling of the Aryl Hydrocarbon Receptor (AhR) Ligand Binding Domain and Its Utility in Virtual Ligand Screening to Predict New AhR Ligands. J. Med. Chem. 2009, 52, 5635–5641. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hankinson, O. Functional Involvement of the Brahma/SWI2-related Gene 1 Protein in Cytochrome P4501A1 Transcription Mediated by the Aryl Hydrocarbon Receptor Complex. J. Biol. Chem. 2002, 277, 11821–11827. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2005, 433, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Watzky, M.; Huard, S.; Juricek, L.; Dairou, J.; Chauvet, C.; Coumoul, X.; Letessier, A.; Miotto, B. Hexokinase 2 is a transcriptional target and a positive modulator of AHR signalling. Nucleic Acids Res. 2022, 50, 5545–5564. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.R.; Karchner, S.I.; Allan, L.L.; Pollenz, R.S.; Tanguay, R.L.; Jenny, M.J.; Sherr, D.H.; Hahn, M.E. Repression of Aryl Hydrocarbon Receptor (AHR) Signaling by AHR Repressor: Role of DNA Binding and Competition for AHR Nuclear Translocator. Mol. Pharmacol. 2008, 73, 387–398. [Google Scholar] [CrossRef]
- Ahmed, S.; Bott, D.; Gomez, A.; Tamblyn, L.; Rasheed, A.; Cho, T.; MacPherson, L.; Sugamori, K.S.; Yang, Y.; Grant, D.M.; et al. Loss of the Mono-ADP-ribosyltransferase, Tiparp, Increases Sensitivity to Dioxin-induced Steatohepatitis and Lethality. J. Biol. Chem. 2015, 290, 16824–16840. [Google Scholar] [CrossRef]
- Grimaldi, G.; Rajendra, S.; Matthews, J. The aryl hydrocarbon receptor regulates the expression of TIPARP and its cis long non-coding RNA, TIPARP-AS1. Biochem. Biophys. Res. Commun. 2018, 495, 2356–2362. [Google Scholar] [CrossRef]
- Hutin, D.; Tamblyn, L.; Gomez, A.; Grimaldi, G.; Soedling, H.; Cho, T.; Ahmed, S.; Lucas, C.; Kanduri, C.; Grant, D.M.; et al. Hepatocyte-Specific Deletion of TIPARP, a Negative Regulator of the Aryl Hydrocarbon Receptor, Is Sufficient to Increase Sensitivity to Dioxin-Induced Wasting Syndrome. Toxicol. Sci. 2018, 165, 347–360. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, J.; Dong, L.; Lin, H. TiPARP forms nuclear condensates to degrade HIF-1α and suppress tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 13447–13456. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Cai, X.; Guo, Y.; Xu, M.; Tian, J.; Locker, J.; Xie, W. Constitutive Activation of the Human Aryl Hydrocarbon Receptor in Mice Promotes Hepatocarcinogenesis Independent of Its Coactivator Gadd45b. Toxicol. Sci. 2019, 167, 581–592. [Google Scholar] [CrossRef]
- Weinert, B.T.; Narita, T.; Satpathy, S.; Srinivasan, B.; Hansen, B.K.; Schölz, C.; Hamilton, W.B.; Zucconi, B.E.; Wang, W.W.; Liu, W.R.; et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018, 174, 231–244.e12. [Google Scholar] [CrossRef] [PubMed]
- Nakano, N.; Sakata, N.; Katsu, Y.; Nochise, D.; Sato, E.; Takahashi, Y.; Yamaguchi, S.; Haga, Y.; Ikeno, S.; Motizuki, M.; et al. Dissociation of the AhR/ARNT complex by TGF-β/Smad signaling represses CYP1A1 gene expression and inhibits benze[a]pyrene-mediated cytotoxicity. J. Biol. Chem. 2020, 295, 9033–9051. [Google Scholar] [CrossRef]
- Solaimani, P.; Damoiseaux, R.; Hankinson, O. Genome-Wide RNAi High-Throughput Screen Identifies Proteins Necessary for the AHR-Dependent Induction of CYP1A1 by 2,3,7,8-Tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2013, 136, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Solaimani, P.; Wang, F.; Hankinson, O. SIN3A, Generally Regarded as a Transcriptional Repressor, Is Required for Induction of Gene Transcription by the Aryl Hydrocarbon Receptor. J. Biol. Chem. 2014, 289, 33655–33662. [Google Scholar] [CrossRef] [PubMed]
- Bourner, L.A.; Muro, I.; Cooper, A.M.; Choudhury, B.K.; Bailey, A.O.; Russell, W.K.; Khanipov, K.; Golovko, G.; Wright, C.W. AhR promotes phosphorylation of ARNT isoform 1 in human T cell malignancies as a switch for optimal AhR activity. Proc. Natl. Acad. Sci. USA 2022, 119, e2114336119. [Google Scholar] [CrossRef] [PubMed]
- Elson, D.J.; Nguyen, B.D.; Wood, R.; Zhang, Y.; Puig-Sanvicens, V.; Kolluri, S.K. The cyclin-dependent kinase inhibitor p27 Kip1 interacts with the aryl hydrocarbon receptor and negatively regulates its transcriptional activity. FEBS Lett. 2022, 596, 2056–2071. [Google Scholar] [CrossRef]
- Orlando, S.; Gallastegui, E.; Besson, A.; Abril, G.; Aligué, R.; Pujol, M.J.; Bachs, O. p27Kip1and p21Cip1collaborate in the regulation of transcription by recruiting cyclin–Cdk complexes on the promoters of target genes. Nucleic Acids Res. 2015, 43, 6860–6873. [Google Scholar] [CrossRef]
- Pippa, R.; Espinosa, L.; Gundem, G.; García-Escudero, R.; Dominguez, A.; Orlando, S.; Gallastegui, E.; Saiz, C.; Besson, A.; Pujol, M.J.; et al. p27Kip1 represses transcription by direct interaction with p130/E2F4 at the promoters of target genes. Oncogene 2012, 31, 4207–4220. [Google Scholar] [CrossRef]
- Puga, A.; Barnes, S.J.; Dalton, T.P.; Chang, C.-Y.; Knudsen, E.S.; Maier, M.A. Aromatic Hydrocarbon Receptor Interaction with the Retinoblastoma Protein Potentiates Repression of E2F-dependent Transcription and Cell Cycle Arrest. J. Biol. Chem. 2000, 275, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Elferink, C.J.; Ge, N.-L.; Levine, A. Maximal Aryl Hydrocarbon Receptor Activity Depends on an Interaction with the Retinoblastoma Protein. Mol. Pharmacol. 2001, 59, 664–673. [Google Scholar] [CrossRef]
- Perearnau, A.; Orlando, S.; Islam, A.B.; Gallastegui, E.; Martínez, J.; Jordan, A.; Bigas, A.; Aligué, R.; Pujol, M.J.; Bachs, O. p27Kip1, PCAF and PAX5 cooperate in the transcriptional regulation of specific target genes. Nucleic Acids Res. 2017, 45, 5086–5099. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Luna, M.; Aguasca, M.; Perearnau, A.; Serratosa, J.; Martínez-Balbas, M.; Pujol, M.J.; Bachs, O. PCAF regulates the stability of the transcriptional regulator and cyclin-dependent kinase inhibitor p27Kip1. Nucleic Acids Res. 2012, 40, 6520–6533. [Google Scholar] [CrossRef]
- Yang, X.; Liu, D.; Murray, T.J.; Mitchell, G.C.; Hesterman, E.V.; Karchner, S.I.; Merson, R.R.; Hahn, M.E.; Sherr, D.H. The aryl hydrocarbon receptor constitutively represses c-myc transcription in human mammary tumor cells. Oncogene 2005, 24, 7869–7881. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Al-Saigh, S.; Matthews, J. FOXA1 Is Essential for Aryl Hydrocarbon Receptor–Dependent Regulation of Cyclin G2. Mol. Cancer Res. 2012, 10, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Kolluri, S.K.; Weiss, C.; Koff, A.; Göttlicher, M. p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev. 1999, 13, 1742–1753. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Jang, H.S.; Pearce, M.; Kerkvliet, N.I.; Kolluri, S.K. The aryl hydrocarbon receptor is required for induction of p21cip1/waf1 expression and growth inhibition by SU5416 in hepatoma cells. Oncotarget 2017, 8, 25211–25225. [Google Scholar] [CrossRef]
- Sartor, M.A.; Schnekenburger, M.; Marlowe, J.L.; Reichard, J.F.; Wang, Y.; Fan, Y.; Ma, C.; Karyala, S.; Halbleib, D.; Liu, X.; et al. Genomewide Analysis of Aryl Hydrocarbon Receptor Binding Targets Reveals an Extensive Array of Gene Clusters that Control Morphogenetic and Developmental Programs. Environ. Health Perspect. 2009, 117, 1139–1146. [Google Scholar] [CrossRef]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl Hydrocarbon Receptor Antagonists Promote the Expansion of Human Hematopoietic Stem Cells. Science 2010, 329, 1345–1348, Correction in Science 2011, 332, 664. [Google Scholar] [CrossRef]
- Marin, N.M.; Merino, J.M.; Alvarez-Barrientos, A.; Patel, D.P.; Takahashi, S.; Sancho, J.M.G.; Gandolfo, P.; Rios, R.M.; Munoz, A.; Gonzalez, F.J.; et al. Aryl Hydrocarbon Receptor Promotes Liver Polyploidization and Inhibits PI3K, ERK, and Wnt/β-Catenin Signaling. iScience 2018, 4, 44–63. [Google Scholar] [CrossRef] [PubMed]
- Kawajiri, K.; Kobayashi, Y.; Ohtake, F.; Ikuta, T.; Matsushima, Y.; Mimura, J.; Pettersson, S.; Pollenz, R.S.; Sakaki, T.; Hirokawa, T.; et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in Apc Min/+ mice with natural ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 13481–13486. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Davidson, L.A.; Hensel, M.; Yoon, G.; Landrock, K.; Allred, C.; Jayaraman, A.; Ivanov, I.; Safe, S.H.; Chapkin, R.S. Loss of Aryl Hydrocarbon Receptor Promotes Colon Tumorigenesis in ApcS580/+; KrasG12D/+ Mice. Mol. Cancer Res. 2021, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Fan, Y.; Karyala, S.; Schwemberger, S.; Tomlinson, C.R.; Sartor, M.A.; Puga, A. Ligand-Independent Regulation of Transforming Growth Factor β1 Expression and Cell Cycle Progression by the Aryl Hydrocarbon Receptor. Mol. Cell. Biol. 2007, 27, 6127–6139. [Google Scholar] [CrossRef]
- Sarić, N.; Selby, M.; Ramaswamy, V.; Kool, M.; Stockinger, B.; Hogstrand, C.; Williamson, D.; Marino, S.; Taylor, M.D.; Clifford, S.C.; et al. The AHR pathway represses TGFβ-SMAD3 signalling and has a potent tumour suppressive role in SHH medulloblastoma. Sci. Rep. 2020, 10, 148. [Google Scholar] [CrossRef]
- Wu, P.-Y.; Liao, Y.-F.; Juan, H.-F.; Huang, H.-C.; Wang, B.-J.; Lu, Y.-L.; Yu, I.-S.; Shih, Y.-Y.; Jeng, Y.-M.; Hsu, W.-M.; et al. Aryl hydrocarbon receptor downregulates MYCN expression and promotes cell differentiation of neuroblastoma. PLoS ONE 2014, 9, e88795. [Google Scholar] [CrossRef]
- Cheng, J.; Li, W.; Kang, B.; Zhou, Y.; Song, J.; Dan, S.; Yang, Y.; Zhang, X.; Li, J.; Yin, S.; et al. Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat. Commun. 2015, 6, 7209. [Google Scholar] [CrossRef]
- Bunaciu, R.P.; Yen, A. Activation of the Aryl Hydrocarbon Receptor AhR Promotes Retinoic Acid–Induced Differentiation of Myeloblastic Leukemia Cells by Restricting Expression of the Stem Cell Transcription Factor Oct4. Cancer Res. 2011, 71, 2371–2380. [Google Scholar] [CrossRef]
- Romine, K.A.; Nechiporuk, T.; Bottomly, D.; Jeng, S.; McWeeney, S.K.; Kaempf, A.; Corces, M.R.; Majeti, R.; Tyner, J.W. Monocytic Differentiation and AHR Signaling as Primary Nodes of BET Inhibitor Response in Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 518–531. [Google Scholar] [CrossRef]
- Luecke-Johansson, S.; Gralla, M.; Rundqvist, H.; Ho, J.C.; Johnson, R.S.; Gradin, K.; Poellinger, L. A Molecular Mechanism to Switch the Aryl Hydrocarbon Receptor from a Transcription Factor to an E3 Ubiquitin Ligase. Mol. Cell. Biol. 2017, 37, e00630-16. [Google Scholar] [CrossRef]
- Huang, B.; Butler, R.; Miao, Y.; Dai, Y.; Wu, W.; Su, W.; Fujii-Kuriyama, Y.; Warner, M.; Gustafsson, J. Dysregulation of Notch and ERα signaling in AhR −/− male mice. Proc. Natl. Acad. Sci. USA 2016, 113, 11883–11888. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, J.L.; Knudsen, E.S.; Schwemberger, S.; Puga, A. The Aryl Hydrocarbon Receptor Displaces p300 from E2F-dependent Promoters and Represses S Phase-specific Gene Expression. J. Biol. Chem. 2004, 279, 29013–29022. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.D.; Stevens, B.L.; Elson, D.J.; Finlay, D.; Gamble, J.T.; Kopparapu, P.R.; Tanguay, R.L.; Buermeyer, A.B.; Kerkvliet, N.I.; Kolluri, S.K. 11-Cl-BBQ, a select modulator of AhR-regulated transcription, suppresses lung cancer cell growth via activation of p53 and p27 Kip1. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salguero, P.; Pineau, T.; Hilbert, D.M.; McPhail, T.; Lee, S.S.T.; Kimura, S.; Nebert, D.W.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Immune System Impairment and Hepatic Fibrosis in Mice Lacking the Dioxin-Binding Ah Receptor. Science 1995, 268, 722–726. [Google Scholar] [CrossRef]
- Schmidt, J.V.; Su, G.H.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6736. [Google Scholar] [CrossRef]
- Abbott, B.D.; Schmid, J.E.; Pitt, J.A.; Buckalew, A.R.; Wood, C.R.; Held, G.A.; Diliberto, J.J. Adverse Reproductive Outcomes in the Transgenic Ah Receptor-Deficient Mouse. Toxicol. Appl. Pharmacol. 1999, 155, 62–70. [Google Scholar] [CrossRef]
- Baba, T.; Mimura, J.; Nakamura, N.; Harada, N.; Yamamoto, M.; Morohashi, K.-I.; Fujii-Kuriyama, Y. Intrinsic Function of the Aryl Hydrocarbon (Dioxin) Receptor as a Key Factor in Female Reproduction. Mol. Cell. Biol. 2005, 25, 10040–10051. [Google Scholar] [CrossRef]
- Kerkvliet, N.I. TCDD: An Environmental Immunotoxicant Reveals a Novel Pathway of Immunoregulation—A 30-Year Odyssey. Toxicol. Pathol. 2011, 40, 138–142. [Google Scholar] [CrossRef]
- Gonzalez, F.J.; Fernandez-Salguero, P. The aryl hydrocarbon receptor. Studies using the AHR-null mice. Drug Metab. Dispos. 1998, 26, 1194–1198. [Google Scholar]
- Fritz, W.A.; Lin, T.-M.; Cardiff, R.D.; Peterson, R.E. The aryl hydrocarbon receptor inhibits prostate carcinogenesis in TRAMP mice. Carcinogenesis 2007, 28, 497–505. [Google Scholar] [CrossRef]
- Fritz, W.A.; Lin, T.-M.; Peterson, R.E. The aryl hydrocarbon receptor (AhR) inhibits vanadate-induced vascular endothelial growth factor (VEGF) production in TRAMP prostates. Carcinogenesis 2008, 29, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Fritz, W.A.; Lin, T.-M.; Safe, S.; Moore, R.W.; Peterson, R.E. The selective aryl hydrocarbon receptor modulator 6-methyl-1,3,8-trichlorodibenzofuran inhibits prostate tumor metastasis in TRAMP mice. Biochem. Pharmacol. 2009, 77, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Aloufi, N.; Traboulsi, H.; Trempe, J.-F.; Eidelman, D.H.; Baglole, C.J. Endogenous regulation of the Akt pathway by the aryl hydrocarbon receptor (AhR) in lung fibroblasts. Sci. Rep. 2021, 11, 23189. [Google Scholar] [CrossRef] [PubMed]
- Nacarino-Palma, A.; Rejano-Gordillo, C.M.; González-Rico, F.J.; Ordiales-Talavero, A.; Román, C.; Cuadrado, M.; Bustelo, X.R.; Merino, J.M.; Fernández-Salguero, P.M. Loss of Aryl Hydrocarbon Receptor Favors K-RasG12D-Driven Non-Small Cell Lung Cancer. Cancers 2021, 13, 4071. [Google Scholar] [CrossRef] [PubMed]
- Nothdurft, S.; Thumser-Henner, C.; Breitenbücher, F.; Okimoto, R.A.; Dorsch, M.; Opitz, C.A.; Sadik, A.; Esser, C.; Hölzel, M.; Asthana, S.; et al. Functional screening identifies aryl hydrocarbon receptor as suppressor of lung cancer metastasis. Oncogenesis 2020, 9, 102. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Li, C.-H.; Cheng, Y.-W.; Lee, C.-C.; Liao, P.-L.; Lin, C.-H.; Huang, S.-H.; Kang, J.-J. The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci. Rep. 2017, 7, srep41927. [Google Scholar] [CrossRef]
- Lee, C.-C.; Yang, W.-H.; Li, C.-H.; Cheng, Y.-W.; Tsai, C.-H.; Kang, J.-J. Ligand independent aryl hydrocarbon receptor inhibits lung cancer cell invasion by degradation of Smad4. Cancer Lett. 2016, 376, 211–217. [Google Scholar] [CrossRef]
- Wang, H.-C.; Zhou, Y.; Huang, S.-K. SHP-2 phosphatase controls aryl hydrocarbon receptor-mediated ER stress response in mast cells. Arch. Toxicol. 2017, 91, 1739–1748. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Pennington, J.M.; Wang, X.; Rohlman, D.; Punj, S.; Löhr, C.V.; Newman, M.T.; Kolluri, S.K.; Kerkvliet, N.I. Activation of the Aryl Hydrocarbon Receptor by 10-Cl-BBQ Prevents Insulitis and Effector T Cell Development Independently of Foxp3+ Regulatory T Cells in Nonobese Diabetic Mice. J. Immunol. 2016, 196, 264–273. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Pennington, J.M.; Tilton, S.; Wang, X.; Marshall, N.B.; Rohlman, D.; Funatake, C.; Punj, S.; O’Donnell, E.; Yu, Z.; et al. AhR activation increases IL-2 production by alloreactive CD4+T cells initiating the differentiation of mucosal-homing Tim3+Lag3+Tr1 cells. Eur. J. Immunol. 2017, 47, 1989–2001. [Google Scholar] [CrossRef]
- Punj, S.; Kopparapu, P.; Jang, H.S.; Phillips, J.L.; Pennington, J.; Rohlman, D.; O’Donnell, E.; Iversen, P.L.; Kolluri, S.K.; Kerkvliet, N.I. Benzimidazoisoquinolines: A New Class of Rapidly Metabolized Aryl Hydrocarbon Receptor (AhR) Ligands that Induce AhR-Dependent Tregs and Prevent Murine Graft-Versus-Host Disease. PLoS ONE 2014, 9, e88726. [Google Scholar] [CrossRef] [PubMed]
- Arnason, T.; Pino, M.S.; Yilmaz, O.; Kirley, S.D.; Rueda, B.R.; Chung, D.C.; Zukerberg, L.R. Cables1 is a tumor suppressor gene that regulates intestinal tumor progression in ApcMin mice. Cancer Biol. Ther. 2013, 14, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Park, H.R.; Du, Y.; Li, Z.; Cheng, K.; Sun, S.Y.; Li, Z.; Fu, H.; Khuri, F.R. Cables1 complex couples survival signaling to the cell death machinery. Cancer Res. 2015, 75, 147–158. [Google Scholar] [CrossRef]
- Han, H.; Davidson, L.A.; Fan, Y.; Goldsby, J.S.; Yoon, G.; Jin, U.; Wright, G.A.; Landrock, K.K.; Weeks, B.R.; Wright, R.C.; et al. Loss of aryl hydrocarbon receptor potentiates FoxM1 signaling to enhance self-renewal of colonic stem and progenitor cells. EMBO J. 2020, 39, e104319. [Google Scholar] [CrossRef]
- Garcia-Villatoro, E.L.; DeLuca, J.A.A.; Callaway, E.S.; Allred, K.F.; Davidson, L.A.; Hensel, M.E.; Menon, R.; Ivanov, I.; Safe, S.H.; Jayaraman, A.; et al. Effects of high-fat diet and intestinal aryl hydrocarbon receptor deletion on colon carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G451–G463. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Davidson, L.A.; Fan, Y.-Y.; Landrock, K.K.; Jayaraman, A.; Safe, S.H.; Chapkin, R.S. Loss of aryl hydrocarbon receptor suppresses the response of colonic epithelial cells to IL22 signaling by upregulating SOCS3. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G93–G106. [Google Scholar] [CrossRef]
- Ikuta, T.; Kobayashi, Y.; Fujii-Kuriyama, Y.; Kawajiri, K. Aryl Hydrocarbon Receptor Suppresses Cecal Carcinogenesis. Fifty Years Cytochrome P450 Res. 2014, 1, 233–245. [Google Scholar] [CrossRef]
- Matoba, H.; Takamoto, M.; Fujii, C.; Kawakubo, M.; Kasuga, E.; Matsumura, T.; Natori, T.; Misawa, K.; Taniguchi, S.; Nakayama, J. Cecal Tumorigenesis in Aryl Hydrocarbon Receptor–Deficient Mice Depends on Cecum-Specific Mitogen-Activated Protein Kinase Pathway Activation and Inflammation. Am. J. Pathol. 2020, 190, 453–468. [Google Scholar] [CrossRef]
- Yakkundi, P.; Gonsalves, E.; Galou-Lameyer, M.; Selby, M.J.; Chan, W.K. Aryl hydrocarbon receptor acts as a tumor suppressor in a syngeneic MC38 colon carcinoma tumor model. Hypoxia 2019, 7, 1–16. [Google Scholar] [CrossRef]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous Stimuli Maintain Intraepithelial Lymphocytes via Aryl Hydrocarbon Receptor Activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef]
- Shah, K.; Maradana, M.R.; Delàs, M.J.; Metidji, A.; Graelmann, F.; Llorian, M.; Chakravarty, P.; Li, Y.; Tolaini, M.; Shapiro, M.; et al. Cell-intrinsic Aryl Hydrocarbon Receptor signalling is required for the resolution of injury-induced colonic stem cells. Nat. Commun. 2022, 13, 1827. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Safe, S.; Jayaraman, A.; Chapkin, R.S. Diet–Host–Microbiota Interactions Shape Aryl Hydrocarbon Receptor Ligand Production to Modulate Intestinal Homeostasis. Annu. Rev. Nutr. 2021, 41, 455–478. [Google Scholar] [CrossRef] [PubMed]
- Furumatsu, K.; Nishiumi, S.; Kawano, Y.; Ooi, M.; Yoshie, T.; Shiomi, Y.; Kutsumi, H.; Ashida, H.; Fujii-Kuriyama, Y.; Azuma, T.; et al. A Role of the Aryl Hydrocarbon Receptor in Attenuation of Colitis. Dig. Dis. Sci. 2011, 56, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362.e5. [Google Scholar] [CrossRef]
- Yang, Y.; Osorio, D.; Davidson, L.A.; Han, H.; Mullens, D.A.; Jayaraman, A.; Safe, S.; Ivanov, I.; Cai, J.J.; Chapkin, R.S. Single-cell RNA Sequencing Reveals How the Aryl Hydrocarbon Receptor Shapes Cellular Differentiation Potency in the Mouse Colon. Cancer Prev. Res. 2022, 15, 17–28. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Ellis, T.; Markant, S.L.; Read, T.-A.; Kessler, J.D.; Bourboulas, M.; Schüller, U.; Machold, R.; Fishell, G.; Rowitch, D.H.; et al. Medulloblastoma Can Be Initiated by Deletion of Patched in Lineage-Restricted Progenitors or Stem Cells. Cancer Cell 2008, 14, 135–145. [Google Scholar] [CrossRef]
- Wu, P.-Y.; Yu, I.-S.; Lin, Y.-C.; Chang, Y.-T.; Chen, C.-C.; Lin, K.-H.; Tseng, T.-H.; Kargren, M.; Tai, Y.-L.; Shen, T.-L.; et al. Activation of Aryl Hydrocarbon Receptor by Kynurenine Impairs Progression and Metastasis of Neuroblastoma. Cancer Res. 2019, 79, 5550–5562. [Google Scholar] [CrossRef]
- Wu, P.-Y.; Chuang, P.-Y.; Chang, G.-D.; Chan, Y.-Y.; Tsai, T.-C.; Wang, B.-J.; Lin, K.-H.; Hsu, W.-M.; Liao, Y.-F.; Lee, H. Novel Endogenous Ligands of Aryl Hydrocarbon Receptor Mediate Neural Development and Differentiation of Neuroblastoma. ACS Chem. Neurosci. 2019, 10, 4031–4042. [Google Scholar] [CrossRef]
- Jin, U.-H.; Karki, K.; Cheng, Y.; Michelhaugh, S.K.; Mittal, S.; Safe, S. The aryl hydrocarbon receptor is a tumor suppressor–like gene in glioblastoma. J. Biol. Chem. 2019, 294, 11342–11353. [Google Scholar] [CrossRef]
- Moreno-Marín, N.; Barrasa, E.; Morales-Hernández, A.; Paniagua, B.; Blanco-Fernández, G.; Merino, J.M.; Fernández-Salguero, P.M. Dioxin Receptor Adjusts Liver Regeneration After Acute Toxic Injury and Protects Against Liver Carcinogenesis. Sci. Rep. 2017, 7, 10420. [Google Scholar] [CrossRef]
- Khanal, T.; Choi, K.; Leung, Y.-K.; Wang, J.; Kim, D.; Janakiram, V.; Cho, S.-G.; Puga, A.; Ho, S.-M.; Kim, K. Loss of NR2E3 represses AHR by LSD1 reprogramming, is associated with poor prognosis in liver cancer. Sci. Rep. 2017, 7, 10662. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Boivin, G.P.; Knudsen, E.S.; Nebert, D.W.; Xia, Y.; Puga, A. The Aryl Hydrocarbon Receptor Functions as a Tumor Suppressor of Liver Carcinogenesis. Cancer Res. 2010, 70, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Koide, R.; Kulkeaw, K.; Tanaka, Y.; Swain, A.; Nakanishi, Y.; Sugiyama, D. Aryl Hydrocarbon Receptor Antagonist StemRegenin 1 Promotes the Expansion of Human Promyelocytic Leukemia Cell Line, NB4. Anticancer Res. 2016, 36, 3635–3643. [Google Scholar] [PubMed]
- Ly, M.; Rentas, S.; Vujovic, A.; Wong, N.; Moreira, S.; Xu, J.; Holzapfel, N.; Bhatia, S.; Tran, D.; Minden, M.D.; et al. Diminished AhR signaling drives human acute myeloid leukemia stem cell maintenance. Cancer Res. 2019, 79, 5799–5811. [Google Scholar] [CrossRef]
- Bunaciu, R.P.; Jensen, H.A.; MacDonald, R.J.; LaTocha, D.H.; Varner, J.D.; Yen, A. 6-Formylindolo(3,2-b)Carbazole (FICZ) Modulates the Signalsome Responsible for RA-Induced Differentiation of HL-60 Myeloblastic Leukemia Cells. PLoS ONE 2015, 10, e0135668. [Google Scholar] [CrossRef]
- Ibabao, C.N.; Bunaciu, R.P.; Schaefer, D.M.; Yen, A. The AhR agonist VAF347 augments retinoic acid-induced differentiation in leukemia cells. FEBS Open Bio 2015, 5, 308–318. [Google Scholar] [CrossRef]
- Contador-Troca, M.; Alvarez-Barrientos, A.; Merino, J.M.; Morales-Hernández, A.; Rodríguez, M.I.; Rey-Barroso, J.; Barrasa, E.; Cerezo-Guisado, M.I.; Catalina-Fernández, I.; Sáenz-Santamaría, J.; et al. Dioxin receptor regulates aldehyde dehydrogenase to block melanoma tumorigenesis and metastasis. Mol. Cancer 2015, 14, 148. [Google Scholar] [CrossRef]
- Contador-Troca, M.; Alvarez-Barrientos, A.; Barrasa, E.; Rico-Leo, E.M.; Catalina-Fernández, I.; Menacho-Márquez, M.; Bustelo, X.R.; Garcia-Borron, J.C.; Gómez-Durán, A.; Sáenz-Santamaría, J.; et al. The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis 2013, 34, 2683–2693. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Saili, K.S.; Koch, D.C.; Kopparapu, P.R.; Farrer, D.; Bisson, W.H.; Mathew, L.K.; Sengupta, S.; Kerkvliet, N.I.; Tanguay, R.L.; et al. The Anti-Inflammatory Drug Leflunomide Is an Agonist of the Aryl Hydrocarbon Receptor. PLoS ONE 2010, 5, e13128. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Kopparapu, P.R.; Koch, D.C.; Jang, H.S.; Phillips, J.L.; Tanguay, R.L.; Kerkvliet, N.I.; Kolluri, S.K. The Aryl Hydrocarbon Receptor Mediates Leflunomide-Induced Growth Inhibition of Melanoma Cells. PLoS ONE 2012, 7, e40926. [Google Scholar] [CrossRef]
- Safe, S.; Zhang, L. The Role of the Aryl Hydrocarbon Receptor (AhR) and Its Ligands in Breast Cancer. Cancers 2022, 14, 5574. [Google Scholar] [CrossRef] [PubMed]
- Belton, K.R.; Tian, Y.; Zhang, L.; Anitha, M.; Smith, P.B.; Perdew, G.H.; Patterson, A.D. Metabolomics Reveals Aryl Hydrocarbon Receptor Activation Induces Liver and Mammary Gland Metabolic Dysfunction in Lactating Mice. J. Proteome Res. 2018, 17, 1375–1382. [Google Scholar] [CrossRef]
- Wormke, M.; Stoner, M.; Saville, B.; Safe, S. Crosstalk between estrogen receptor α and the aryl hydrocarbon receptor in breast cancer cells involves unidirectional activation of proteasomes. FEBS Lett. 2000, 478, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Pearce, M.; O’Donnell, E.F.; Nguyen, B.D.; Truong, L.; Mueller, M.J.; Bisson, W.H.; Kerkvliet, N.I.; Tanguay, R.L.; Kolluri, S.K. Identification of a Raloxifene Analog That Promotes AhR-Mediated Apoptosis in Cancer Cells. Biology 2017, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, E.F., III; Jang, H.S.; Liefwalker, D.F.; Kerkvliet, N.I.; Kolluri, S.K. Discovery and Mechanistic Characterization of a Select Modulator of AhR-regulated Transcription (SMAhRT) with Anti-cancer Effects. Apoptosis 2021, 26, 307–322. [Google Scholar] [CrossRef]
- Elson, D.; Nguyen, B.; Bernales, S.; Chakravarty, J.; Jang, H.S.; Korjeff, N.; Zhang, Y.; Wilferd, S.; Castro, D.; Plaiser, C.; et al. Identification of Analog 523 as an Aryl hydrocarbon receptor agonist that induces apoptosis in triple-negative breast cancer. ACS Pharmacol. Transl. Sci. in review.
- Vogel, C.F.A.; Lazennec, G.; Kado, S.Y.; Dahlem, C.; He, Y.; Castaneda, A.; Ishihara, Y.; Vogeley, C.; Rossi, A.; Haarmann-Stemmann, T.; et al. Targeting the Aryl Hydrocarbon Receptor Signaling Pathway in Breast Cancer Development. Front. Immunol. 2021, 12, 625346. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Smith, Y.E.; Cao, Y.; Burrows, A.D.; Cross, R.S.N.; Ling, X.; Redvers, R.P.; Doherty, J.P.; Eckhardt, B.L.; Natoli, A.L.; et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Model. Mech. 2015, 8, 237–251. [Google Scholar] [CrossRef]
- Zudaire, E.; Cuesta, N.; Murty, V.; Woodson, K.; Adams, L.; Gonzalez, N.; Martínez, A.; Narayan, G.; Kirsch, I.; Franklin, W.; et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J. Clin. Investig. 2008, 118, 640–650. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2021, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, S.; Lutz, C.; Henneman, L.; Bhin, J.; Wong, K.; Siteur, B.; van Gerwen, B.; de Korte-Grimmerink, R.; Zafra, M.P.; Schatoff, E.M.; et al. In situ CRISPR-Cas9 base editing for the development of genetically engineered mouse models of breast cancer. EMBO J. 2020, 39, e102169. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, S.; de Ruiter, J.R.; Henneman, L.; Brambillasca, C.S.; Lutz, C.; Vaillant, F.; Ferrante, F.; Drenth, A.P.; van der Burg, E.; Siteur, B.; et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat. Commun. 2019, 10, 397. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.L.; Löhr, C.V.; Nguyen, B.D.; Buermeyer, A.B.; Kolluri, S.K. Loss of the aryl hydrocarbon receptor increases tumorigenesis in p53-deficient mice. Toxicol. Appl. Pharmacol. 2022, 454, 116191. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, C.; Chan, C.; Kang, W.; Sun, Y.; Emerson, R.; Robins, H.; Levine, A.J. The evolution of thymic lymphomas in p53 knockout mice. Genes Dev. 2014, 28, 2613–2620. [Google Scholar] [CrossRef]
- Drainas, A.P.; Lambuta, R.A.; Ivanova, I.; Serçin, Ö.; Sarropoulos, I.; Smith, M.L.; Efthymiopoulos, T.; Raeder, B.; Stütz, A.M.; Waszak, S.M.; et al. Genome-wide Screens Implicate Loss of Cullin Ring Ligase 3 in Persistent Proliferation and Genome Instability in TP53-Deficient Cells. Cell Rep. 2020, 31, 107465. [Google Scholar] [CrossRef]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef]
- Matikainen, T.; Perez, G.I.; Jurisicova, A.; Pru, J.K.; Schlezinger, J.J.; Ryu, H.Y.; Laine, J.; Sakai, T.; Korsmeyer, S.J.; Casper, R.F.; et al. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat. Genet. 2001, 28, 355–360. [Google Scholar] [CrossRef]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Nickerson, T.; Huynh, H.; Pollak, M. Insulin-like growth factor binding protein-3 induces apoptosis in MCF7 breast cancer cells. Biochem. Biophys. Res. Commun. 1997, 237, 690–693. [Google Scholar] [CrossRef]
- Kerkvliet, N.; Shepherd, D.M.; Baecher-Steppan, L. T Lymphocytes Are Direct, Aryl Hydrocarbon Receptor (AhR)-Dependent Targets of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD): AhR Expression in Both CD4+ and CD8+ T Cells Is Necessary for Full Suppression of a Cytotoxic T Lymphocyte Response by TCDD. Toxicol. Appl. Pharmacol. 2002, 185, 146–152. [Google Scholar] [CrossRef]
- Wang, Z.; Li, T.; Mao, C.; Liu, W.; Tao, Y. IL4I1-driven AHR signature: A new avenue for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Sadik, A.; Patterson, L.F.S.; Öztürk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfänder, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed]
- van den Eynde, B.J.; van Baren, N.; Baurain, J.F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef]
- Shin, J.H.; Zhang, L.; Murillo-Sauca, O.; Kim, J.; Kohrt, H.E.K.; Bui, J.D.; Sunwoo, J.B. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 12391–12396. [Google Scholar] [CrossRef]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof Is a Natural AhR Agonist that Resolves Skin Inflammation in Mice and Humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef]
- Bissonnette, R.; Gold, L.S.; Rubenstein, D.S.; Tallman, A.M.; Armstrong, A. Tapinarof in the treatment of psoriasis: A review of the unique mechanism of action of a novel therapeutic aryl hydrocarbon receptor–modulating agent. J. Am. Acad. Dermatol. 2021, 84, 1059–1067. [Google Scholar] [CrossRef]
- Lebwohl, M.G.; Gold, L.S.; Strober, B.; Papp, K.A.; Armstrong, A.W.; Bagel, J.; Kircik, L.; Ehst, B.; Hong, H.C.-H.; Soung, J.; et al. Phase 3 Trials of Tapinarof Cream for Plaque Psoriasis. N. Engl. J. Med. 2021, 385, 2219–2229. [Google Scholar] [CrossRef]
- Timothy, M.; Schroeder Jen. Ikena Oncology. Oral AHR Antagonist in Combination with Nivolumab in Patients with PD-1 Resistant Metastatic or Recurrent Head and Neck Cancer. NCT05472506. 25 July 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05472506 (accessed on 23 March 2023).
- National Library of Medicine. Bayer. A Study to Learn How Safe the Study Drug BAY 2416964 (AhR Inhibitor) in Combination with the Treatment Pembrolizumab Is, How This Combination Affects the Body, the Maximum Amount That Can Be Given, How It Moves into, through and out of the Body and Its Action against Advanced Solid Cancers in Adults. NCT04999202. 10 August 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04999202 (accessed on 23 March 2023).
- Wagner, J.E., Jr.; Brunstein, C.G.; Boitano, A.E.; DeFor, T.E.; McKenna, D.; Sumstad, D.; Blazar, B.R.; Tolar, J.; Le, C.; Jones, J.; et al. Phase I/II Trial of StemRegenin-1 Expanded Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell 2016, 18, 144–155. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. McMaster University. The Role of Dietary Tryptophan on Aryl Hydrocarbon Receptor Activation (Aryl-IMMUNE). NCT03059862. 25 February 2017–20 December 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03059862 (accessed on 20 March 2023).
- Koch, D.C.; Jang, H.S.; O’Donnell, E.F.; Punj, S.; Kopparapu, P.R.; Bisson, W.H.; Kerkvliet, N.I.; Kolluri, S.K. Anti-androgen flutamide suppresses hepatocellular carcinoma cell proliferation via the aryl hydrocarbon receptor mediated induction of transforming growth factor-β1. Oncogene 2015, 34, 6092–6104. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.-H.; Lee, S.-O.; Pfent, C.; Safe, S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer 2014, 14, 498. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.-H.; Lee, S.-O.; Safe, S. Aryl Hydrocarbon Receptor (AHR)-Active Pharmaceuticals Are Selective AHR Modulators in MDA-MB-468 and BT474 Breast Cancer Cells. J. Pharmacol. Exp. Ther. 2012, 343, 333–341. [Google Scholar] [CrossRef]
- Wincent, E.; Amini, N.; Luecke, S.; Glatt, H.; Bergman, J.; Crescenzi, C.; Rannug, A.; Rannug, U. The Suggested Physiologic Aryl Hydrocarbon Receptor Activator and Cytochrome P4501 Substrate 6-Formylindolo[3,2-b]carbazole Is Present in Humans. J. Biol. Chem. 2009, 284, 2690–2696. [Google Scholar] [CrossRef]
- Abron, J.D.; Singh, N.P.; Mishra, M.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, U.P. An endogenous aryl hydrocarbon receptor ligand, ITE, induces regulatory T cells and ameliorates experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G220–G230. [Google Scholar] [CrossRef]
- Caruso, J.A.; Campana, R.; Wei, C.; Su, C.-H.; Hanks, A.M.; Bornmann, W.G.; Keyomarsi, K. Indole-3-carbinol and its N-alkoxy derivatives preferentially target ERα-positive breast cancer cells. Cell Cycle 2014, 13, 2587–2599. [Google Scholar] [CrossRef]
- Weng, J.-R.; Tsai, C.-H.; Omar, H.A.; Sargeant, A.M.; Wang, D.; Kulp, S.K.; Shapiro, C.L.; Chen, C.-S. OSU-A9, a potent indole-3-carbinol derivative, suppresses breast tumor growth by targeting the Akt-NF-κB pathway and stress response signaling. Carcinogenesis 2009, 30, 1702–1709. [Google Scholar] [CrossRef]
- Mohammadi, S.; Seyedhosseini, F.S.; Behnampour, N.; Yazdani, Y. Indole-3-carbinol induces G1 cell cycle arrest and apoptosis through aryl hydrocarbon receptor in THP-1 monocytic cell line. J. Recept. Signal Transduct. 2017, 37, 506–514. [Google Scholar] [CrossRef]
- Rannug, A. 6-Formylindolo[3,2-b]carbazole, a Potent Ligand for the Aryl Hydrocarbon Receptor Produced Both Endogenously and by Microorganisms, can Either Promote or Restrain Inflammatory Responses. Front. Toxicol. 2022, 4, 775010. [Google Scholar] [CrossRef]
- Garcia, G.R.; Bugel, S.M.; Truong, L.; Spagnoli, S.; Tanguay, R.L. AHR2 required for normal behavioral responses and proper development of the skeletal and reproductive systems in zebrafish. PLoS ONE 2018, 13, e0193484. [Google Scholar] [CrossRef] [PubMed]
- Goodale, B.C.; La Du, J.K.; Bisson, W.H.; Janszen, D.B.; Waters, K.M.; Tanguay, R.L. AHR2 Mutant Reveals Functional Diversity of Aryl Hydrocarbon Receptors in Zebrafish. PLoS ONE 2012, 7, e29346. [Google Scholar] [CrossRef] [PubMed]
- Shankar, P.; Dasgupta, S.; Hahn, M.E.; Tanguay, R.L. A Review of the Functional Roles of the Zebrafish Aryl Hydrocarbon Receptors. Toxicol. Sci. 2020, 178, 215–238. [Google Scholar] [CrossRef] [PubMed]
- Bill, B.R.; Petzold, A.M.; Clark, K.J.; Schimmenti, L.A.; Ekker, S.C. A Primer for Morpholino Use in Zebrafish. Zebrafish 2009, 6, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef]
- Zhao, B.; DeGroot, D.E.; Hayashi, A.; He, G.; Denison, M.S. CH223191 Is a Ligand-Selective Antagonist of the Ah (Dioxin) Receptor. Toxicol. Sci. 2010, 117, 393–403. [Google Scholar] [CrossRef]
- Kim, S.-H.; Henry, E.C.; Kim, D.-K.; Shin, K.J.; Han, M.S.; Lee, T.G.; Kang, J.-K.; Gasiewicz, T.A.; Ryu, S.H.; Suh, P.-G. Novel Compound 2-Methyl-2H-pyrazole-3-carboxylic Acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) Prevents 2,3,7,8-TCDD-Induced Toxicity by Antagonizing the Aryl Hydrocarbon Receptor. Mol. Pharmacol. 2006, 69, 1871–1878. [Google Scholar] [CrossRef]
- Zgheib, E.; Limonciel, A.; Jiang, X.; Wilmes, A.; Wink, S.; Van De Water, B.; Kopp-Schneider, A.; Bois, F.Y.; Jennings, P. Investigation of Nrf2, AhR and ATF4 Activation in Toxicogenomic Databases. Front. Genet. 2018, 9, 429. [Google Scholar] [CrossRef]
- Ohoka, N.; Tsuji, G.; Shoda, T.; Fujisato, T.; Kurihara, M.; Demizu, Y.; Naito, M. Development of Small Molecule Chimeras That Recruit AhR E3 Ligase to Target Proteins. ACS Chem. Biol. 2019, 14, 2822–2832. [Google Scholar] [CrossRef]
- Tian, X.; Ahsan, N.; Lulla, A.; Lev, A.; Abbosh, P.; Dicker, D.T.; Zhang, S.; El-Deiry, W.S. P53-independent partial restoration of the p53 pathway in tumors with mutated p53 through ATF4 transcriptional modulation by ERK1/2 and CDK9. Neoplasia 2021, 23, 304–325. [Google Scholar] [CrossRef]
- Sharma, K.; Vu, T.; Cook, W.; Naseri, M.; Zhan, K.; Nakajima, W.; Harada, H. p53-independent Noxa induction by cisplatin is regulated by ATF3/ATF4 in head and neck squamous cell carcinoma cells. Mol. Oncol. 2018, 12, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Jin, U.-H.; Park, H.; Chapkin, R.; Jayaraman, A. Aryl Hydrocarbon Receptor (AHR) Ligands as Selective AHR Modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; De Iuliis, G.N.; McCluskey, A.; Sakoff, J.A. A novel naphthalimide that selectively targets breast cancer via the arylhydrocarbon receptor pathway. Sci. Rep. 2020, 10, 13978. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elson, D.J.; Kolluri, S.K. Tumor-Suppressive Functions of the Aryl Hydrocarbon Receptor (AhR) and AhR as a Therapeutic Target in Cancer. Biology 2023, 12, 526. https://doi.org/10.3390/biology12040526
Elson DJ, Kolluri SK. Tumor-Suppressive Functions of the Aryl Hydrocarbon Receptor (AhR) and AhR as a Therapeutic Target in Cancer. Biology. 2023; 12(4):526. https://doi.org/10.3390/biology12040526
Chicago/Turabian StyleElson, Daniel J., and Siva K. Kolluri. 2023. "Tumor-Suppressive Functions of the Aryl Hydrocarbon Receptor (AhR) and AhR as a Therapeutic Target in Cancer" Biology 12, no. 4: 526. https://doi.org/10.3390/biology12040526
APA StyleElson, D. J., & Kolluri, S. K. (2023). Tumor-Suppressive Functions of the Aryl Hydrocarbon Receptor (AhR) and AhR as a Therapeutic Target in Cancer. Biology, 12(4), 526. https://doi.org/10.3390/biology12040526